DECR Deficiency (mitochondrial 2,4-dienoyl‑CoA reductase deficiency): Disease Characteristics Research Report
Executive summary
“Mitochondrial 2,4‑dienoyl‑CoA reductase (DECR) deficiency” is an extremely rare inborn error of metabolism classically defined biochemically by impaired oxidation of polyunsaturated fatty acids (PUFAs) with accumulation of the acylcarnitine C10:2 (decadienoylcarnitine) and low DECR enzymatic activity. In the best-characterized modern molecular cases, the apparent DECR defect was secondary to autosomal recessive NADK2 deficiency, which causes mitochondrial NADP(H) deficiency and thereby disables NADPH-dependent mitochondrial enzymes including DECR and AASS (lysine pathway), producing a combined signature of C10:2 elevation + hyperlysinemia and severe neurometabolic disease. (houten2014mitochondrialnadp(h)deficiency pages 9-10, houten2014mitochondrialnadp(h)deficiency pages 5-7, houten2014mitochondrialnadp(h)deficiency pages 1-2)
A primary, Mendelian DECR1 (DECR)-mutant human disorder is suggested by historical biochemistry-first reports and by strong mouse genetic evidence, but coding DECR1 mutations were excluded in the NADK2-related human cases described in the key Human Molecular Genetics report, leaving the extent of confirmed human DECR1‑biallelic disease incompletely resolved in the tool-accessible literature used here. (houten2014mitochondrialnadp(h)deficiency pages 4-5, houten2014mitochondrialnadp(h)deficiency pages 5-7)
1. Disease information
What is the disease?
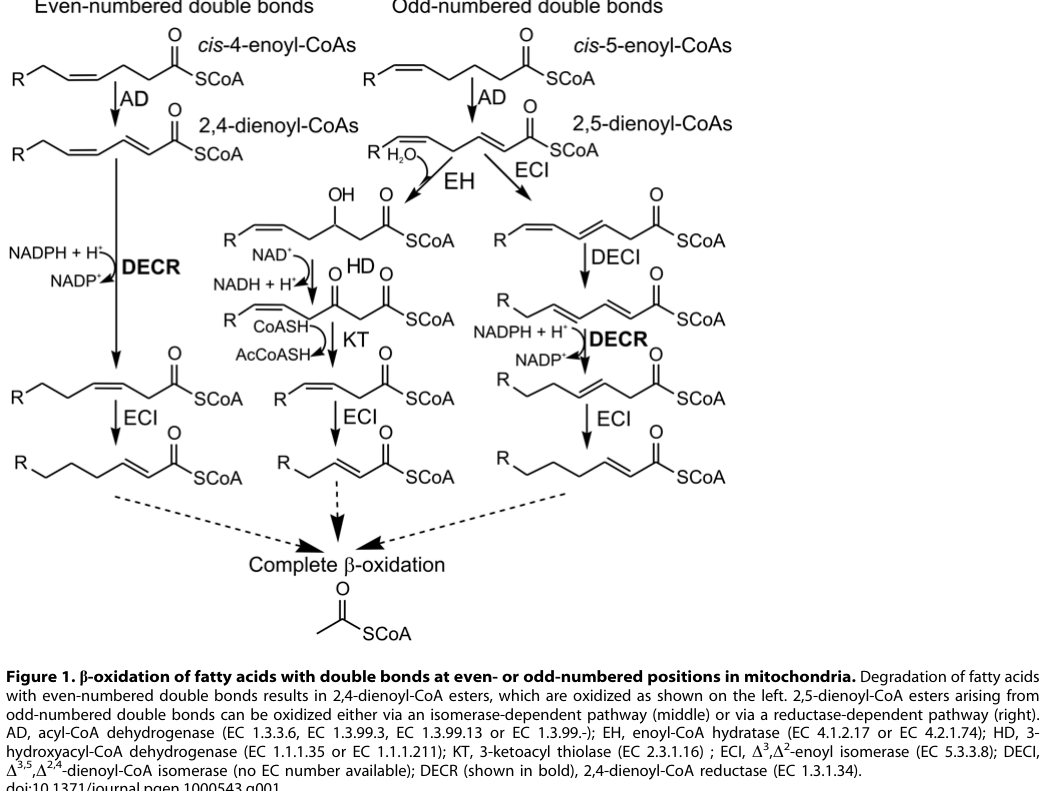
DECR deficiency refers to deficient activity of mitochondrial 2,4‑dienoyl‑CoA reductase (DECR), an auxiliary enzyme required for complete mitochondrial β‑oxidation of polyunsaturated fatty acids by reducing 2,4‑dienoyl‑CoA intermediates. Loss of activity leads to accumulation of characteristic PUFA-derived intermediates (detected as C10:2 acylcarnitine) and clinical decompensation under metabolic stress. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
Key identifiers (OMIM/Orphanet/MeSH/MONDO)
Within the tool-accessible corpus for this run, I could not reliably retrieve OMIM/Orphanet/MONDO identifiers for “DECR deficiency/DECR1 deficiency.” Accordingly, identifier mapping should be validated directly in OMIM/Orphanet/MONDO during knowledge-base curation.
