Mucolipidosis Type II
Mucolipidosis type II (I-cell disease) is a severe, infantile-onset autosomal recessive lysosomal storage disorder caused by biallelic GNPTAB variants that abolish GlcNAc-1-phosphotransferase, the enzyme that generates the mannose-6-phosphate recognition marker directing acid hydrolases to the lysosome. Without the marker, multiple lysosomal enzymes are missorted and hypersecreted instead of delivered to lysosomes, leaving lysosomes functionally deficient and causing multisystem substrate accumulation with coarse facies, gingival hypertrophy, severe skeletal dysplasia (dysostosis multiplex), growth failure, cardiorespiratory complications, and death in early childhood.
Ask OpenScientist
Ask a research question about Mucolipidosis Type II. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Mappings
Definitions
1Show evidence (1 reference)
Inheritance
1Show evidence (1 reference)
Pathophysiology
5Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Pathograph
Phenotypes
10Head and Neck 1
Show evidence (1 reference)
Integument 1
Show evidence (1 reference)
Musculoskeletal 3
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Nervous System 1
Show evidence (1 reference)
Respiratory 1
Show evidence (1 reference)
Growth 1
Show evidence (1 reference)
Other 2
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
1Show evidence (1 reference)
Medical Actions
2Differential Diagnoses
1Conditions with similar clinical presentations that must be differentiated from Mucolipidosis Type II:
- GNPTG defects with attenuated, slowly progressive connective-tissue disease, versus the severe fatal infantile GNPTAB form.
Show evidence (1 reference)
Source YAML
click to showname: Mucolipidosis Type II
creation_date: "2026-06-13T00:00:00Z"
description: >-
Mucolipidosis type II (I-cell disease) is a severe, infantile-onset autosomal recessive
lysosomal storage disorder caused by biallelic GNPTAB variants that abolish
GlcNAc-1-phosphotransferase, the enzyme that generates the mannose-6-phosphate recognition
marker directing acid hydrolases to the lysosome. Without the marker, multiple lysosomal
enzymes are missorted and hypersecreted instead of delivered to lysosomes, leaving lysosomes
functionally deficient and causing multisystem substrate accumulation with coarse facies,
gingival hypertrophy, severe skeletal dysplasia (dysostosis multiplex), growth failure,
cardiorespiratory complications, and death in early childhood.
category: Mendelian
disease_term:
preferred_term: mucolipidosis type II
term:
id: MONDO:0009650
label: mucolipidosis type II
mappings:
mondo_mappings:
- term:
id: MONDO:0009650
label: mucolipidosis type II
mapping_predicate: skos:exactMatch
mapping_source: MONDO
mapping_justification: Primary MONDO disease identifier for this mucolipidosis type II (I-cell disease) entry.
references:
- reference: PMID:20301728
title: "GNPTAB-Related Disorders."
tags:
- GeneReviews
synonyms:
- I-cell disease
- Mucolipidosis II alpha/beta
- Inclusion-cell disease
- GNPTAB deficiency
parents:
- Lysosomal Storage Disorder
pathophysiology:
- name: GlcNAc-1-Phosphotransferase Deficiency and Mannose-6-Phosphate Targeting Failure
conforms_to: "lysosomal_substrate_accumulation#Lysosomal Hydrolase or Cofactor Deficiency"
description: >-
Biallelic GNPTAB variants abolish GlcNAc-1-phosphotransferase, preventing formation of the
mannose-6-phosphate marker that targets soluble acid hydrolases to the lysosome. Untagged
enzymes are missorted and hypersecreted rather than delivered, leaving the lysosome
functionally deficient in multiple hydrolases.
gene:
preferred_term: GNPTAB
term:
id: hgnc:29670
label: GNPTAB
biological_processes:
- preferred_term: protein targeting to lysosome
term:
id: GO:0006622
label: protein targeting to lysosome
modifier: DECREASED
cell_types:
- preferred_term: fibroblast
term:
id: CL:0000057
label: fibroblast
evidence:
- reference: PMID:30882951
reference_title: "The lysosomal storage disorders mucolipidosis type II, type III alpha/beta, and type III gamma: Update on GNPTAB and GNPTG mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Mutations in the GNPTAB and GNPTG genes cause mucolipidosis (ML) type II"
explanation: "GNPTAB mutations cause mucolipidosis type II via loss of GlcNAc-1-phosphotransferase."
downstream:
- target: Multisystem Lysosomal Substrate Accumulation
description: Functional lysosomal hydrolase deficiency leads to multisystem substrate accumulation.
