Temtamy Syndrome
Temtamy syndrome is a rare autosomal recessive neurodevelopmental disorder caused by biallelic loss-of-function variants in C12orf57 (GRCC10). It is a syndromic form of intellectual disability characterized by dysgenesis, hypoplasia, or agenesis of the corpus callosum, ocular coloboma and microphthalmia, intractable seizures, craniofacial dysmorphism, and, in a substantial fraction of patients, congenital heart disease. A recurrent start-loss founder mutation (c.1A>G; p.M1?) is among the most common recessive causes of intellectual disability in the Saudi population. C12orf57 encodes a small, evolutionarily conserved protein implicated in corpus callosum development and, more recently, in synaptic AMPA-receptor homeostatic downscaling in excitatory neurons.
Ask OpenScientist
Ask a research question about Temtamy Syndrome. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (1 reference)
Pathophysiology
4Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (2 references)
Pathograph
Phenotypes
12Cardiovascular 1
Show evidence (1 reference)
Eye 3
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Head and Neck 1
Show evidence (1 reference)
Nervous System 7
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
1Show evidence (3 references)
Medical Actions
3Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Differential Diagnoses
1Conditions with similar clinical presentations that must be differentiated from Temtamy Syndrome:
- Biallelic C12orf57 variants confirm Temtamy syndrome.
- Kabuki syndrome is caused by KMT2D or KDM6A variants.
Show evidence (1 reference)
Source YAML
click to showname: Temtamy Syndrome
creation_date: "2026-06-04T00:00:00Z"
category: Mendelian
description: >-
Temtamy syndrome is a rare autosomal recessive neurodevelopmental disorder
caused by biallelic loss-of-function variants in C12orf57 (GRCC10). It is a
syndromic form of intellectual disability characterized by dysgenesis,
hypoplasia, or agenesis of the corpus callosum, ocular coloboma and
microphthalmia, intractable seizures, craniofacial dysmorphism, and, in a
substantial fraction of patients, congenital heart disease. A recurrent
start-loss founder mutation (c.1A>G; p.M1?) is among the most common
recessive causes of intellectual disability in the Saudi population. C12orf57
encodes a small, evolutionarily conserved protein implicated in corpus
callosum development and, more recently, in synaptic AMPA-receptor
homeostatic downscaling in excitatory neurons.
disease_term:
preferred_term: Temtamy syndrome
term:
id: MONDO:0009033
label: temtamy syndrome
parents:
- Neurodevelopmental disorder
- Syndromic intellectual disability
synonyms:
- C12orf57-related disorder
- corpus callosum, agenesis of, with intellectual disability, ocular coloboma, and micrognathia
- Temtamy syndrome of corpus callosum and ocular abnormalities
inheritance:
- name: Autosomal recessive inheritance
inheritance_term:
preferred_term: autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:24798461
reference_title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Homozygous and compound heterozygous mutations in C12orf57 have recently
been described to cause an autosomal recessive syndromic form of
intellectual disability
explanation: >-
The report explicitly classifies C12orf57-related disease as autosomal
recessive.
pathophysiology:
- name: C12orf57 Loss of Function
description: >-
Biallelic loss-of-function variants in C12orf57 (GRCC10) — most commonly an
ancestral start-loss founder mutation (c.1A>G; p.M1?) and other truncating
or missense changes — abolish or severely reduce function of a small,
poorly annotated 126-amino-acid protein that is ubiquitously expressed and
tightly conserved across evolution. The protein is required for normal
development of the human corpus callosum and is expressed in tissues
including the eye and brain, linking its loss to the combined forebrain
commissural and ocular developmental phenotype.
genes:
- preferred_term: C12orf57
term:
id: hgnc:29521
label: C12orf57
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: corpus callosum development
term:
id: GO:0022038
label: corpus callosum development
modifier: ABNORMAL
- preferred_term: eye development
term:
id: GO:0001654
label: eye development
modifier: ABNORMAL
- preferred_term: nervous system development
term:
id: GO:0007399
label: nervous system development
modifier: ABNORMAL
evidence:
- reference: PMID:23453666
reference_title: "Whole-exome sequencing identifies mutated c12orf57 in recessive corpus callosum hypoplasia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
C12orf57 is ubiquitously expressed and encodes a poorly annotated 126
amino acid protein of unknown function. This protein is without
significant paralogs but has been tightly conserved across evolution. Our
data suggest that this conserved gene is required for development of the
human corpus callosum.
explanation: >-
The whole-exome study identifies C12orf57 as a conserved gene required for
corpus callosum development, establishing the primary developmental defect.
- reference: PMID:23453665
reference_title: "Mutations in c12orf57 cause a syndromic form of colobomatous microphthalmia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Little is known about C12orf57 but we show that it is expressed in several
mouse tissues, including the eye and brain.
explanation: >-
Expression in eye and brain links loss of C12orf57 to the combined ocular
and CNS developmental phenotype.
downstream:
- target: Impaired Corpus Callosum and Forebrain Commissural Development
description: >-
Loss of the conserved C12orf57 protein impairs midline forebrain
commissural development, producing agenesis, hypoplasia, or dysgenesis of
the corpus callosum.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:23453666
reference_title: "Whole-exome sequencing identifies mutated c12orf57 in recessive corpus callosum hypoplasia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Our data suggest that this conserved gene is required for development of
the human corpus callosum.
explanation: >-
The founding cohort directly links C12orf57 loss to defective corpus
callosum development, although the molecular intermediates remain
unresolved.
- target: Impaired Ocular Development
description: >-
C12orf57 expression in the developing eye and its association with
colobomatous microphthalmia implicate its loss in defective ocular
morphogenesis.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:23453665
reference_title: "Mutations in c12orf57 cause a syndromic form of colobomatous microphthalmia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Our data strongly implicate mutations in C12orf57 in the pathogenesis of
a clinically distinct autosomal-recessive syndromic form of colobomatous
microphthalmia.
explanation: >-
The founding paper links C12orf57 loss to colobomatous microphthalmia,
an ocular developmental defect.
- target: Disrupted Synaptic AMPA-Receptor Homeostasis
description: >-
Beyond its developmental role, loss of C12orf57/GRCC10 disrupts activity
dependent synaptic homeostatic downscaling in excitatory neurons,
providing a candidate mechanism for the epilepsy and intellectual
disability of the syndrome.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- CAMK4 activity modulation
- CREB and ARC expression regulation
- AMPA receptor downscaling
evidence:
- reference: PMID:39974932
reference_title: "C12ORF57: a novel principal regulator of synaptic AMPA currents and excitatory neuronal homeostasis."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
We further found that GRCC10/C12ORF57 modulates the activity of
calcium/calmodulin dependent kinase 4 (CAMK4) and thereby regulates the
expression of CREB and ARC.
explanation: >-
The mouse-model and in vitro study defines a molecular pathway
(CAMK4-CREB-ARC) connecting C12orf57 loss to disrupted synaptic AMPA
receptor homeostasis.
- name: Impaired Corpus Callosum and Forebrain Commissural Development
description: >-
Defective midline commissural development produces a spectrum of corpus
callosum abnormalities ranging from complete agenesis to hypoplasia or
dysgenesis, often accompanied by cerebral white matter abnormalities.
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: corpus callosum development
term:
id: GO:0022038
label: corpus callosum development
modifier: ABNORMAL
- preferred_term: commissural neuron axon guidance
term:
id: GO:0071679
label: commissural neuron axon guidance
modifier: ABNORMAL
locations:
- preferred_term: corpus callosum
term:
id: UBERON:0002336
label: corpus callosum
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
63% (34/54) had corpus callosal abnormalities
explanation: >-
The combined cohort quantifies corpus callosal abnormalities as a frequent
feature of the syndrome.
downstream:
- target: Corpus callosum abnormality
description: >-
Defective commissural development manifests as agenesis, hypoplasia, or
dysgenesis of the corpus callosum on neuroimaging.
causal_link_type: DIRECT
evidence:
- reference: PMID:24798461
reference_title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
an autosomal recessive syndromic form of intellectual disability,
including agenesis/hypoplasia of the corpus callosum, optic coloboma, and
intractable seizures
explanation: >-
The clinical description directly links the syndrome to
agenesis/hypoplasia of the corpus callosum.
- target: Cerebral white matter abnormality
description: >-
Brain white matter abnormalities accompany the commissural defect in a
substantial fraction of patients.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
a high frequency of less recognized features such as congenital heart
disease (51.4%), and brain white matter abnormalities (38%, 19/50)
explanation: >-
The delineation cohort documents brain white matter abnormalities as a
recurrent imaging feature.
- name: Impaired Ocular Development
description: >-
Disrupted ocular morphogenesis produces a spectrum of eye malformations,
including coloboma (iris, chorioretinal, optic disc) and microphthalmia,
that defined the original colobomatous-microphthalmia presentation.
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: eye development
term:
id: GO:0001654
label: eye development
modifier: ABNORMAL
locations:
- preferred_term: eye
term:
id: UBERON:0000970
label: eye
evidence:
- reference: PMID:23453665
reference_title: "Mutations in c12orf57 cause a syndromic form of colobomatous microphthalmia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
colobomatous microphthalmia is associated with profound global
developmental delay, intractable seizures, and corpus callosum
abnormalities
explanation: >-
The founding paper documents colobomatous microphthalmia as the defining
ocular phenotype.
downstream:
- target: Coloboma
description: >-
Ocular maldevelopment manifests as coloboma affecting the iris, retina, or
optic disc.
causal_link_type: DIRECT
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
14.5% (8/55) had coloboma
explanation: >-
The delineation cohort quantifies coloboma as a recurring ocular feature.
- target: Microphthalmia
description: >-
Ocular maldevelopment can produce microphthalmia, the original defining
ocular feature of the syndrome.
causal_link_type: DIRECT
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
16.4% (9/55) had microphthalmia
explanation: >-
The delineation cohort quantifies microphthalmia among affected patients.
- name: Disrupted Synaptic AMPA-Receptor Homeostasis
description: >-
Loss of C12orf57/GRCC10 disrupts the homeostatic downscaling of synaptic
AMPA receptors that normally accompanies elevated neuronal activity. In a
Grcc10 knockout mouse, hippocampal neurons show increased AMPA receptor
expression and larger miniature excitatory postsynaptic currents, with
increased epileptiform activity, via modulation of CAMK4 and downstream CREB
and ARC expression.
cell_types:
- preferred_term: glutamatergic neuron
term:
id: CL:0000679
label: glutamatergic neuron
- preferred_term: hippocampal neuron
term:
id: CL:0002608
label: hippocampal neuron
genes:
- preferred_term: CAMK4
term:
id: hgnc:1464
label: CAMK4
biological_processes:
- preferred_term: regulation of AMPA receptor activity
term:
id: GO:2000311
label: regulation of AMPA receptor activity
modifier: ABNORMAL
- preferred_term: regulation of synaptic plasticity
term:
id: GO:0048167
label: regulation of synaptic plasticity
modifier: ABNORMAL
evidence:
- reference: PMID:39974932
reference_title: "C12ORF57: a novel principal regulator of synaptic AMPA currents and excitatory neuronal homeostasis."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
hippocampal neurons in these mice exhibited significantly increased AMPA
receptor expression levels and higher amplitude of miniature excitatory
postsynaptic currents (mEPSCs)
explanation: >-
The knockout mouse model demonstrates the AMPA-receptor and synaptic
current abnormalities downstream of Grcc10/C12orf57 loss.