Common synonyms / alternative names
- Mitochondrial 2,4‑dienoyl‑CoA reductase deficiency
- DECR deficiency
- (Biochemical signature) C10:2 (decadienoyl)carnitine elevation suggestive of impaired PUFA oxidation (wanders2019translationalmetabolisma pages 3-3, houten2014mitochondrialnadp(h)deficiency pages 4-5)
Evidence sources
Evidence is derived from: * Individual patient reports/series with deep biochemical phenotyping and genetics (notably NADK2-associated cases). (houten2014mitochondrialnadp(h)deficiency pages 4-5, houten2014mitochondrialnadp(h)deficiency pages 5-7, houten2014mitochondrialnadp(h)deficiency pages 1-2) * Aggregated biochemical screening considerations (newborn screening marker interpretation) discussed within primary case literature. (houten2014mitochondrialnadp(h)deficiency pages 10-12) * A mechanistic Decr knockout mouse model that supports pathway understanding and biomarker interpretation. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
2. Etiology
Disease causal factors
Genetic causes (current evidence)
1) Secondary DECR deficiency due to NADK2 deficiency (autosomal recessive) * A homozygous nonsense NADK2 variant (reported as c.1018C>T; p.R340X) caused mitochondrial NADP(H) deficiency, which in turn impaired NADPH-dependent mitochondrial enzymes including DECR and AASS (lysine pathway), explaining C10:2 elevation plus hyperlysinemia. (houten2014mitochondrialnadp(h)deficiency pages 5-7) * In this context, the DECR biochemical phenotype is downstream of NADK2 and reflects combined mitochondrial redox cofactor deficiency rather than a structural DECR1 defect. (houten2014mitochondrialnadp(h)deficiency pages 9-10, houten2014mitochondrialnadp(h)deficiency pages 10-12)
2) Primary DECR deficiency (putative DECR1-related) * Historical biochemical cases of “DECR deficiency” predate current sequencing and reported low enzyme activity in tissues. (houten2014mitochondrialnadp(h)deficiency pages 2-4) * A Decr knockout mouse strongly supports that loss of mitochondrial DECR activity is sufficient to cause the characteristic C10:2 biomarker and stress-intolerance phenotype. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2) * However, in the best-characterized modern human cases in the available evidence, targeted sequencing excluded DECR1 coding mutations. (houten2014mitochondrialnadp(h)deficiency pages 4-5, houten2014mitochondrialnadp(h)deficiency pages 5-7)
Risk factors
- Fasting / metabolic stress is a key precipitant of hypoglycemia and decompensation in Decr−/− mice and is mechanistically plausible as a trigger in humans with FAO auxiliary enzyme deficiency. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
Protective factors
No DECR- or NADK2-specific protective factors were identified in the available evidence.
Gene–environment interactions
- A plausible interaction is genetic impairment of PUFA β‑oxidation (DECR pathway defect) with environmental/physiologic stressors (fasting, illness, cold exposure), which increases energy demand and reliance on fatty acid oxidation and may precipitate decompensation. In Decr−/− mice, acute cold stress and fasting precipitated severe phenotypes. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
3. Phenotypes
Core phenotype spectrum (humans; NADK2-associated secondary DECR deficiency)
Clinical presentation can be severe, early-onset, and progressive: * Failure to thrive (HP:0001508), microcephaly (HP:0000252), hypotonia (HP:0001252) (houten2014mitochondrialnadp(h)deficiency pages 2-4) * Developmental delay (HP:0001263) and progressive neurologic decline (houten2014mitochondrialnadp(h)deficiency pages 1-2) * Movement disorder including choreoathetosis/dystonia (HP:0001266/HP:0001332) (houten2014mitochondrialnadp(h)deficiency pages 4-5) * Epilepsy (HP:0001250), cortical blindness/visual loss (HP:0000608/HP:0000505) in severe cases (houten2014mitochondrialnadp(h)deficiency pages 4-5) * Lactic acidosis (HP:0003128) and renal tubular acidosis (HP:0001947) consistent with multisystem mitochondrial dysfunction (houten2014mitochondrialnadp(h)deficiency pages 4-5) * Neuroimaging: progressive leukodystrophy / white matter disease (HP:0002415), cerebral atrophy (HP:0002059), basal ganglia lesions (HP:0002134) (houten2014mitochondrialnadp(h)deficiency pages 4-5)
Age of onset: reported as early infancy (e.g., presentation at 8 weeks) (houten2014mitochondrialnadp(h)deficiency pages 2-4).