- name: Multisystem Lysosomal Substrate Accumulation
conforms_to: "lysosomal_substrate_accumulation#Lysosomal Substrate Accumulation"
description: >-
Functional deficiency of multiple lysosomal hydrolases causes accumulation of undegraded
substrates across many tissues (skeleton, connective tissue, heart, brain), producing the
severe multisystem disease of I-cell disease.
biological_processes:
- preferred_term: lysosomal transport
term:
id: GO:0007041
label: lysosomal transport
modifier: ABNORMAL
cell_types:
- preferred_term: fibroblast
term:
id: CL:0000057
label: fibroblast
cellular_components:
- preferred_term: lysosome
term:
id: GO:0005764
label: lysosome
evidence:
- reference: PMID:34172897
reference_title: "Mucolipidosis type II and type III: a systematic review of 843 published cases."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Mucolipidosis (ML) II, MLIII alpha/beta, and MLIII gamma are rare\nautosomal recessive lysosomal storage disorders."
explanation: "Mucolipidosis II is a lysosomal storage disorder with multisystem substrate accumulation."
downstream:
- target: Skeletal and Connective-Tissue Storage Disease
causal_link_type: DIRECT
description: Multisystem storage in connective tissues and cartilage produces the severe skeletal and soft-tissue phenotype.
- target: Cardiorespiratory Storage Disease
causal_link_type: DIRECT
description: Multisystem storage affects cardiac valves, mucosa, and the thoracic cage, producing cardiorespiratory disease.
- target: Neuromotor Developmental Arrest
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
description: Multisystem lysosomal dysfunction is associated with cessation of neuromotor development.
- name: Skeletal and Connective-Tissue Storage Disease
description: >-
Storage in skeletal and connective tissues produces the severe dysostosis
multiplex pattern, growth failure, contractures, kyphosis, thickened skin,
coarse facies, and gingival overgrowth of ML II.
biological_processes:
- preferred_term: extracellular matrix organization
term:
id: GO:0030198
label: extracellular matrix organization
modifier: DYSREGULATED
cell_types:
- preferred_term: fibroblast
term:
id: CL:0000057
label: fibroblast
- preferred_term: chondrocyte
term:
id: CL:0000138
label: chondrocyte
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "clubfeet, deformed long bones, and/or dislocation of the hip(s)"
explanation: GeneReviews documents the birth-present orthopedic component of the ML II storage phenotype.
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "skin is thickened, facial features are coarse, and gingiva are hypertrophic"
explanation: GeneReviews documents the connective-tissue and craniofacial storage phenotype.
downstream:
- target: Coarse facial features
causal_link_type: DIRECT
description: Craniofacial soft-tissue storage produces coarse facial features.
- target: Gingival overgrowth
causal_link_type: DIRECT
description: Gingival connective-tissue storage produces gingival hypertrophy.
- target: Dysostosis multiplex
causal_link_type: DIRECT
description: Skeletal storage produces dysostosis multiplex and deformed long bones.
- target: Growth failure
causal_link_type: DIRECT
description: Severe skeletal storage disease contributes to cessation of statural growth.
- target: Joint contractures
causal_link_type: DIRECT
description: Connective-tissue storage produces large-joint contractures.
- target: Kyphosis
causal_link_type: DIRECT
description: Skeletal storage produces thoracic deformity and kyphosis.