- reference: PMID:39974932
reference_title: "C12ORF57: a novel principal regulator of synaptic AMPA currents and excitatory neuronal homeostasis."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
deletion of Grcc10 disrupts the characteristic synaptic AMPA receptor
downscaling that accompanies increased activity in glutamatergic neurons
explanation: >-
The study identifies disrupted AMPA-receptor homeostatic downscaling as the
core synaptic mechanism.
downstream:
- target: Seizure
description: >-
Disrupted AMPA-receptor downscaling increases excitatory drive and seizure
susceptibility, recapitulated as increased epileptiform activity in the
knockout model.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- increased AMPA receptor expression
- increased excitatory postsynaptic currents
evidence:
- reference: PMID:39974932
reference_title: "C12ORF57: a novel principal regulator of synaptic AMPA currents and excitatory neuronal homeostasis."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Grcc10 KO mice exhibit the characteristic phenotypic features seen in
human TS patients, including increased epileptiform activity.
explanation: >-
The knockout model links disrupted synaptic homeostasis to increased
epileptiform activity, paralleling the seizures seen in patients.
phenotypes:
- category: Clinical
name: Intellectual disability
description: >-
Intellectual disability / developmental delay is a constant feature, present
in all reported patients, and can be severe.
phenotype_term:
preferred_term: Intellectual disability
term:
id: HP:0001249
label: Intellectual disability
frequency: OBLIGATE
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
While all patients presented with intellectual disability/developmental
delay, the frequency of other phenotypic features was variable
explanation: >-
The combined cohort of 56 patients reports intellectual disability in all
affected individuals.
- category: Clinical
name: Global developmental delay
description: >-
Affected children show profound global developmental delay.
phenotype_term:
preferred_term: Global developmental delay
term:
id: HP:0001263
label: Global developmental delay
frequency: VERY_FREQUENT
evidence:
- reference: PMID:23453665
reference_title: "Mutations in c12orf57 cause a syndromic form of colobomatous microphthalmia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
colobomatous microphthalmia is associated with profound global
developmental delay, intractable seizures, and corpus callosum
abnormalities
explanation: >-
The founding report documents profound global developmental delay in
affected patients.
- category: Clinical
name: Seizure
description: >-
Seizures are frequent and often intractable, occurring in roughly
three-quarters of patients.
phenotype_term:
preferred_term: Seizure
term:
id: HP:0001250
label: Seizure
frequency: FREQUENT
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
73.2% (41/56) had epilepsy
explanation: >-
The delineation cohort quantifies epilepsy/seizures in 73.2% of patients,
supporting a FREQUENT frequency band.
- reference: PMID:24798461
reference_title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
including agenesis/hypoplasia of the corpus callosum, optic coloboma, and
intractable seizures
explanation: >-
The report documents intractable seizures as part of the syndrome.
- category: Clinical
name: Corpus callosum abnormality
description: >-
Agenesis, hypoplasia, or dysgenesis of the corpus callosum is a defining
neuroanatomic feature, present in roughly two-thirds of patients.
phenotype_term:
preferred_term: Hypoplasia of the corpus callosum
term:
id: HP:0002079
label: Hypoplasia of the corpus callosum
frequency: FREQUENT
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
63% (34/54) had corpus callosal abnormalities
explanation: >-
The delineation cohort quantifies corpus callosal abnormalities in 63% of
patients, supporting a FREQUENT frequency band.
- reference: PMID:24798461
reference_title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
including agenesis/hypoplasia of the corpus callosum, optic coloboma, and
intractable seizures
explanation: >-
The report documents agenesis/hypoplasia of the corpus callosum as part of
the syndrome.
- category: Clinical
name: Agenesis of corpus callosum
description: >-
Complete agenesis of the corpus callosum occurs at the severe end of the
commissural-defect spectrum.
phenotype_term:
preferred_term: Agenesis of corpus callosum
term:
id: HP:0001274
label: Agenesis of corpus callosum
evidence:
- reference: PMID:35791610
reference_title: "C12orf57 pathogenic variants: a unique cause of developmental encephalopathy in a south Indian child."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Temtamy syndrome, an extremely rare disorder characterized by intellectual
disability, seizures, facial dysmorphism and agenesis of corpus callosum
explanation: >-
This case report lists agenesis of the corpus callosum among the defining
features of the syndrome.
- category: Clinical
name: Coloboma
description: >-
Ocular coloboma (iris, chorioretinal, or optic disc) is a characteristic eye
malformation, present in roughly 15% of patients.
phenotype_term:
preferred_term: Coloboma
term:
id: HP:0000589
label: Coloboma
frequency: OCCASIONAL
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
14.5% (8/55) had coloboma
explanation: >-
The delineation cohort quantifies coloboma in 14.5% of patients, supporting
an OCCASIONAL frequency band.
- category: Clinical
name: Chorioretinal coloboma
description: >-
Chorioretinal coloboma is among the specific ocular coloboma subtypes
reported in the syndrome.
phenotype_term:
preferred_term: Chorioretinal coloboma
term:
id: HP:0000567
label: Chorioretinal coloboma
evidence:
- reference: PMID:24798461
reference_title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
two siblings with severe intellectual disability, hypoplasia of the corpus
callosum, chorioretinal coloboma, and intractable seizures
explanation: >-
The report documents chorioretinal coloboma in two affected siblings.
- category: Clinical
name: Microphthalmia
description: >-
Microphthalmia (small eye) was the original defining ocular feature and
occurs in roughly 16% of patients.
phenotype_term:
preferred_term: Microphthalmia
term:
id: HP:0000568
label: Microphthalmia
frequency: OCCASIONAL
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
16.4% (9/55) had microphthalmia
explanation: >-
The delineation cohort quantifies microphthalmia in 16.4% of patients,
supporting an OCCASIONAL frequency band.
- category: Clinical
name: Facial dysmorphism
description: >-
Craniofacial dysmorphism is a recurrent feature of the syndrome.
phenotype_term:
preferred_term: Abnormal facial shape
term:
id: HP:0001999
label: Abnormal facial shape
evidence:
- reference: PMID:31853307
reference_title: "Temtamy syndrome caused by a new C12orf57 variant in a Chinese boy, including pedigree analysis and literature review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
C12orf57 pathogenic variants are mainly associated with global
developmental delay, epilepsy and dysmorphic facial appearances
explanation: >-
The literature-review case report identifies dysmorphic facial appearance
as a main associated feature.
- category: Clinical
name: Congenital heart disease
description: >-
Congenital heart disease is a less recognized but frequent feature,
documented in roughly half of patients in the largest delineation cohort.
phenotype_term:
preferred_term: Abnormal heart morphology
term:
id: HP:0001627
label: Abnormal heart morphology
frequency: FREQUENT
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
a high frequency of less recognized features such as congenital heart
disease (51.4%), and brain white matter abnormalities (38%, 19/50)
explanation: >-
The combined cohort reports congenital heart disease in 51.4% of patients,
supporting a FREQUENT frequency band.
- category: Clinical
name: Cerebral white matter abnormality
description: >-
Brain white matter abnormalities accompany the commissural defect in a
substantial fraction of patients.
phenotype_term:
preferred_term: Abnormal cerebral white matter morphology
term:
id: HP:0002500
label: Abnormal cerebral white matter morphology
frequency: FREQUENT
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
brain white matter abnormalities (38%, 19/50)
explanation: >-
The delineation cohort documents brain white matter abnormalities in 38% of
patients, supporting a FREQUENT frequency band.
- category: Clinical
name: Autistic behavior
description: >-
Autism / autistic behavior has been reported as part of the
neurodevelopmental phenotype.
phenotype_term:
preferred_term: Autistic behavior
term:
id: HP:0000729
label: Autistic behavior
evidence:
- reference: PMID:37451886
reference_title: "A novel pathogenic compound heterozygous variant in C12orf57 gene in a child with Temtamy syndrome presenting with overlapping phenotypic features of Kabuki-like syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Temtamy syndrome is a rare syndromic intellectual developmental disorder
that presents with global developmental delay, autism, seizures, and
agenesis/dysgenesis of the corpus callosum
explanation: >-
The case report describes autism as part of the Temtamy syndrome
phenotype.
genetic:
- name: C12orf57
gene_term:
preferred_term: C12orf57
term:
id: hgnc:29521
label: C12orf57
association: Pathogenic Variants
inheritance:
- name: Autosomal recessive inheritance
inheritance_term:
preferred_term: autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:23453666
reference_title: "Whole-exome sequencing identifies mutated c12orf57 in recessive corpus callosum hypoplasia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
members affected with corpus callosum hypoplasia (CCH) lacked syndromic
features and had consanguineous parents, suggesting recessive causes
explanation: >-
Affected individuals with consanguineous parents establish autosomal
recessive inheritance of the C12orf57-related syndrome.
evidence:
- reference: PMID:23453666
reference_title: "Whole-exome sequencing identifies mutated c12orf57 in recessive corpus callosum hypoplasia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Exome sequence analysis identified C12orf57 mutations at the initiator
methionine codon in four different families.
explanation: >-
The founding study identifies recurrent C12orf57 start-loss mutations in
multiple families with the syndrome.
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
we noted a high carrier frequency of an ancient startloss founder mutation
explanation: >-
The delineation study identifies a recurrent ancestral start-loss founder
variant as a common cause of the syndrome.