Severity/progression: severe, progressive encephalopathy with death in childhood reported in the detailed NADK2-associated cases. (houten2014mitochondrialnadp(h)deficiency pages 4-5)
Phenotypes (mouse model; primary Decr deficiency)
Decr−/− mice demonstrate: * Severe fasting/stress intolerance with profound hypoglycemia and “unimpaired ketogenesis” (consistent with preserved ketone production despite impaired PUFA oxidation) (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2) * Hepatic steatosis (fatty liver) and accumulation of unsaturated fatty acids in hepatic lipids (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2) * Cold intolerance / fatal hypothermia with impaired thermogenesis under acute cold challenge (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
Quality of life impact
Human cases described are consistent with severe neurodevelopmental disability and progressive neurologic impairment, implying major quality-of-life impact; disease-specific QoL instruments were not identified in the accessible evidence. (houten2014mitochondrialnadp(h)deficiency pages 4-5)
4. Genetic / molecular information
Causal genes
- NADK2 (mitochondrial NAD kinase): confirmed cause of mitochondrial NADP(H) deficiency leading to secondary DECR deficiency with hyperlysinemia. (houten2014mitochondrialnadp(h)deficiency pages 5-7, houten2014mitochondrialnadp(h)deficiency pages 1-2)
- DECR1 (mitochondrial 2,4‑dienoyl‑CoA reductase): strongly supported by mouse knockout as essential for PUFA β‑oxidation and the C10:2 biomarker, but definitive human DECR1-biallelic disease was not established in the key human case series available here. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2, houten2014mitochondrialnadp(h)deficiency pages 5-7)
Pathogenic variants (reported in accessible evidence)
- NADK2 c.1018C>T (p.R340X), homozygous in the proband and heterozygous in parents; associated with absent NADK2 protein and low mitochondrial NADP(H). (houten2014mitochondrialnadp(h)deficiency pages 5-7)
Functional consequences
- NADK2 deficiency reduces mitochondrial NADP(H) and thereby disables NADPH-dependent mitochondrial enzymes; the paper emphasizes NADPH as both a cosubstrate and a stabilizing/chaperoning factor for enzymes like DECR and AASS, explaining combined biochemical signatures (C10:2 elevation + hyperlysinemia). (houten2014mitochondrialnadp(h)deficiency pages 1-2)
Suggested GO / pathway terms (for curation; not exhaustive)
- GO:0006635 fatty acid beta-oxidation
- GO:0006636 unsaturated fatty acid metabolic process
- GO:0055114 oxidation-reduction process
- Reactome/KEGG concept: PUFA β‑oxidation auxiliary enzymes requiring DECR for 2,4‑dienoyl‑CoA intermediates (supported by pathway schematic evidence). (miinalainen2009mitochondrial24dienoylcoareductase media db369249)
Suggested cell types (CL terms) and tissues
Based on energy-demand and reported pathology: * Hepatocyte (CL:0000182) / liver involvement (steatosis; biochemical FAO organ) (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2) * Neuron (CL:0000540) and oligodendrocyte (CL:0000128) as plausible cell types in progressive leukodystrophy/encephalopathy (houten2014mitochondrialnadp(h)deficiency pages 4-5) * Renal tubular epithelial cell (CL:0000066) (renal tubular acidosis) (houten2014mitochondrialnadp(h)deficiency pages 4-5)
5. Environmental information
No specific toxins/lifestyle infectious triggers were identified. The main non-genetic precipitants described are metabolic stressors (fasting, cold exposure) in the mouse model. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
6. Mechanism / pathophysiology
Molecular pathway and causal chain
Upstream trigger: Loss of DECR activity (primary Decr knockout) or loss of mitochondrial NADP(H) (NADK2 deficiency) → reduced ability to reduce 2,4‑dienoyl‑CoA intermediates during mitochondrial PUFA β‑oxidation. (houten2014mitochondrialnadp(h)deficiency pages 1-2, miinalainen2009mitochondrial24dienoylcoareductase media db369249)
Biochemical block: Incomplete PUFA β‑oxidation → accumulation of PUFA-derived intermediates that appear in blood as C10:2 (decadienoyl)carnitine; mouse fasting acylcarnitine profiles show marked C10:2 accumulation in Decr−/− mice. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2, miinalainen2009mitochondrial24dienoylcoareductase media f7dcc2b0)
Downstream metabolic effects: Energy stress response impairment under fasting/cold challenge → severe hypoglycemia and stress intolerance; liver lipid accumulation/steatosis; compensatory upregulation of peroxisomal β‑oxidation/ω‑oxidation in mice. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