- target: Thickened skin
causal_link_type: DIRECT
description: Dermal connective-tissue storage produces thickened skin.
- name: Cardiorespiratory Storage Disease
description: >-
ML II storage affects cardiac valves, mucosa, and the thoracic cage,
producing valve thickening/insufficiency and progressive respiratory
insufficiency.
biological_processes:
- preferred_term: extracellular matrix organization
term:
id: GO:0030198
label: extracellular matrix organization
modifier: DYSREGULATED
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "children have cardiac involvement, most commonly thickening and insufficiency of"
explanation: GeneReviews documents cardiac-valve involvement in all children with ML II.
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "contributes to respiratory insufficiency, the most common cause of death"
explanation: GeneReviews links mucosal and thoracic-cage storage disease to respiratory insufficiency.
downstream:
- target: Cardiac valve disease
causal_link_type: DIRECT
description: Cardiac valve thickening and insufficiency manifest as valve disease.
- target: Respiratory insufficiency
causal_link_type: DIRECT
description: Airway narrowing and thoracic-cage stiffening manifest as respiratory insufficiency.
- name: Neuromotor Developmental Arrest
description: >-
Severe ML II includes cessation of neuromotor development downstream of the
lysosomal enzyme-targeting defect and multisystem storage burden.
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
evidence:
- reference: PMID:30882951
reference_title: "The lysosomal storage disorders mucolipidosis type II, type III alpha/beta, and type III gamma: Update on GNPTAB and GNPTG mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "cessation of statural growth and neuromotor development"
explanation: This review directly documents cessation of neuromotor development in the ML II phenotype.
downstream:
- target: Developmental delay
causal_link_type: DIRECT
description: Neuromotor developmental arrest manifests clinically as global developmental delay.
phenotypes:
- name: Coarse facial features
description: Coarse facies, evident from early infancy.
phenotype_term:
preferred_term: Coarse facial features
term:
id: HP:0000280
label: Coarse facial features
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "facial features are coarse, and gingiva are hypertrophic"
explanation: "Coarse facial features are characteristic of I-cell disease."
- name: Gingival overgrowth
description: Gingival (gum) hypertrophy is a recognizable feature.
phenotype_term:
preferred_term: Gingival overgrowth
term:
id: HP:0000212
label: Gingival overgrowth
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "facial features are coarse, and gingiva are hypertrophic"
explanation: "Gingival hypertrophy is a characteristic oral finding."
- name: Dysostosis multiplex
description: Severe skeletal dysplasia with deformed long bones and joint involvement.
phenotype_term:
preferred_term: Dysostosis multiplex
term:
id: HP:0000943
label: Dysostosis multiplex
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "clubfeet, deformed long bones, and/or dislocation of the hip(s)"
explanation: "Severe skeletal dysplasia (dysostosis multiplex) is a hallmark."
- name: Cardiac valve disease
description: Cardiac involvement affects all children, most commonly mitral valve thickening and insufficiency.
phenotype_term:
preferred_term: Mitral regurgitation
term:
id: HP:0001653
label: Mitral regurgitation
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "All\nchildren have cardiac involvement, most commonly thickening and insufficiency of\nthe mitral valve"
explanation: All children with ML II have cardiac (mitral valve) involvement.
- name: Respiratory insufficiency
description: Thoracic-cage stiffening produces respiratory insufficiency, the most common cause of death.
phenotype_term:
preferred_term: Respiratory insufficiency
term:
id: HP:0002093
label: Respiratory insufficiency
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "gradual stiffening of the thoracic cage\ncontributes to respiratory insufficiency, the most common cause of death"
explanation: Respiratory insufficiency is the most common cause of death in ML II.
- name: Growth failure
description: Growth often ceases in the second year of life.
phenotype_term:
preferred_term: Growth delay
term:
id: HP:0001510
label: Growth delay
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Growth often\nceases in the second year of life"
explanation: Growth failure with cessation of growth in the second year is characteristic.