- reference: PMID:24798461
reference_title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
two compound heterozygous loss-of-function mutations in C12orf57
identified by exome sequencing, including a novel nonsense mutation
explanation: >-
The report documents compound heterozygous loss-of-function variants,
supporting a loss-of-function disease mechanism.
diagnosis:
- name: Molecular Genetic Testing
diagnosis_term:
preferred_term: molecular genetic testing
term:
id: MAXO:0000533
label: molecular genetic testing
description: >-
Exome or targeted sequencing of C12orf57 confirms the diagnosis by
identifying biallelic pathogenic variants, frequently the recurrent
start-loss founder allele.
evidence:
- reference: PMID:24798461
reference_title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
In patients with genetically heterogeneous disorders such as intellectual
disability or epilepsy, exome sequencing is a powerful tool to elucidate
the underlying genetic cause.
explanation: >-
Exome sequencing is the diagnostic modality used to identify C12orf57
variants in these patients.
differential_diagnoses:
- name: Kabuki Syndrome
disease_term:
preferred_term: Kabuki syndrome
term:
id: MONDO:0016512
label: Kabuki syndrome
description: >-
Temtamy syndrome can present with overlapping features of Kabuki-like
syndrome, including long palpebral fissures with eversion of the lateral
lower eyelid and persistent fetal fingertip pads, making Kabuki syndrome an
important clinical mimic.
distinguishing_features:
- Biallelic C12orf57 variants confirm Temtamy syndrome.
- Kabuki syndrome is caused by KMT2D or KDM6A variants.
evidence:
- reference: PMID:37451886
reference_title: "A novel pathogenic compound heterozygous variant in C12orf57 gene in a child with Temtamy syndrome presenting with overlapping phenotypic features of Kabuki-like syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The clinical features were in favor of Kabuki-like syndrome.
explanation: >-
The case report documents phenotypic overlap with Kabuki-like syndrome,
supporting it as a differential diagnosis.
clinical_trials: []
datasets: []
treatments:
- name: Antiseizure Medication
description: >-
Seizures in Temtamy syndrome are often intractable and managed with
antiseizure (antiepileptic) drugs as supportive symptomatic therapy; no
disease-modifying treatment exists.
therapeutic_modality: SMALL_MOLECULE
treatment_term:
preferred_term: pharmacotherapy
term:
id: MAXO:0000058
label: pharmacotherapy
therapeutic_agent:
- preferred_term: anticonvulsant agent
term:
id: NCIT:C264
label: Anticonvulsant Agent
target_mechanisms:
- target: Seizure
treatment_effect: MODULATES
description: >-
Antiseizure medication suppresses the seizures arising from increased
excitatory drive, but does not address the underlying genetic defect.
evidence:
- reference: PMID:24798461
reference_title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
including agenesis/hypoplasia of the corpus callosum, optic coloboma, and
intractable seizures
explanation: >-
The intractable seizures of the syndrome are the target of
symptomatic antiseizure therapy.
evidence:
- reference: PMID:24798461
reference_title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
including agenesis/hypoplasia of the corpus callosum, optic coloboma, and
intractable seizures
explanation: >-
The intractable seizures documented in patients are managed with
antiseizure pharmacotherapy as supportive care.
- name: Supportive and Multidisciplinary Care
description: >-
Management is supportive and multidisciplinary, addressing developmental
delay, intellectual disability, seizures, ophthalmologic involvement, and
cardiac disease.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
While all patients presented with intellectual disability/developmental
delay, the frequency of other phenotypic features was variable
explanation: >-
The multisystem phenotype documented in the cohort requires
multidisciplinary supportive management.
- name: Genetic Counseling
description: >-
Given autosomal recessive inheritance and recurrent founder alleles in
certain populations, genetic counseling and carrier testing are important
for affected families.
treatment_term:
preferred_term: Genetic Counseling
term:
id: NCIT:C15240
label: Genetic Counseling
evidence:
- reference: PMID:29383837
reference_title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
especially in those who trace their ancestry to Saudi Arabia where a
founder C12orf57 mutation is among the most common recessive causes of
intellectual disability
explanation: >-
The high carrier frequency of a recessive founder mutation supports the
value of genetic counseling and carrier testing.
# NOTE: No GeneReviews chapter exists for Temtamy syndrome (C12orf57); the
# founding clinical/molecular descriptions (Zahrani 2013, Akizu 2013) and the
# combined delineation cohort (Alrakaf 2018) serve as the baseline references.
references:
- reference: PMID:23453665
title: "Mutations in c12orf57 cause a syndromic form of colobomatous microphthalmia."
- reference: PMID:23453666
title: "Whole-exome sequencing identifies mutated c12orf57 in recessive corpus callosum hypoplasia."
- reference: PMID:24798461
title: "Exome sequencing identifies compound heterozygous mutations in C12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures."
- reference: PMID:29383837
title: "Further delineation of Temtamy syndrome of corpus callosum and ocular abnormalities."
- reference: PMID:31853307
title: "Temtamy syndrome caused by a new C12orf57 variant in a Chinese boy, including pedigree analysis and literature review."
- reference: PMID:35791610
title: "C12orf57 pathogenic variants: a unique cause of developmental encephalopathy in a south Indian child."
- reference: PMID:37451886
title: "A novel pathogenic compound heterozygous variant in C12orf57 gene in a child with Temtamy syndrome presenting with overlapping phenotypic features of Kabuki-like syndrome."
- reference: PMID:39974932
title: "C12ORF57: a novel principal regulator of synaptic AMPA currents and excitatory neuronal homeostasis."
References & Deep Research
References
8Deep Research
11. Disease Information
1.1 Concise overview
C12orf57-related Temtamy syndrome (MIM 218340) is an extremely rare autosomal recessive syndromic neurodevelopmental disorder characterized by global developmental delay / intellectual disability, epilepsy, frequent corpus callosum hypoplasia/agenesis, and variable ocular anomalies including coloboma, often with autistic features, hypotonia, and dysmorphic facial features. (wang2020temtamysyndromecaused pages 1-2, akizu2013wholeexomesequencingidentifies pages 2-4, platzer2014exomesequencingidentifies pages 3-5)
Temtamy preaxial brachydactyly syndrome (TPBS; MIM 605282) is an autosomal recessive multiple-congenital-anomaly syndrome with hallmark bilateral symmetric preaxial brachydactyly and hyperphalangism, frequently accompanied by hearing loss, dental anomalies, craniofacial dysmorphism, and growth retardation; it is caused by loss-of-function mutations in CHSY1. (li2010temtamypreaxialbrachydactyly pages 1-2, li2010temtamypreaxialbrachydactyly pages 5-7, li2010temtamypreaxialbrachydactyly pages 4-5)
1.2 Key identifiers and synonyms (from retrieved sources)
- C12orf57-related Temtamy syndrome: reported as “Temtamy syndrome (MIM 218340)” in case-series literature. (wang2020temtamysyndromecaused pages 1-2, alfiya2022c12orf57pathogenicvariants pages 1-3)
- TPBS: “Temtamy preaxial brachydactyly syndrome (MIM 605282)”. (li2010temtamypreaxialbrachydactyly pages 1-2)
Common alternative names used in retrieved sources (non-exhaustive; varies by author): - “Temtamy syndrome of corpus callosum and ocular abnormalities” (as cited in a 2024 founder-mutation perspective). (marafi2024foundermutationsand pages 6-7) - “Syndromic form of intellectual disability characterized by agenesis/hypoplasia of the corpus callosum, optic/chorioretinal coloboma, and intractable seizures” (used to describe C12orf57-related disease). (platzer2014exomesequencingidentifies pages 1-2)
1.3 Evidence types
Most available disease characterization in retrieved sources is derived from: - Aggregated case series / literature reviews (e.g., compiled cohorts of 17–56+ patients) (wang2020temtamysyndromecaused pages 2-4, platzer2014exomesequencingidentifies pages 3-5) - Individual case reports (e.g., a Chinese patient with a novel C12orf57 start-codon variant) (wang2020temtamysyndromecaused pages 1-2) - Genetic-discovery family studies (consanguineous multiplex families) including functional assays for one recurrent allele (akizu2013wholeexomesequencingidentifies pages 2-4, akizu2013wholeexomesequencingidentifies pages 5-7)
2. Etiology
2.1 Disease causal factors
A) C12orf57-related Temtamy syndrome (MIM 218340)
- Cause: biallelic pathogenic variants in C12orf57, frequently affecting the translation initiation codon (start-loss). (wang2020temtamysyndromecaused pages 1-2, akizu2013wholeexomesequencingidentifies pages 2-4)
- Inheritance: autosomal recessive with segregation consistent with full penetrance within reported pedigrees. (wang2020temtamysyndromecaused pages 2-4, akizu2013wholeexomesequencingidentifies pages 2-4)
B) TPBS (MIM 605282)
- Cause: biallelic loss-of-function variants in CHSY1, encoding chondroitin synthase 1. (li2010temtamypreaxialbrachydactyly pages 1-2, li2010temtamypreaxialbrachydactyly pages 5-7)
- Inheritance: autosomal recessive, commonly reported in consanguineous families. (li2010temtamypreaxialbrachydactyly pages 1-2, sher2014anovelchsy1 pages 2-4)
2.2 Risk factors
- Consanguinity / endogamy is repeatedly observed in reported C12orf57 cases; in a compiled review of 56 patients, 49/56 (87.7%) were from consanguineous families. (wang2020temtamysyndromecaused pages 2-4)
- Geographic clustering suggests population-specific recurrence (Middle East enrichment): in Wang’s 2020 review, 54/56 (96.4%) of reported patients were from Middle Eastern countries, consistent with founder effects and ascertainment patterns. (wang2020temtamysyndromecaused pages 1-2)
2.3 Protective factors / gene–environment interactions
No protective alleles or gene–environment interactions were identified in the retrieved sources.