Multisystem mitochondrial dysfunction (NADK2 cases): Mitochondrial redox cofactor deficiency also affects additional NADPH-dependent enzymes and is associated with reduced maximal oxygen consumption and increased extracellular acidification in fibroblasts, consistent with broader mitochondrial disease and severe neurodegeneration. (houten2014mitochondrialnadp(h)deficiency pages 9-10)
Key biochemical abnormalities (with suggested CHEBI entities)
- C10:2 acylcarnitine / decadienoylcarnitine (CHEBI mapping to be validated; key diagnostic analyte) (houten2014mitochondrialnadp(h)deficiency pages 4-5, miinalainen2009mitochondrial24dienoylcoareductase media f7dcc2b0)
- Lysine (CHEBI:18019) elevated in NADK2-associated phenotype (hyperlysinemia) (houten2014mitochondrialnadp(h)deficiency pages 4-5)
- Lactate (CHEBI:24996) elevated (lactic acidosis) (houten2014mitochondrialnadp(h)deficiency pages 4-5)
Authoritative expert framing (abstract quote)
A translational metabolism review described the diagnostic clue as: “a very unusual C10:2 acylcarnitine… most likely derived from linoleic acid (C18:2) oxidation.” (wanders2019translationalmetabolisma pages 3-3)
7. Anatomical structures affected
Organs/systems (supported by evidence)
- Central nervous system (UBERON:0001016): progressive encephalopathy/leukodystrophy and basal ganglia lesions in severe human cases. (houten2014mitochondrialnadp(h)deficiency pages 4-5)
- Liver (UBERON:0002107): steatosis and lipid accumulation in Decr−/− mice; clinically relevant FAO organ. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
- Kidney (UBERON:0002113): renal tubular acidosis in human NADK2-associated cases. (houten2014mitochondrialnadp(h)deficiency pages 4-5)
Subcellular localization
- Mitochondrion (GO:0005739): DECR function is mitochondrial; NADK2 generates mitochondrial NADP(H). (houten2014mitochondrialnadp(h)deficiency pages 1-2)
8. Temporal development
- Onset: infancy (e.g., 8 weeks in a reported case) (houten2014mitochondrialnadp(h)deficiency pages 2-4)
- Course: progressive neurologic decline with episodic metabolic derangements (intermittent lactic acidosis described) and early mortality in severe cases. (houten2014mitochondrialnadp(h)deficiency pages 4-5)
9. Inheritance and population
Inheritance
- NADK2-associated secondary DECR deficiency is autosomal recessive, supported by homozygous variant in the proband and heterozygosity in parents. (houten2014mitochondrialnadp(h)deficiency pages 5-7)
Epidemiology
- The disorder is described as very rare: the key report notes only a historical single case (1990) and a second, newly characterized case, underscoring paucity of epidemiologic data. (houten2014mitochondrialnadp(h)deficiency pages 2-4)
- In the broader context of sudden unexpected death (SUD) in infancy/childhood, metabolic disorders are estimated to account for ~3–5% of cases (not specific to DECR). (hung2024driedbloodspot pages 1-2)
10. Diagnostics
Clinical laboratory tests and biomarkers
Front-line biochemical screening * Acylcarnitine profile (tandem MS): elevation of C10:2 (decadienoyl)carnitine is the signature marker discussed for DECR deficiency and was retrospectively present at low levels on newborn blood spot in a NADK2 case. (houten2014mitochondrialnadp(h)deficiency pages 10-12, houten2014mitochondrialnadp(h)deficiency pages 2-4) * Plasma/CSF amino acids: hyperlysinemia can co-occur in NADK2-associated cases. (houten2014mitochondrialnadp(h)deficiency pages 4-5, houten2014mitochondrialnadp(h)deficiency pages 5-7) * Lactate: elevated in blood/CSF in severe neurometabolic presentations. (houten2014mitochondrialnadp(h)deficiency pages 4-5) * Urine organic acids: persistent abnormalities reported (e.g., lactic/pyruvic and other acids), consistent with mitochondrial dysfunction. (houten2014mitochondrialnadp(h)deficiency pages 4-5)
Functional/confirmatory testing * DECR enzyme activity assay in fibroblasts/tissues (reported residual ~10% using sorboyl‑CoA substrate). (houten2014mitochondrialnadp(h)deficiency pages 4-5) * Cellular bioenergetics (oxygen consumption / extracellular acidification) consistent with mitochondrial disorder. (houten2014mitochondrialnadp(h)deficiency pages 9-10)
Genetic testing
- In NADK2-associated cases, exome sequencing identified the causal NADK2 variant after targeted gene testing excluded DECR1/related genes; rescue experiments via wild-type NADK2 expression restored biochemical defects, strengthening causal inference. (houten2014mitochondrialnadp(h)deficiency pages 5-7, houten2014mitochondrialnadp(h)deficiency pages 1-2)