3. Phenotypes
3.1 C12orf57-related Temtamy syndrome: phenotype spectrum (with frequencies)
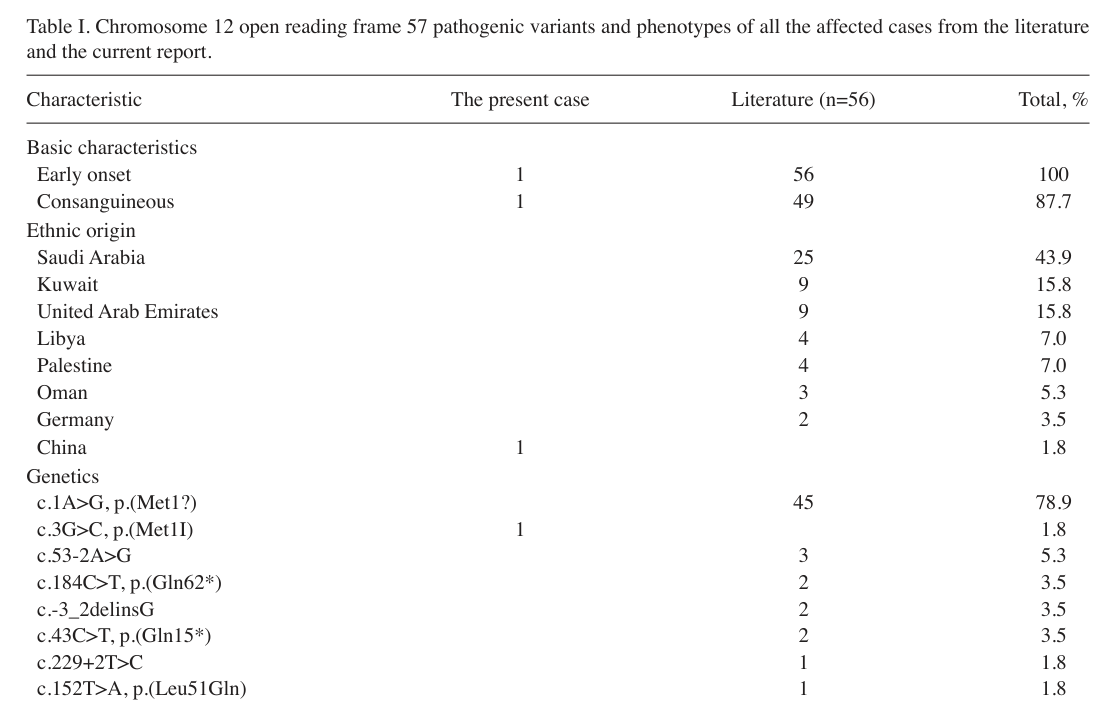
The most quantitative phenotype synthesis in retrieved sources comes from Wang 2020 (n=56 literature review) and Platzer 2014 (n=17 aggregated from 7 families). (wang2020temtamysyndromecaused pages 2-4, platzer2014exomesequencingidentifies pages 3-5)
| Clinical feature | Frequency/notes (with source and n/N) | Suggested HPO term(s) |
|---|---|---|
| Global developmental delay / developmental delay | 56/56 (100%) in literature review summarized by Wang 2020; all 17/17 had developmental delay in Platzer 2014 cohort summary (wang2020temtamysyndromecaused pages 2-4, platzer2014exomesequencingidentifies pages 3-5) | HP:0001263 Global developmental delay; HP:0001268 Mental deterioration / developmental regression not established |
| Intellectual disability, severe | Moderate-to-severe intellectual disability reported in Akizu families; severe ID in 11/11 cases with specified cognitive testing in Platzer 2014 (akizu2013wholeexomesequencingidentifies pages 2-4, platzer2014exomesequencingidentifies pages 3-5) | HP:0010864 Intellectual disability, severe |
| Epilepsy / seizures | 41/56 (73.7%) in Wang 2020 review; 41/56 (~73.2%) in Wang 2020 text; 15/17 (88%) in Platzer 2014 summary; onset by age ≤3 years in 9/9 specified cases in Platzer 2014 (wang2020temtamysyndromecaused pages 2-4, wang2020temtamysyndromecaused pages 4-6, platzer2014exomesequencingidentifies pages 3-5) | HP:0001250 Seizure; HP:0002373 EEG abnormality |
| Refractory / difficult-to-control seizures | Historically 37.5% relatively refractory and only 15.6% seizure-free in Wang 2020 review; difficult to control in 7/9 (78%) in Platzer 2014 despite multiple AED trials (wang2020temtamysyndromecaused pages 4-6, platzer2014exomesequencingidentifies pages 3-5) | HP:0001272 Cerebral seizure resistant to treatment |
| Absent or very limited speech | 41/55 (74.5%) absent speech in Wang 2020 review; 15/17 had no active speech in Platzer 2014 summary (wang2020temtamysyndromecaused pages 2-4, platzer2014exomesequencingidentifies pages 3-5) | HP:0001344 Absent speech; HP:0000750 Delayed speech and language development |
| Generalized hypotonia | 40/56 (71.9%) in Wang 2020 review; hypotonia present in Akizu families (wang2020temtamysyndromecaused pages 2-4, akizu2013wholeexomesequencingidentifies pages 2-4) | HP:0001290 Generalized hypotonia |
| Autistic behavior / autistic features | 40/55 (72.7%) in Wang 2020 review; all 10/10 affected had autistic features in Akizu families; ASD reported in 6/17 (35%) in Platzer 2014 summary (wang2020temtamysyndromecaused pages 2-4, akizu2013wholeexomesequencingidentifies pages 2-4, platzer2014exomesequencingidentifies pages 3-5) | HP:0000729 Autistic behavior |
| Corpus callosum abnormality (hypoplasia/agenesis) | ~34/54 (61.8%) in Wang 2020 review; corpus callosum absent in 3 and hypoplastic in 5 of 8 imaged in Akizu; 12/15 (80%) in Platzer 2014 summary (wang2020temtamysyndromecaused pages 4-6, akizu2013wholeexomesequencingidentifies pages 2-4, platzer2014exomesequencingidentifies pages 3-5) | HP:0001274 Agenesis of corpus callosum; HP:0002079 Hypoplasia of the corpus callosum |
| Ventriculomegaly / enlarged ventricles | 17/50 (35.3%) in Wang 2020 review; thalamic hypoplasia with enlarged V-shaped third ventricle described in Akizu families (wang2020temtamysyndromecaused pages 4-6, akizu2013wholeexomesequencingidentifies pages 2-4) | HP:0002119 Ventriculomegaly; HP:0006842 Abnormality of the third ventricle |
| Ocular anomalies, overall | 26/56 (46.4%) in Wang 2020 review (as summarized in Wang text); visual abnormalities in 4/10 in Akizu; visual impairment in 9/17 (53%) in Platzer 2014 summary (wang2020temtamysyndromecaused pages 2-4, akizu2013wholeexomesequencingidentifies pages 2-4, platzer2014exomesequencingidentifies pages 3-5) | HP:0000478 Abnormality of the eye |
| Coloboma / chorioretinal coloboma | 8/55 (14.5%) coloboma in Wang 2020 review; optic/chorioretinal coloboma in 5/17 (29%) in Platzer 2014 summary (wang2020temtamysyndromecaused pages 4-6, platzer2014exomesequencingidentifies pages 3-5) | HP:0000589 Coloboma of optic disc; HP:0000490 Chorioretinal coloboma; HP:0000486 Strabismus not specifically established |
| Dysmorphic facial features | 36/55 (66.1%) in Wang 2020 review; Wang abstract/text also notes dysmorphic craniofacial appearance as common (wang2020temtamysyndromecaused pages 4-6, wang2020temtamysyndromecaused pages 1-2) | HP:0001999 Facial dysmorphism |
| Atrial septal defect / cardiac defect | 16/55 (30.4%) atrial septal defect in Wang 2020 review; cardiac defects variably reported in case literature (wang2020temtamysyndromecaused pages 4-6) | HP:0001631 Atrial septal defect |
| Spasticity | 10/17 (59%) in Platzer 2014 summary (platzer2014exomesequencingidentifies pages 3-5) | HP:0001257 Spasticity |
| Visual impairment | 9/17 (53%) in Platzer 2014 summary; abnormal visual function in 4/10 in Akizu families (akizu2013wholeexomesequencingidentifies pages 2-4, platzer2014exomesequencingidentifies pages 3-5) | HP:0000505 Visual impairment |
Table: This table summarizes the main reported phenotypes of C12orf57-related Temtamy syndrome using frequencies from Wang 2020 and Platzer 2014, with related HPO suggestions. It is useful for structured phenotype annotation and for comparing feature prevalence across published case series.
Key clinical concepts (current understanding): - Neurodevelopmental impairment is universal (developmental delay 100% in Wang review). (wang2020temtamysyndromecaused pages 2-4) - Epilepsy is common (73.7% in Wang review; 88% in Platzer summary) and may be treatment-resistant in a substantial subset. (wang2020temtamysyndromecaused pages 4-6, platzer2014exomesequencingidentifies pages 3-5) - Brain imaging abnormalities frequently involve the corpus callosum; Akizu described variable severity from hypoplasia to agenesis within and across families. (akizu2013wholeexomesequencingidentifies pages 2-4) - Ocular involvement may include coloboma (14.5% in Wang review; 29% in Platzer summary). (wang2020temtamysyndromecaused pages 4-6, platzer2014exomesequencingidentifies pages 3-5)
3.2 Age of onset, progression, severity
- C12orf57-related disease is typically early onset. In the Platzer aggregated series, where specified, seizure onset occurred by ≤3 years (9/9). (platzer2014exomesequencingidentifies pages 3-5)
- Severity is variable, including within families, which has been proposed to relate to hypomorphic effects of start-codon variants that reduce (rather than abolish) protein production. (akizu2013wholeexomesequencingidentifies pages 5-7)
3.3 TPBS: phenotype spectrum (qualitative)
TPBS has a distinct phenotype dominated by limb development anomalies: - “Typical preaxial brachydactyly of digits 1–3” with hyper- and symphalangism, duplicated phalanges/metatarsals, and additional skeletal anomalies (radio-ulnar synostosis, carpal/tarsal fusions). (li2010temtamypreaxialbrachydactyly pages 2-4, li2010temtamypreaxialbrachydactyly pages 4-5) - Syndromic features include facial dysmorphism, dental anomalies, growth retardation/short stature, and frequent sensorineural hearing loss. (li2010temtamypreaxialbrachydactyly pages 1-2, sher2014anovelchsy1 pages 2-4)
3.4 Quality-of-life impact
No standardized QoL instruments (e.g., EQ-5D, PROMIS) were identified in retrieved sources. However, the high rates of absent speech, severe ID, and refractory seizures indicate substantial functional impact in C12orf57-related disease. (wang2020temtamysyndromecaused pages 2-4, platzer2014exomesequencingidentifies pages 3-5)
4. Genetic / Molecular Information
4.1 Causal genes
- C12orf57 (Temtamy syndrome; MIM 218340). (wang2020temtamysyndromecaused pages 1-2, akizu2013wholeexomesequencingidentifies pages 2-4)
- CHSY1 (TPBS; MIM 605282). (li2010temtamypreaxialbrachydactyly pages 1-2)
4.2 Pathogenic variants (examples with evidence)
| Disease entity | Gene | Variant (c.; p.) | Variant type | Evidence/notes (founder, segregation, functional) | Reported in (paper, year) | URL |
|---|---|---|---|---|---|---|
| Temtamy syndrome (C12orf57-related) | C12orf57 | c.1A>G; p.Met1? / p.M1V | Start-loss / initiator codon variant | Homozygous in multiple consanguineous Arab families; segregated with disease under AR inheritance; absent from >1,400 exomes and ethnically matched controls in Akizu; recurrent in Arab patients and suggested founder effect; functional data show AUG→GUG can still initiate translation but with markedly reduced protein levels; 2024 ASD study again found the homozygous variant in affected brothers (akizu2013wholeexomesequencingidentifies pages 2-4, akizu2013wholeexomesequencingidentifies pages 5-7, alsarraj2024thegeneticlandscape pages 10-11, platzer2014exomesequencingidentifies pages 3-5, alfiya2022c12orf57pathogenicvariants pages 3-4) | Akizu et al., 2013; Platzer et al., 2014; Al-Sarraj et al., 2024; Alfiya et al., 2022 | https://doi.org/10.1016/j.ajhg.2013.02.004; https://doi.org/10.1002/ajmg.a.36592; https://doi.org/10.3389/fgene.2024.1363849; https://doi.org/10.1007/s12041-022-01371-0 |
| Temtamy syndrome (C12orf57-related) | C12orf57 | c.3G>C; p.Met1Ile | Start-loss / start-codon variant | Novel homozygous variant in a Chinese boy; segregated with AR inheritance and full penetrance in pedigree; predicted to abolish translation / cause loss of function; expanded ethnic spectrum beyond predominantly Middle Eastern cases (wang2020temtamysyndromecaused pages 1-2, wang2020temtamysyndromecaused pages 4-6, wang2020temtamysyndromecaused pages 2-4) | Wang et al., 2020 | https://doi.org/10.3892/etm.2019.8183 |
| Temtamy syndrome (C12orf57-related) | C12orf57 | c.184C>T; p.Gln62* | Nonsense / stop-gain | Novel nonsense allele reported in trans with c.1A>G in two siblings from nonconsanguineous German parents; compound heterozygous loss-of-function genotype confirmed by parental studies; associated with severe ID, callosal hypoplasia, chorioretinal coloboma, and intractable seizures (platzer2014exomesequencingidentifies pages 3-5, platzer2014exomesequencingidentifies pages 1-2, platzer2014exomesequencingidentifies pages 2-3) | Platzer et al., 2014 | https://doi.org/10.1002/ajmg.a.36592 |
| Temtamy syndrome (C12orf57-related) | C12orf57 | c.C43T; p.Q15X | Nonsense / stop-gain | Premature stop codon; reported as compound heterozygous with c.1A>G in a South Indian child; Sanger-confirmed in proband and parents; interpreted as truncating loss-of-function under ACMG framework (alfiya2022c12orf57pathogenicvariants pages 3-4, alfiya2022c12orf57pathogenicvariants pages 4-5) | Alfiya et al., 2022 | https://doi.org/10.1007/s12041-022-01371-0 |
| Temtamy preaxial brachydactyly syndrome (TPBS) | CHSY1 | c.14delG; p.G5AfsX30 | Frameshift | Homozygous LOF allele in TPBS families; cosegregated with autosomal recessive disease; predicted truncation / nonfunctional protein (li2010temtamypreaxialbrachydactyly pages 5-7, li2010temtamypreaxialbrachydactyly pages 4-5) | Li et al., 2010 | https://doi.org/10.1016/j.ajhg.2010.10.003 |
| Temtamy preaxial brachydactyly syndrome (TPBS) | CHSY1 | c.55-84del30; p.G19_L28del | In-frame deletion | Reported exon 1 pathogenic deletion in TPBS; part of recurrent CHSY1 loss-of-function spectrum in consanguineous families; absent from controls in original study (li2010temtamypreaxialbrachydactyly pages 1-2, li2010temtamypreaxialbrachydactyly pages 2-4, li2010temtamypreaxialbrachydactyly pages 4-5) | Li et al., 2010 | https://doi.org/10.1016/j.ajhg.2010.10.003 |
| Temtamy preaxial brachydactyly syndrome (TPBS) | CHSY1 | c.205C>T; p.Q69X | Nonsense | Protein-truncating LOF allele identified in TPBS families with AR segregation; supports CHSY1 haploinsufficiency is not mechanism, but biallelic loss is pathogenic (li2010temtamypreaxialbrachydactyly pages 1-2, li2010temtamypreaxialbrachydactyly pages 5-7, li2010temtamypreaxialbrachydactyly pages 4-5) | Li et al., 2010 | https://doi.org/10.1016/j.ajhg.2010.10.003 |
| Temtamy preaxial brachydactyly syndrome (TPBS) | CHSY1 | c.321-3C>G | Splice-site | Acceptor splice variant causing exon 2 skipping, frameshift and premature truncation; strong functional evidence for loss of function (li2010temtamypreaxialbrachydactyly pages 5-7) | Li et al., 2010 | https://doi.org/10.1016/j.ajhg.2010.10.003 |
| Temtamy preaxial brachydactyly syndrome (TPBS) | CHSY1 | c.1616C>G; p.P539R | Missense | Affects highly conserved residue in CHSY1; interpreted as deleterious and disruptive of protein function; part of pathogenic CHSY1 spectrum in TPBS (li2010temtamypreaxialbrachydactyly pages 5-7, sher2014anovelchsy1 pages 2-4) | Li et al., 2010 | https://doi.org/10.1016/j.ajhg.2010.10.003 |
| Temtamy preaxial brachydactyly syndrome (TPBS) | CHSY1 | c.1897G>A; p.D633N | Missense | Homozygous in Pakistani family; parents heterozygous carriers; absent in 100 matched controls; alters conserved Asp633 within DXD motif required for glycosyltransferase activity, supporting enzymatic loss of function (sher2014anovelchsy1 pages 4-4, sher2014anovelchsy1 pages 2-4) | Sher & Naeem, 2014 | https://doi.org/10.1016/j.ejmg.2013.11.001 |
Table: This table summarizes key pathogenic variants reported for the two distinct entities often called Temtamy syndrome: C12orf57-related Temtamy syndrome and CHSY1-related Temtamy preaxial brachydactyly syndrome. It highlights variant class, segregation, founder evidence, and functional support using only the gathered evidence snippets.
Notable quantitative variant statistics (C12orf57-related): - In Wang’s 2020 review of 56 patients, c.1A>G was the most frequent reported variant (45/56; 80.3%). (wang2020temtamysyndromecaused pages 4-6)
Evidence supporting loss-of-function: - For C12orf57 start-codon variant c.1A>G, Akizu showed the mutant AUG→GUG start can still initiate translation but produces markedly reduced protein levels, consistent with a loss-of-function/hypomorphic mechanism. (akizu2013wholeexomesequencingidentifies pages 5-7)
4.3 Modifier genes / epigenetic information
No modifier genes or epigenetic mechanisms specific to Temtamy syndrome were identified in retrieved sources.
4.4 Chromosomal abnormalities (Temtamy-like)
A 2003 report described a Temtamy-like phenotype (callosal agenesis, colobomas, profound ID, hearing loss) with a de novo balanced translocation t(2;9)(p24;q32), highlighting historical locus-mapping approaches and the possibility of chromosomal disruption in Temtamy-like presentations. (talisetti2003temtamy‐likesyndromeassociated pages 1-3)
5. Environmental Information
No environmental, lifestyle, toxicant, or infectious causal factors were identified in the retrieved sources. These syndromes are primarily genetic. (wang2020temtamysyndromecaused pages 1-2, li2010temtamypreaxialbrachydactyly pages 1-2)
6. Mechanism / Pathophysiology
6.1 C12orf57-related Temtamy syndrome (proposed mechanism; limited mechanistic detail available)
Causal chain (supported components): 1. Biallelic C12orf57 variants (often start-loss) → 2. Reduced C12orf57 protein levels (experimental evidence for c.1A>G) and cytoplasmic localization of the protein → 3. Disrupted neurodevelopmental processes required for corpus callosum development and broader brain development → 4. Clinical manifestations: callosal hypoplasia/agenesis, seizures/epilepsy, developmental delay/ID, autistic features, and ocular anomalies. (akizu2013wholeexomesequencingidentifies pages 5-7, akizu2013wholeexomesequencingidentifies pages 2-4)
Functional notes: - Akizu found the major neural transcript to be highly enriched in fetal brain and concluded the gene is “required for development of the human corpus callosum,” but molecular pathways remain poorly defined. (akizu2013wholeexomesequencingidentifies pages 5-7, akizu2013wholeexomesequencingidentifies pages 1-2) - A 2024 cerebral organoid/ribosome study (preprint) described C12orf57 as “an important factor for early brain development” and noted that its mRNA contains a TOP-like element, making its translation sensitive to ribosome availability and global translation state; this is a mechanistic clue at the level of translational regulation rather than disease-specific causation. (ni2024aninappropriatedecline pages 13-17)
Suggested GO / CL terms (hypothesis-generating; not explicitly asserted in sources): - GO (process): corpus callosum development; regulation of translation; neurogenesis. - CL (cell types, based on organoid discussion): radial glia / neural progenitor cells (mentioned as impacted in organoid study). (ni2024aninappropriatedecline pages 13-17)
6.2 TPBS (CHSY1) mechanism: chondroitin sulfate biosynthesis with BMP/NOTCH crosstalk
Causal chain (supported components): 1. Biallelic CHSY1 loss-of-function → 2. Impaired chondroitin sulfate biosynthesis (CHSY1 provides enzymatic activities needed to build CS repeating disaccharides) → 3. Perturbed extracellular matrix/proteoglycan-mediated developmental signaling and morphogenesis → 4. Limb/digit, craniofacial, and inner-ear developmental anomalies consistent with TPBS. (li2010temtamypreaxialbrachydactyly pages 5-7)
Pathways and processes: - BMP signaling: CHSY1/chsy1 is described as a “potential target of BMP signaling,” with zebrafish data indicating BMP signaling negatively regulates chsy1 expression and BMP pathway perturbations phenocopy chsy1 knockdown. (li2010temtamypreaxialbrachydactyly pages 5-7, li2010temtamypreaxialbrachydactyly pages 1-2) - NOTCH signaling: A separate 2010 study proposed that CHSY1 inhibits NOTCH extracellularly via a Fringe domain and that loss leads to increased Notch signaling (e.g., jag2 upregulation; lim1 silencing) contributing to abnormal ossification/patterning. (tian2010lossofchsy1 pages 9-10, tian2010lossofchsy1 pages 8-9)
Suggested GO / CL / pathway terms (supported directionally by the above): - GO (process): glycosaminoglycan biosynthetic process; cartilage development; limb development; Notch signaling pathway; BMP signaling pathway. - CL (cell types): chondrocytes; inner-ear sensory epithelium/hair-cell–adjacent epithelium (zebrafish expression in chondrocytes and inner ear). (li2010temtamypreaxialbrachydactyly pages 5-7)
7. Anatomical Structures Affected
C12orf57-related Temtamy syndrome
- CNS: corpus callosum (hypoplasia/agenesis), thalamic/ventricular abnormalities described in family studies. (akizu2013wholeexomesequencingidentifies pages 2-4)
- Suggested UBERON: corpus callosum; thalamus; lateral ventricle; third ventricle.