Newborn screening and screening limitations
- Newborn screening programs have used C10:2‑carnitine as the primary marker; the key report emphasizes that slight C10:2 elevations are neither fully specific nor sensitive, since other FAO disorders can also elevate C10:2. (houten2014mitochondrialnadp(h)deficiency pages 10-12)
- The authors suggested that adding lysine could improve screening specificity for the NADK2-associated combined phenotype (C10:2 + hyperlysinemia). (houten2014mitochondrialnadp(h)deficiency pages 10-12)
Differential diagnosis (examples)
- Other fatty acid oxidation disorders (FAODs) with overlapping acylcarnitine patterns and stress-induced hypoglycemia; in NADK2-associated disease, broader mitochondrial dysfunction is also present. (houten2014mitochondrialnadp(h)deficiency pages 10-12, houten2014mitochondrialnadp(h)deficiency pages 9-10)
- Primary hyperlysinemia due to lysine pathway defects (contextualized in the NADK2 paper as a confounder). (houten2014mitochondrialnadp(h)deficiency pages 2-4)
11. Outcome / prognosis
- Severe NADK2-associated secondary DECR deficiency can be progressive and fatal in childhood, with progressive leukodystrophy/encephalopathy and multisystem involvement. (houten2014mitochondrialnadp(h)deficiency pages 4-5)
- Prognosis for putative primary DECR1-related human disease cannot be estimated from the limited accessible evidence; in mice, baseline compensation exists but metabolic stress causes severe hypoglycemia and intolerance. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
12. Treatment
Reported management attempts (human cases)
In the detailed NADK2-associated report, interventions attempted included: * Dietary lysine restriction (MAXO suggestion: dietary amino acid restriction) * Caloric support (MAXO: nutritional support therapy) * Medium-chain fatty acids (often used in FAO disorders to bypass some β‑oxidation steps) * Carnitine supplementation These are described as management attempts rather than established effective therapy. (houten2014mitochondrialnadp(h)deficiency pages 4-5)
Clinical trials
No DECR/NADK2-specific interventional clinical trials were identified in this run’s clinical trial retrieval.
13. Prevention
- Primary prevention is not established; given autosomal recessive inheritance for NADK2-associated disease, genetic counseling (MAXO: genetic counseling) and carrier testing/cascade testing are the most actionable preventive strategies once a familial variant is known. (houten2014mitochondrialnadp(h)deficiency pages 5-7)
- Secondary prevention: early recognition of suggestive metabolic signatures (isolated or predominant C10:2 elevation; C10:2 + hyperlysinemia) and prompt genetic testing may reduce diagnostic delay, although evidence for improved outcomes is not available here. (houten2014mitochondrialnadp(h)deficiency pages 10-12)
14. Other species / natural disease
No naturally occurring veterinary DECR deficiency evidence was identified in the accessible corpus.
15. Model organisms
Mouse
A targeted Decr−/− knockout mouse is a key model demonstrating: * Accumulation of the diagnostic biomarker C10:2 (decadienoylcarnitine) during fasting * Stress-induced hypoglycemia with preserved ketogenesis * Hepatic steatosis and cold intolerance This model is directly relevant for biomarker interpretation and mechanistic studies of PUFA β‑oxidation auxiliary enzymes. (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2, miinalainen2009mitochondrial24dienoylcoareductase media f7dcc2b0)
Recent developments (prioritizing 2023–2024)
2024: Postmortem “metabolic autopsy” operationalization for IEMs (contextual, not DECR-specific)
A 2024 retrospective study from Hong Kong reinforces modern real-world implementation of IEM detection in sudden unexpected deaths via combined dried blood spot acylcarnitines/amino acids, urine organic acids when available, and NGS panels, and cites an estimated 3–5% contribution of metabolic disorders to SUD. This supports the utility of maintaining FAO/IEM competence in forensic/pediatric pathology workflows, even though DECR deficiency was not a highlighted diagnosis in the excerpted findings. (hung2024driedbloodspot pages 1-2)
Evidence gap
In this run, I did not retrieve 2023–2024 primary publications that add new confirmed human DECR1-biallelic cases, nor updated disease-specific consensus guidelines. The most disease-defining human molecular mechanism remained the 2014 NADK2 report, with the strongest mechanistic support from earlier mouse genetics.