- Eye: optic/chorioretinal coloboma and other ocular anomalies. (platzer2014exomesequencingidentifies pages 3-5, wang2020temtamysyndromecaused pages 4-6)
- Suggested UBERON: retina; choroid; optic nerve.
- Heart: atrial septal defects reported in an aggregated case review. (wang2020temtamysyndromecaused pages 4-6)
TPBS (CHSY1)
- Limbs/digits: preaxial digits 1–3 (hands/feet), metacarpals/metatarsals, carpal/tarsal bones. (li2010temtamypreaxialbrachydactyly pages 4-5)
- Auditory system: sensorineural hearing impairment is frequent. (li2010temtamypreaxialbrachydactyly pages 4-5)
8. Temporal Development
C12orf57-related Temtamy syndrome
- Onset: congenital/infancy presentation with developmental delay; seizures typically begin in early childhood and may start in infancy. (wang2020temtamysyndromecaused pages 1-2, platzer2014exomesequencingidentifies pages 3-5)
- Course: chronic lifelong neurodevelopmental disability; seizure control is variable, with a substantial refractory subset. (wang2020temtamysyndromecaused pages 4-6, platzer2014exomesequencingidentifies pages 3-5)
TPBS
- Onset: congenital limb malformations are present at birth (by definition of brachydactyly/hyperphalangism syndrome). (li2010temtamypreaxialbrachydactyly pages 1-2)
9. Inheritance and Population
C12orf57-related Temtamy syndrome
- Inheritance: autosomal recessive with frequent consanguinity. (akizu2013wholeexomesequencingidentifies pages 2-4, wang2020temtamysyndromecaused pages 2-4)
- Founder effect: Platzer explicitly states that the recurrent c.1A>G (p.Met1?) observed in Arab-descent patients “strongly suggests a founder effect within the Arab population.” (platzer2014exomesequencingidentifies pages 3-5)
- Epidemiology: prevalence/incidence not available in retrieved sources. However, case aggregation provides a minimal evidence base:
- Wang 2020 summarized 56 reported patients with C12orf57 pathogenic variants at that time. (wang2020temtamysyndromecaused pages 2-4)
- Platzer 2014 summarized 17 patients from 7 families (as-of their publication). (platzer2014exomesequencingidentifies pages 3-5)
TPBS
- Inheritance: autosomal recessive. (li2010temtamypreaxialbrachydactyly pages 1-2)
- Epidemiology: not available in retrieved sources.
10. Diagnostics
10.1 Clinical evaluation (C12orf57-related)
Common diagnostic components described across reports include: - Neurologic assessment and EEG for seizures. (wang2020temtamysyndromecaused pages 1-2, talisetti2003temtamy‐likesyndromeassociated pages 1-3) - Brain MRI to assess corpus callosum and ventricles. (wang2020temtamysyndromecaused pages 1-2, akizu2013wholeexomesequencingidentifies pages 2-4) - Ophthalmologic evaluation for coloboma/microphthalmia. (wang2020temtamysyndromecaused pages 4-6, talisetti2003temtamy‐likesyndromeassociated pages 1-3) - Cardiac evaluation (e.g., ASD/VSD) when indicated. (wang2020temtamysyndromecaused pages 4-6)
10.2 Genetic testing
- Whole-exome sequencing (WES) is repeatedly used to identify causal C12orf57 variants in affected children and families, with segregation analysis by Sanger sequencing. (wang2020temtamysyndromecaused pages 2-4, akizu2013wholeexomesequencingidentifies pages 2-4)
- A representative filtering/interpretation pipeline (depth thresholds, population frequency filters, in silico predictors, Sanger confirmation) is described in Wang 2020. (wang2020temtamysyndromecaused pages 2-4)
10.3 Differential diagnosis
Not comprehensively addressed in retrieved sources; however, historical “Temtamy-like” reports stress that overlapping syndromes with corpus callosum agenesis and ocular colobomas exist, and chromosomal abnormalities can produce similar phenotypes. (talisetti2003temtamy‐likesyndromeassociated pages 1-3)
11. Outcome / Prognosis
C12orf57-related Temtamy syndrome
- Quantitative outcome data are limited, but severe neurodevelopmental impairment is common: severe ID in 11/11 where specified in Platzer’s cohort summary and absent speech in a majority. (platzer2014exomesequencingidentifies pages 3-5)
- Seizure prognosis is variable: Wang 2020 cites a substantial refractory proportion and a relatively small seizure-free fraction in historical cases. (wang2020temtamysyndromecaused pages 4-6)
No survival curves or life expectancy estimates were identified in retrieved sources.
12. Treatment
12.1 Pharmacotherapy
- Antiseizure medications are the primary disease-directed therapy described. In a single C12orf57-related case report, oxcarbazepine dosing was escalated and the child was reported “seizure-free for 1 month” during follow-up; this illustrates symptomatic management rather than disease modification. (wang2020temtamysyndromecaused pages 1-2)
MAXO suggestions (general, not explicitly in sources): anticonvulsant therapy; developmental therapy; supportive care.
12.2 Supportive/rehabilitative care
- Given the high rates of developmental delay, hypotonia, and absent speech, supportive therapies (PT/OT/speech therapy) are implied but not described quantitatively in retrieved sources.
12.3 Clinical trials / advanced therapeutics
- No disease-specific interventional clinical trials were identified in this run. (clinical-trials search returned none relevant)
13. Prevention
- Primary prevention is not applicable for a monogenic disorder in the usual sense, but genetic counseling and carrier testing are directly relevant due to autosomal recessive inheritance, particularly in consanguineous families. (wang2020temtamysyndromecaused pages 2-4)
- Secondary prevention: early identification of seizures and developmental issues to initiate symptomatic therapies.
- Reproductive options: prenatal/preimplantation genetic testing is not explicitly discussed in retrieved sources, but the literature emphasizes segregation testing and recurrence risk awareness. (wang2020temtamysyndromecaused pages 2-4)
14. Other Species / Natural Disease
No naturally occurring non-human disease analogs were identified in retrieved sources.
15. Model Organisms
C12orf57-related Temtamy syndrome
- Akizu reports conservation and notes a fly RNAi screen where knockdown of the fly ortholog produced a nonspecific “malformation death” phenotype in ~50% of treated flies, but without a detailed phenotypic match to human disease. (akizu2013wholeexomesequencingidentifies pages 7-8)
- A 2024 human cerebral organoid study (preprint) provides mechanistic context regarding translation sensitivity of TOP-like transcripts including C12orf57, but is not a Temtamy syndrome disease model per se. (ni2024aninappropriatedecline pages 13-17)
TPBS (CHSY1)
- Zebrafish morphants: multiple studies show chsy1 knockdown produces developmental defects, including skeletal/jaw defects, pectoral fin defects (~65%), and ocular anomalies (>50%), partially phenocopying human TPBS and supporting conserved developmental roles. (li2010temtamypreaxialbrachydactyly pages 1-2, tian2010lossofchsy1 pages 8-9)
Recent developments (prioritized 2023–2024)
- Founder mutation framing (2024): A Disease Models & Mechanisms Perspective highlights that some rare diseases in Arab populations may be largely driven by founder variants and lists C12ORF57 among such examples; it also cites “Temtamy syndrome of corpus callosum and ocular abnormalities” in its reference list. This is interpretive/public-health context rather than new mechanistic or variant discovery. (marafi2024foundermutationsand pages 4-5, marafi2024foundermutationsand pages 6-7)
- Variant recurrence in modern cohorts (2024): A Frontiers in Genetics ASD cohort reports segregation of a homozygous C12orf57 start-codon variant (c.A1G/p.M1V; CADD 21.9) in affected siblings and notes prior reporting in consanguineous Saudi/Kuwaiti patients with global developmental delay, autism, and epilepsy—demonstrating continuing clinical relevance of this recurrent allele in Middle Eastern populations. (alsarraj2024thegeneticlandscape pages 10-11)
- Systems-level translational regulation (2024 preprint): A bioRxiv study suggests C12orf57 is among transcripts with TOP-like motifs whose translation is sensitive to ribosome availability during early neurodevelopment in cerebral organoids. This provides a plausible mechanistic clue for why reduced C12orf57 dosage might be impactful in neurodevelopment, but it does not establish a Temtamy-specific pathway. (ni2024aninappropriatedecline pages 13-17)
- High-throughput 5′UTR functional screening (2023 preprint): A medRxiv study included C12orf57 among genes screened for 5′UTR variant effects on translation, but the authors report they could not validate endogenous protein changes for C12orf57 due to antibody limitations (no correct-sized band), so it does not provide definitive new functional findings for C12orf57. (plassmeyer2023amassivelyparallel pages 21-23, plassmeyer2023amassivelyparallel pages 32-35)
Real-world applications / implementations
- Clinical genomics (WES) as standard-of-care for heterogeneous syndromic neurodevelopmental disorders: Multiple Temtamy syndrome diagnoses in the retrieved literature were enabled by WES with segregation testing, illustrating real-world deployment of genomic diagnostics in rare disease. (wang2020temtamysyndromecaused pages 2-4, akizu2013wholeexomesequencingidentifies pages 2-4)
- Population genetics / founder mutation programs: The 2024 founder-mutation perspective and the continued observation of recurrent C12orf57 start-codon variants in Middle Eastern pedigrees support the utility of region-specific carrier screening and counseling strategies (conceptual; implementation details not provided in retrieved sources). (marafi2024foundermutationsand pages 4-5, platzer2014exomesequencingidentifies pages 3-5)
Data gaps / limitations of this report
- MONDO/Orphanet/ICD/MeSH identifiers and prevalence/incidence were not available from the retrieved texts in this run.
- No disease-specific guidelines, standardized clinical criteria, or interventional trials were identified in the retrieved sources.
- Mechanistic understanding of C12orf57 remains limited; available evidence primarily supports loss-of-function via reduced protein dosage, with emerging hints about translation regulation sensitivity. (akizu2013wholeexomesequencingidentifies pages 5-7, ni2024aninappropriatedecline pages 13-17)
Appendix: Key quoted statements from abstracts / key excerpts (as requested)
- C12orf57 start-codon functional effect: the mutant allele “was able to produce some protein, although less efficiently than the wild-type” and “Cells transduced with the mutant construct show notably reduced protein levels.” (akizu2013wholeexomesequencingidentifies pages 5-7)
- Founder effect statement (C12orf57 c.1A>G): recurrent observation “strongly suggests a founder effect within the Arab population.” (platzer2014exomesequencingidentifies pages 3-5)
- Diagnostic yield and phenotype frequencies (Wang 2020 review): developmental delay 56/56 (100%), seizures 41/56 (73.7%), hypotonia 40/56 (71.9%), autistic behavior 40/55 (72.7%). (wang2020temtamysyndromecaused pages 2-4)
Retrieved figure/table evidence
- Wang 2020 includes a table summarizing clinical-feature frequencies and variants across 56 reported cases, and a pedigree/variant figure; these were retrieved as images in this run. (wang2020temtamysyndromecaused media b3d8bfd5, wang2020temtamysyndromecaused media 805b1c71, wang2020temtamysyndromecaused media 21e24220)
References
-
(wang2020temtamysyndromecaused pages 1-2): Yanqin Wang, Ming Li, Yuanyuan Luo, Xin Zhao, Shuang Liao, Li Jiang, Xiujuan Li, and Min Zhong. Temtamy syndrome caused by a new c12orf57 variant in a chinese boy, including pedigree analysis and literature review. Experimental and therapeutic medicine, 19 1:327-332, Nov 2020. URL: https://doi.org/10.3892/etm.2019.8183, doi:10.3892/etm.2019.8183. This article has 8 citations and is from a peer-reviewed journal.
-
(li2010temtamypreaxialbrachydactyly pages 1-2): Yun Li, Kathrin Laue, Samia Temtamy, Mona Aglan, L. Damla Kotan, Gökhan Yigit, Husniye Canan, Barbara Pawlik, Gudrun Nürnberg, Emma L. Wakeling, Oliver W. Quarrell, Ingelore Baessmann, Matthew B. Lanktree, Mustafa Yilmaz, Robert A. Hegele, Khalda Amr, Klaus W. May, Peter Nürnberg, A. Kemal Topaloglu, Matthias Hammerschmidt, and Bernd Wollnik. Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of bmp signaling. The American Journal of Human Genetics, 87:757-767, Dec 2010. URL: https://doi.org/10.1016/j.ajhg.2010.10.003, doi:10.1016/j.ajhg.2010.10.003. This article has 89 citations.
-
(akizu2013wholeexomesequencingidentifies pages 2-4): Naiara Akizu, Nuri M. Shembesh, Tawfeg Ben-Omran, Laila Bastaki, Asma Al-Tawari, Maha S. Zaki, Roshan Koul, Emily Spencer, Rasim Ozgur Rosti, Eric Scott, Elizabeth Nickerson, Stacey Gabriel, Gilberto da Gente, Jiang Li, Matthew A. Deardorff, Laura K. Conlin, Margaret A. Horton, Elaine H. Zackai, Elliott H. Sherr, and Joseph G. Gleeson. Whole-exome sequencing identifies mutated c12orf57 in recessive corpus callosum hypoplasia. American journal of human genetics, 92 3:392-400, Mar 2013. URL: https://doi.org/10.1016/j.ajhg.2013.02.004, doi:10.1016/j.ajhg.2013.02.004. This article has 43 citations and is from a highest quality peer-reviewed journal.
-
(platzer2014exomesequencingidentifies pages 3-5): Konrad Platzer, Irina Hüning, Carolin Obieglo, Thomas Schwarzmayr, Rainer Gabriel, Tim M. Strom, Gabriele Gillessen‐Kaesbach, and Frank J. Kaiser. Exome sequencing identifies compound heterozygous mutations in c12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures. American Journal of Medical Genetics Part A, 164:1976-1980, Aug 2014. URL: https://doi.org/10.1002/ajmg.a.36592, doi:10.1002/ajmg.a.36592. This article has 14 citations.
-
(akizu2013wholeexomesequencingidentifies pages 5-7): Naiara Akizu, Nuri M. Shembesh, Tawfeg Ben-Omran, Laila Bastaki, Asma Al-Tawari, Maha S. Zaki, Roshan Koul, Emily Spencer, Rasim Ozgur Rosti, Eric Scott, Elizabeth Nickerson, Stacey Gabriel, Gilberto da Gente, Jiang Li, Matthew A. Deardorff, Laura K. Conlin, Margaret A. Horton, Elaine H. Zackai, Elliott H. Sherr, and Joseph G. Gleeson. Whole-exome sequencing identifies mutated c12orf57 in recessive corpus callosum hypoplasia. American journal of human genetics, 92 3:392-400, Mar 2013. URL: https://doi.org/10.1016/j.ajhg.2013.02.004, doi:10.1016/j.ajhg.2013.02.004. This article has 43 citations and is from a highest quality peer-reviewed journal.
-
(wang2020temtamysyndromecaused pages 4-6): Yanqin Wang, Ming Li, Yuanyuan Luo, Xin Zhao, Shuang Liao, Li Jiang, Xiujuan Li, and Min Zhong. Temtamy syndrome caused by a new c12orf57 variant in a chinese boy, including pedigree analysis and literature review. Experimental and therapeutic medicine, 19 1:327-332, Nov 2020. URL: https://doi.org/10.3892/etm.2019.8183, doi:10.3892/etm.2019.8183. This article has 8 citations and is from a peer-reviewed journal.
-
(platzer2014exomesequencingidentifies pages 2-3): Konrad Platzer, Irina Hüning, Carolin Obieglo, Thomas Schwarzmayr, Rainer Gabriel, Tim M. Strom, Gabriele Gillessen‐Kaesbach, and Frank J. Kaiser. Exome sequencing identifies compound heterozygous mutations in c12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures. American Journal of Medical Genetics Part A, 164:1976-1980, Aug 2014. URL: https://doi.org/10.1002/ajmg.a.36592, doi:10.1002/ajmg.a.36592. This article has 14 citations.
-
(wang2020temtamysyndromecaused pages 2-4): Yanqin Wang, Ming Li, Yuanyuan Luo, Xin Zhao, Shuang Liao, Li Jiang, Xiujuan Li, and Min Zhong. Temtamy syndrome caused by a new c12orf57 variant in a chinese boy, including pedigree analysis and literature review. Experimental and therapeutic medicine, 19 1:327-332, Nov 2020. URL: https://doi.org/10.3892/etm.2019.8183, doi:10.3892/etm.2019.8183. This article has 8 citations and is from a peer-reviewed journal.
-
(li2010temtamypreaxialbrachydactyly pages 2-4): Yun Li, Kathrin Laue, Samia Temtamy, Mona Aglan, L. Damla Kotan, Gökhan Yigit, Husniye Canan, Barbara Pawlik, Gudrun Nürnberg, Emma L. Wakeling, Oliver W. Quarrell, Ingelore Baessmann, Matthew B. Lanktree, Mustafa Yilmaz, Robert A. Hegele, Khalda Amr, Klaus W. May, Peter Nürnberg, A. Kemal Topaloglu, Matthias Hammerschmidt, and Bernd Wollnik. Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of bmp signaling. The American Journal of Human Genetics, 87:757-767, Dec 2010. URL: https://doi.org/10.1016/j.ajhg.2010.10.003, doi:10.1016/j.ajhg.2010.10.003. This article has 89 citations.
-
(li2010temtamypreaxialbrachydactyly pages 4-5): Yun Li, Kathrin Laue, Samia Temtamy, Mona Aglan, L. Damla Kotan, Gökhan Yigit, Husniye Canan, Barbara Pawlik, Gudrun Nürnberg, Emma L. Wakeling, Oliver W. Quarrell, Ingelore Baessmann, Matthew B. Lanktree, Mustafa Yilmaz, Robert A. Hegele, Khalda Amr, Klaus W. May, Peter Nürnberg, A. Kemal Topaloglu, Matthias Hammerschmidt, and Bernd Wollnik. Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of bmp signaling. The American Journal of Human Genetics, 87:757-767, Dec 2010. URL: https://doi.org/10.1016/j.ajhg.2010.10.003, doi:10.1016/j.ajhg.2010.10.003. This article has 89 citations.
-
(li2010temtamypreaxialbrachydactyly pages 5-7): Yun Li, Kathrin Laue, Samia Temtamy, Mona Aglan, L. Damla Kotan, Gökhan Yigit, Husniye Canan, Barbara Pawlik, Gudrun Nürnberg, Emma L. Wakeling, Oliver W. Quarrell, Ingelore Baessmann, Matthew B. Lanktree, Mustafa Yilmaz, Robert A. Hegele, Khalda Amr, Klaus W. May, Peter Nürnberg, A. Kemal Topaloglu, Matthias Hammerschmidt, and Bernd Wollnik. Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of bmp signaling. The American Journal of Human Genetics, 87:757-767, Dec 2010. URL: https://doi.org/10.1016/j.ajhg.2010.10.003, doi:10.1016/j.ajhg.2010.10.003. This article has 89 citations.
-
(sher2014anovelchsy1 pages 2-4): Gulab Sher and Muhammad Naeem. A novel chsy1 gene mutation underlies temtamy preaxial brachydactyly syndrome in a pakistani family. European journal of medical genetics, 57 1:21-4, Jan 2014. URL: https://doi.org/10.1016/j.ejmg.2013.11.001, doi:10.1016/j.ejmg.2013.11.001. This article has 28 citations and is from a peer-reviewed journal.
-
(tian2010lossofchsy1 pages 9-10): Jing Tian, Ling Ling, Mohammad Shboul, Hane Lee, Brian O'Connor, Barry Merriman, Stanley F. Nelson, Simon Cool, Osama H. Ababneh, Azmy Al-Hadidy, Amira Masri, Hanan Hamamy, and Bruno Reversade. Loss of chsy1, a secreted fringe enzyme, causes syndromic brachydactyly in humans via increased notch signaling. American journal of human genetics, 87 6:768-78, Dec 2010. URL: https://doi.org/10.1016/j.ajhg.2010.11.005, doi:10.1016/j.ajhg.2010.11.005. This article has 121 citations and is from a highest quality peer-reviewed journal.
-
(sher2014anovelchsy1 pages 4-4): Gulab Sher and Muhammad Naeem. A novel chsy1 gene mutation underlies temtamy preaxial brachydactyly syndrome in a pakistani family. European journal of medical genetics, 57 1:21-4, Jan 2014. URL: https://doi.org/10.1016/j.ejmg.2013.11.001, doi:10.1016/j.ejmg.2013.11.001. This article has 28 citations and is from a peer-reviewed journal.
-
(alfiya2022c12orf57pathogenicvariants pages 1-3): F. Alfiya, Manna Jose, Soumya V. Chandrasekharan, Soumya Sundaram, Madhusoodanan Urulangodi, Bejoy Thomas, Ashalatha Radhakrishnan, Moinak Banerjee, and Ramshekhar N. Menon. C12orf57 pathogenic variants: a unique cause of developmental encephalopathy in a south indian child. Journal of Genetics, Jun 2022. URL: https://doi.org/10.1007/s12041-022-01371-0, doi:10.1007/s12041-022-01371-0. This article has 5 citations and is from a peer-reviewed journal.