Visual evidence (pathway + biomarker)
The following figure extractions show (i) the DECR-dependent PUFA β‑oxidation step and (ii) fasting-associated accumulation of C10:2 acylcarnitine in Decr−/− mice, supporting both mechanistic understanding and biomarker rationale. (miinalainen2009mitochondrial24dienoylcoareductase media db369249, miinalainen2009mitochondrial24dienoylcoareductase media f7dcc2b0)
Summary table
Table (click to expand)
| Entity | Causal gene/defect | Key biomarkers | Core clinical features | Notes on diagnosis/screening | Key citations |
|---|---|---|---|---|---|
| Human NADK2-related secondary DECR deficiency | Autosomal recessive NADK2 loss causing mitochondrial NADP(H) deficiency with secondary impairment of NADPH-dependent DECR activity; DECR1 coding mutations were excluded in the reported modern cases | Elevated C10:2 (decadienoyl)carnitine; hyperlysinemia in plasma/CSF/urine; elevated lactate; low free carnitine; abnormal urinary organic acids; residual fibroblast DECR activity ~10% | Early infancy onset; failure to thrive, developmental delay, hypotonia, progressive encephalopathy, movement disorder/choreoathetosis-dystonia, visual loss/cortical blindness, epilepsy, renal tubular acidosis; death in childhood in severe cases | Diagnosis integrated acylcarnitines, amino acids, fibroblast enzyme assay, and exome sequencing; mild newborn-screen C10:2 elevation may occur but is not fully sensitive/specific; authors suggested lysine might improve screening specificity | (houten2014mitochondrialnadp(h)deficiency pages 9-10, houten2014mitochondrialnadp(h)deficiency pages 4-5, houten2014mitochondrialnadp(h)deficiency pages 5-7, houten2014mitochondrialnadp(h)deficiency pages 10-12) |
| Historical 1990 DECR deficiency case | Biochemical mitochondrial 2,4-dienoyl-CoA reductase deficiency reported before modern molecular diagnosis; residual DECR activity 17% in muscle and 40% in liver; later literature indicates DECR1 mutations were not demonstrated in similar cases | Elevated plasma C10:2-carnitine; hyperlysinemia | Failure to thrive, persistent hypotonia, microcephaly; death at 4 months | Landmark historical case establishing the phenotype/biochemical signature; no definitive molecular cause reported in the available evidence | (houten2014mitochondrialnadp(h)deficiency pages 2-4) |
| Decr1 knockout mouse | Targeted Decr1/Decr disruption causing primary loss of mitochondrial 2,4-dienoyl-CoA reductase and defective PUFA β-oxidation | Increased serum decadienoylcarnitine (C10:2); urinary unsaturated dicarboxylic acids; hepatic steatosis with unsaturated fatty acid accumulation; hypoglycemia during fasting/stress | Usually compensated at baseline, but severe fasting/stress intolerance, profound hypoglycemia, impaired cold tolerance/thermogenesis, fatty liver; ketogenesis preserved | Strong mechanistic model for interpreting human C10:2 biomarker and PUFA β-oxidation block; supports causal role of DECR in metabolic adaptation rather than proving human DECR1-mutant cases | (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2, wanders2019translationalmetabolisma pages 3-3, miinalainen2009mitochondrial24dienoylcoareductase media db369249) |
Table: This table contrasts the historical human DECR phenotype, modern NADK2-related secondary DECR deficiency, and the Decr1 knockout mouse model. It highlights the key biomarker pattern, phenotype, and diagnostic implications useful for rare disease knowledge-base curation.
Curator notes / limitations
1) Ontology identifiers (OMIM/Orphanet/MONDO) were not retrievable with the available tools in this run; they must be filled via direct database lookup. 2) Human evidence in the accessible corpus most strongly supports NADK2-associated secondary DECR deficiency, while definitive DECR1-mutant human disease was not confirmed here. 3) Treatment evidence is limited to reported management attempts and general FAO-disorder logic; no proven disease-modifying therapy was identified in the retrieved evidence.