-
(marafi2024foundermutationsand pages 6-7): Dana Marafi. Founder mutations and rare disease in the arab world. Disease Models & Mechanisms, Jun 2024. URL: https://doi.org/10.1242/dmm.050715, doi:10.1242/dmm.050715. This article has 18 citations and is from a domain leading peer-reviewed journal.
-
(platzer2014exomesequencingidentifies pages 1-2): Konrad Platzer, Irina Hüning, Carolin Obieglo, Thomas Schwarzmayr, Rainer Gabriel, Tim M. Strom, Gabriele Gillessen‐Kaesbach, and Frank J. Kaiser. Exome sequencing identifies compound heterozygous mutations in c12orf57 in two siblings with severe intellectual disability, hypoplasia of the corpus callosum, chorioretinal coloboma, and intractable seizures. American Journal of Medical Genetics Part A, 164:1976-1980, Aug 2014. URL: https://doi.org/10.1002/ajmg.a.36592, doi:10.1002/ajmg.a.36592. This article has 14 citations.
-
(alsarraj2024thegeneticlandscape pages 10-11): Yasser Al-Sarraj, Rowaida Z. Taha, Eman Al-Dous, Dina Ahram, Somayyeh Abbasi, Eman Abuazab, Hibah Shaath, Wesal Habbab, Khaoula Errafii, Yosra Bejaoui, Maryam AlMotawa, Namat Khattab, Yasmin Abu Aqel, Karim E. Shalaby, Amina Al-Ansari, Marios Kambouris, Adel Abouzohri, Iman Ghazal, Mohammed Tolfat, Fouad Alshaban, Hatem El-Shanti, and Omar M. E. Albagha. The genetic landscape of autism spectrum disorder in the middle eastern population. Frontiers in Genetics, Mar 2024. URL: https://doi.org/10.3389/fgene.2024.1363849, doi:10.3389/fgene.2024.1363849. This article has 11 citations and is from a peer-reviewed journal.
-
(alfiya2022c12orf57pathogenicvariants pages 3-4): F. Alfiya, Manna Jose, Soumya V. Chandrasekharan, Soumya Sundaram, Madhusoodanan Urulangodi, Bejoy Thomas, Ashalatha Radhakrishnan, Moinak Banerjee, and Ramshekhar N. Menon. C12orf57 pathogenic variants: a unique cause of developmental encephalopathy in a south indian child. Journal of Genetics, Jun 2022. URL: https://doi.org/10.1007/s12041-022-01371-0, doi:10.1007/s12041-022-01371-0. This article has 5 citations and is from a peer-reviewed journal.
-
(alfiya2022c12orf57pathogenicvariants pages 4-5): F. Alfiya, Manna Jose, Soumya V. Chandrasekharan, Soumya Sundaram, Madhusoodanan Urulangodi, Bejoy Thomas, Ashalatha Radhakrishnan, Moinak Banerjee, and Ramshekhar N. Menon. C12orf57 pathogenic variants: a unique cause of developmental encephalopathy in a south indian child. Journal of Genetics, Jun 2022. URL: https://doi.org/10.1007/s12041-022-01371-0, doi:10.1007/s12041-022-01371-0. This article has 5 citations and is from a peer-reviewed journal.
-
(talisetti2003temtamy‐likesyndromeassociated pages 1-3): Anita Talisetti, Shawnia R. Forrester, David Gregory, Lisa Johnson, Michael C. Schneider, and Virginia E. Kimonis. Temtamy‐like syndrome associated with translocation of 2p24 and 9q32. Clinical Dysmorphology, 12:175–177, Jul 2003. URL: https://doi.org/10.1097/01.mcd.0000072161.33788.56, doi:10.1097/01.mcd.0000072161.33788.56. This article has 20 citations and is from a peer-reviewed journal.
-
(akizu2013wholeexomesequencingidentifies pages 1-2): Naiara Akizu, Nuri M. Shembesh, Tawfeg Ben-Omran, Laila Bastaki, Asma Al-Tawari, Maha S. Zaki, Roshan Koul, Emily Spencer, Rasim Ozgur Rosti, Eric Scott, Elizabeth Nickerson, Stacey Gabriel, Gilberto da Gente, Jiang Li, Matthew A. Deardorff, Laura K. Conlin, Margaret A. Horton, Elaine H. Zackai, Elliott H. Sherr, and Joseph G. Gleeson. Whole-exome sequencing identifies mutated c12orf57 in recessive corpus callosum hypoplasia. American journal of human genetics, 92 3:392-400, Mar 2013. URL: https://doi.org/10.1016/j.ajhg.2013.02.004, doi:10.1016/j.ajhg.2013.02.004. This article has 43 citations and is from a highest quality peer-reviewed journal.
-
(ni2024aninappropriatedecline pages 13-17): Chunyang Ni, Leqian Yu, Barbara Vona, Dayea Park, Yulei Wei, Daniel A Schmitz, Yudong Wei, Yi Ding, Masahiro Sakurai, Emily Ballard, Yan Liu, Ashwani Kumar, Chao Xing, Hyung-Goo Kim, Cumhur Ekmekci, Ehsan Ghayoor Karimiani, Shima Imannezhad, Fatemeh Eghbal, Reza Shervin Badv, Eva Maria Christina Schwaibold, Mohammadreza Dehghani, Mohammad Yahya Vahidi Mehrjardi, Zahra Metanat, Hosein Eslamiyeh, Ebtissal Khouj, Saleh Mohammed Nasser Alhajj, Aziza Chedrawi, César Augusto Pinheiro Ferreira Alves, Henry Houlden, Michael Kruer, Fowzan S. Alkuraya, Can Cenik, Reza Maroofian, Jun Wu, and Michael Buszczak. An inappropriate decline in ribosome levels drives a diverse set of neurodevelopmental disorders. BioRxiv, Jan 2024. URL: https://doi.org/10.1101/2024.01.09.574708, doi:10.1101/2024.01.09.574708. This article has 4 citations.
-
(tian2010lossofchsy1 pages 8-9): Jing Tian, Ling Ling, Mohammad Shboul, Hane Lee, Brian O'Connor, Barry Merriman, Stanley F. Nelson, Simon Cool, Osama H. Ababneh, Azmy Al-Hadidy, Amira Masri, Hanan Hamamy, and Bruno Reversade. Loss of chsy1, a secreted fringe enzyme, causes syndromic brachydactyly in humans via increased notch signaling. American journal of human genetics, 87 6:768-78, Dec 2010. URL: https://doi.org/10.1016/j.ajhg.2010.11.005, doi:10.1016/j.ajhg.2010.11.005. This article has 121 citations and is from a highest quality peer-reviewed journal.
-
(akizu2013wholeexomesequencingidentifies pages 7-8): Naiara Akizu, Nuri M. Shembesh, Tawfeg Ben-Omran, Laila Bastaki, Asma Al-Tawari, Maha S. Zaki, Roshan Koul, Emily Spencer, Rasim Ozgur Rosti, Eric Scott, Elizabeth Nickerson, Stacey Gabriel, Gilberto da Gente, Jiang Li, Matthew A. Deardorff, Laura K. Conlin, Margaret A. Horton, Elaine H. Zackai, Elliott H. Sherr, and Joseph G. Gleeson. Whole-exome sequencing identifies mutated c12orf57 in recessive corpus callosum hypoplasia. American journal of human genetics, 92 3:392-400, Mar 2013. URL: https://doi.org/10.1016/j.ajhg.2013.02.004, doi:10.1016/j.ajhg.2013.02.004. This article has 43 citations and is from a highest quality peer-reviewed journal.
-
(marafi2024foundermutationsand pages 4-5): Dana Marafi. Founder mutations and rare disease in the arab world. Disease Models & Mechanisms, Jun 2024. URL: https://doi.org/10.1242/dmm.050715, doi:10.1242/dmm.050715. This article has 18 citations and is from a domain leading peer-reviewed journal.
-
(plassmeyer2023amassivelyparallel pages 21-23): Stephen P. Plassmeyer, Colin P. Florian, Michael J. Kasper, Rebecca Chase, Shayna Mueller, Yating Liu, Kelli McFarland White, Courtney F. Jungers, Slavica Pavlovic Djuranovic, Sergej Djuranovic, and Joseph D. Dougherty. A massively parallel screen of 5′utr mutations identifies variants impacting translation and protein production in neurodevelopmental disorder genes. MedRxiv, Nov 2023. URL: https://doi.org/10.1101/2023.11.02.23297961, doi:10.1101/2023.11.02.23297961. This article has 15 citations.
-
(plassmeyer2023amassivelyparallel pages 32-35): Stephen P. Plassmeyer, Colin P. Florian, Michael J. Kasper, Rebecca Chase, Shayna Mueller, Yating Liu, Kelli McFarland White, Courtney F. Jungers, Slavica Pavlovic Djuranovic, Sergej Djuranovic, and Joseph D. Dougherty. A massively parallel screen of 5′utr mutations identifies variants impacting translation and protein production in neurodevelopmental disorder genes. MedRxiv, Nov 2023. URL: https://doi.org/10.1101/2023.11.02.23297961, doi:10.1101/2023.11.02.23297961. This article has 15 citations.
-
(wang2020temtamysyndromecaused media b3d8bfd5): Yanqin Wang, Ming Li, Yuanyuan Luo, Xin Zhao, Shuang Liao, Li Jiang, Xiujuan Li, and Min Zhong. Temtamy syndrome caused by a new c12orf57 variant in a chinese boy, including pedigree analysis and literature review. Experimental and therapeutic medicine, 19 1:327-332, Nov 2020. URL: https://doi.org/10.3892/etm.2019.8183, doi:10.3892/etm.2019.8183. This article has 8 citations and is from a peer-reviewed journal.
-
(wang2020temtamysyndromecaused media 805b1c71): Yanqin Wang, Ming Li, Yuanyuan Luo, Xin Zhao, Shuang Liao, Li Jiang, Xiujuan Li, and Min Zhong. Temtamy syndrome caused by a new c12orf57 variant in a chinese boy, including pedigree analysis and literature review. Experimental and therapeutic medicine, 19 1:327-332, Nov 2020. URL: https://doi.org/10.3892/etm.2019.8183, doi:10.3892/etm.2019.8183. This article has 8 citations and is from a peer-reviewed journal.
-
(wang2020temtamysyndromecaused media 21e24220): Yanqin Wang, Ming Li, Yuanyuan Luo, Xin Zhao, Shuang Liao, Li Jiang, Xiujuan Li, and Min Zhong. Temtamy syndrome caused by a new c12orf57 variant in a chinese boy, including pedigree analysis and literature review. Experimental and therapeutic medicine, 19 1:327-332, Nov 2020. URL: https://doi.org/10.3892/etm.2019.8183, doi:10.3892/etm.2019.8183. This article has 8 citations and is from a peer-reviewed journal.