Key primary sources (with URLs and publication dates)
- Miinalainen IJ et al. PLoS Genetics (2009-07). “Mitochondrial 2,4-dienoyl-CoA reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis.” https://doi.org/10.1371/journal.pgen.1000543 (miinalainen2009mitochondrial24dienoylcoareductase pages 1-2)
- Houten SM et al. Human Molecular Genetics (2014-09). “Mitochondrial NADP(H) deficiency due to a mutation in NADK2 causes dienoyl-CoA reductase deficiency with hyperlysinemia.” https://doi.org/10.1093/hmg/ddu218 (houten2014mitochondrialnadp(h)deficiency pages 1-2)
- Wanders RJA et al. Journal of Inherited Metabolic Disease (2019-02). “Translational Metabolism: A multidisciplinary approach towards precision diagnosis of inborn errors of metabolism in the omics era.” https://doi.org/10.1002/jimd.12008 (wanders2019translationalmetabolisma pages 3-3)
- Hung LY et al. Cureus (2024-06). “Dried Blood Spot Postmortem Metabolic Autopsy With Genotype Validation for Sudden Unexpected Deaths in Infancy and Childhood in Hong Kong.” https://doi.org/10.7759/cureus.62347 (hung2024driedbloodspot pages 1-2)
References
-
(houten2014mitochondrialnadp(h)deficiency pages 9-10): Sander M. Houten, Simone Denis, Heleen te Brinke, Aldo Jongejan, Antoine H.C. van Kampen, Edward J. Bradley, Frank Baas, Raoul C.M. Hennekam, David S. Millington, Sarah P. Young, Dianne M. Frazier, Muge Gucsavas-Calikoglu, and Ronald J.A. Wanders. Mitochondrial nadp(h) deficiency due to a mutation in nadk2 causes dienoyl-coa reductase deficiency with hyperlysinemia. Human molecular genetics, 23 18:5009-16, Sep 2014. URL: https://doi.org/10.1093/hmg/ddu218, doi:10.1093/hmg/ddu218. This article has 89 citations and is from a domain leading peer-reviewed journal.
-
(houten2014mitochondrialnadp(h)deficiency pages 5-7): Sander M. Houten, Simone Denis, Heleen te Brinke, Aldo Jongejan, Antoine H.C. van Kampen, Edward J. Bradley, Frank Baas, Raoul C.M. Hennekam, David S. Millington, Sarah P. Young, Dianne M. Frazier, Muge Gucsavas-Calikoglu, and Ronald J.A. Wanders. Mitochondrial nadp(h) deficiency due to a mutation in nadk2 causes dienoyl-coa reductase deficiency with hyperlysinemia. Human molecular genetics, 23 18:5009-16, Sep 2014. URL: https://doi.org/10.1093/hmg/ddu218, doi:10.1093/hmg/ddu218. This article has 89 citations and is from a domain leading peer-reviewed journal.
-
(houten2014mitochondrialnadp(h)deficiency pages 1-2): Sander M. Houten, Simone Denis, Heleen te Brinke, Aldo Jongejan, Antoine H.C. van Kampen, Edward J. Bradley, Frank Baas, Raoul C.M. Hennekam, David S. Millington, Sarah P. Young, Dianne M. Frazier, Muge Gucsavas-Calikoglu, and Ronald J.A. Wanders. Mitochondrial nadp(h) deficiency due to a mutation in nadk2 causes dienoyl-coa reductase deficiency with hyperlysinemia. Human molecular genetics, 23 18:5009-16, Sep 2014. URL: https://doi.org/10.1093/hmg/ddu218, doi:10.1093/hmg/ddu218. This article has 89 citations and is from a domain leading peer-reviewed journal.
-
(houten2014mitochondrialnadp(h)deficiency pages 4-5): Sander M. Houten, Simone Denis, Heleen te Brinke, Aldo Jongejan, Antoine H.C. van Kampen, Edward J. Bradley, Frank Baas, Raoul C.M. Hennekam, David S. Millington, Sarah P. Young, Dianne M. Frazier, Muge Gucsavas-Calikoglu, and Ronald J.A. Wanders. Mitochondrial nadp(h) deficiency due to a mutation in nadk2 causes dienoyl-coa reductase deficiency with hyperlysinemia. Human molecular genetics, 23 18:5009-16, Sep 2014. URL: https://doi.org/10.1093/hmg/ddu218, doi:10.1093/hmg/ddu218. This article has 89 citations and is from a domain leading peer-reviewed journal.
-
(miinalainen2009mitochondrial24dienoylcoareductase pages 1-2): Ilkka J. Miinalainen, Werner Schmitz, Anne Huotari, Kaija J. Autio, Raija Soininen, Emiel Ver Loren van Themaat, Myriam Baes, Karl-Heinz Herzig, Ernst Conzelmann, and J. Kalervo Hiltunen. Mitochondrial 2,4-dienoyl-coa reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis. Jul 2009. URL: https://doi.org/10.1371/journal.pgen.1000543, doi:10.1371/journal.pgen.1000543. This article has 78 citations and is from a domain leading peer-reviewed journal.
-
(wanders2019translationalmetabolisma pages 3-3): Ronald J. A. Wanders, Frederic M. Vaz, Sacha Ferdinandusse, André B. P. van Kuilenburg, Stephan Kemp, Clara D. van Karnebeek, Hans R. Waterham, and Riekelt H. Houtkooper. Translational metabolism: a multidisciplinary approach towards precision diagnosis of inborn errors of metabolism in the omics era. Journal of Inherited Metabolic Disease, 42:197-208, Feb 2019. URL: https://doi.org/10.1002/jimd.12008, doi:10.1002/jimd.12008. This article has 29 citations and is from a peer-reviewed journal.
-
(houten2014mitochondrialnadp(h)deficiency pages 10-12): Sander M. Houten, Simone Denis, Heleen te Brinke, Aldo Jongejan, Antoine H.C. van Kampen, Edward J. Bradley, Frank Baas, Raoul C.M. Hennekam, David S. Millington, Sarah P. Young, Dianne M. Frazier, Muge Gucsavas-Calikoglu, and Ronald J.A. Wanders. Mitochondrial nadp(h) deficiency due to a mutation in nadk2 causes dienoyl-coa reductase deficiency with hyperlysinemia. Human molecular genetics, 23 18:5009-16, Sep 2014. URL: https://doi.org/10.1093/hmg/ddu218, doi:10.1093/hmg/ddu218. This article has 89 citations and is from a domain leading peer-reviewed journal.
-
(houten2014mitochondrialnadp(h)deficiency pages 2-4): Sander M. Houten, Simone Denis, Heleen te Brinke, Aldo Jongejan, Antoine H.C. van Kampen, Edward J. Bradley, Frank Baas, Raoul C.M. Hennekam, David S. Millington, Sarah P. Young, Dianne M. Frazier, Muge Gucsavas-Calikoglu, and Ronald J.A. Wanders. Mitochondrial nadp(h) deficiency due to a mutation in nadk2 causes dienoyl-coa reductase deficiency with hyperlysinemia. Human molecular genetics, 23 18:5009-16, Sep 2014. URL: https://doi.org/10.1093/hmg/ddu218, doi:10.1093/hmg/ddu218. This article has 89 citations and is from a domain leading peer-reviewed journal.
-
(miinalainen2009mitochondrial24dienoylcoareductase media db369249): Ilkka J. Miinalainen, Werner Schmitz, Anne Huotari, Kaija J. Autio, Raija Soininen, Emiel Ver Loren van Themaat, Myriam Baes, Karl-Heinz Herzig, Ernst Conzelmann, and J. Kalervo Hiltunen. Mitochondrial 2,4-dienoyl-coa reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis. Jul 2009. URL: https://doi.org/10.1371/journal.pgen.1000543, doi:10.1371/journal.pgen.1000543. This article has 78 citations and is from a domain leading peer-reviewed journal.
-
(miinalainen2009mitochondrial24dienoylcoareductase media f7dcc2b0): Ilkka J. Miinalainen, Werner Schmitz, Anne Huotari, Kaija J. Autio, Raija Soininen, Emiel Ver Loren van Themaat, Myriam Baes, Karl-Heinz Herzig, Ernst Conzelmann, and J. Kalervo Hiltunen. Mitochondrial 2,4-dienoyl-coa reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis. Jul 2009. URL: https://doi.org/10.1371/journal.pgen.1000543, doi:10.1371/journal.pgen.1000543. This article has 78 citations and is from a domain leading peer-reviewed journal.
-
(hung2024driedbloodspot pages 1-2): Ling Yin Hung, Chloe M Mak, Ka Chung Foo, Chun Hei Toby Chan, Hok-Fung Tong, Tsz Ki Wong, Hoi Shan Leung, Ka Chai Cheung, Han Chih Hencher Lee, and Chor Kwan Ching. Dried blood spot postmortem metabolic autopsy with genotype validation for sudden unexpected deaths in infancy and childhood in hong kong. Cureus, Jun 2024. URL: https://doi.org/10.7759/cureus.62347, doi:10.7759/cureus.62347. This article has 2 citations.