- name: Joint contractures
description: Contractures develop in all large joints.
phenotype_term:
preferred_term: Joint contracture
term:
id: HP:0001371
label: Flexion contracture
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "contractures develop in all large joints"
explanation: Large-joint contractures are a consistent feature of ML II.
- name: Kyphosis
description: Thoracic deformity and kyphosis are present at birth.
phenotype_term:
preferred_term: Kyphosis
term:
id: HP:0002808
label: Kyphosis
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Orthopedic\nabnormalities present at birth may include thoracic deformity, kyphosis"
explanation: Kyphosis is among the orthopedic abnormalities present at birth.
- name: Thickened skin
description: The skin is thickened, part of the somatic storage phenotype.
phenotype_term:
preferred_term: Thickened skin
term:
id: HP:0001072
label: Thickened skin
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The\nskin is thickened, facial features are coarse"
explanation: Thickened skin is part of the ML II somatic phenotype.
- name: Developmental delay
description: Cessation of statural growth and neuromotor development.
phenotype_term:
preferred_term: Global developmental delay
term:
id: HP:0001263
label: Global developmental delay
evidence:
- reference: PMID:30882951
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "cessation of statural growth and neuromotor development"
explanation: Neuromotor developmental arrest is part of the severe ML II phenotype.
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:34172897
reference_title: "Mucolipidosis type II and type III: a systematic review of 843 published cases."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "rare\nautosomal recessive lysosomal storage disorders"
explanation: "Mucolipidosis II is autosomal recessive."
genetic:
- name: GNPTAB
association: Biallelic GNPTAB variants abolishing GlcNAc-1-phosphotransferase
relationship_type: CAUSATIVE
variant_origin: GERMLINE
gene_term:
preferred_term: GNPTAB
term:
id: hgnc:29670
label: GNPTAB
evidence:
- reference: PMID:30882951
reference_title: "The lysosomal storage disorders mucolipidosis type II, type III alpha/beta, and type III gamma: Update on GNPTAB and GNPTG mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Mutations in the GNPTAB and GNPTG genes cause mucolipidosis (ML) type II"
explanation: "GNPTAB mutations are the genetic cause of mucolipidosis type II."
progression:
- phase: Fatal childhood course
notes: >-
Mucolipidosis II is progressive with death most often occurring in early childhood.
evidence:
- reference: PMID:20301728
reference_title: "GNPTAB-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "death most often occurs in early childhood"
explanation: "Documents the fatal early-childhood prognosis of I-cell disease."
diagnosis:
- name: Plasma lysosomal enzyme activity and GNPTAB sequencing
diagnosis_term:
preferred_term: clinical laboratory procedure
term:
id: MAXO:0000006
label: clinical laboratory procedure
description: >-
Markedly elevated activities of multiple lysosomal enzymes in plasma/serum (from
hypersecretion) with reduced intracellular activity suggests a mannose-6-phosphate
targeting defect; diagnosis is confirmed by GNPTAB sequencing.
markers: Elevated plasma lysosomal enzyme activities with reduced intracellular activity.
evidence:
- reference: PMID:20301728
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The diagnosis of a GNPTAB-related disorder is established in\na proband with suggestive clinical, radiographic, and biochemical findings and\nbiallelic pathogenic (or likely pathogenic) variants in GNPTAB"
explanation: "Diagnosis combines suggestive clinical/radiographic/biochemical findings with biallelic GNPTAB variants."
- name: GNPTAB molecular genetic testing
diagnosis_term:
preferred_term: genetic testing
term:
id: MAXO:0000127
label: genetic testing
description: Confirmatory biallelic GNPTAB sequencing.
evidence:
- reference: PMID:20301728
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "biallelic pathogenic (or likely pathogenic) variants in GNPTAB identified on molecular genetic testing"
explanation: "Biallelic GNPTAB variants on molecular testing provide diagnostic confirmation."
differential_diagnoses:
- name: GNPTG-mucolipidosis
description: >-
The attenuated form of GlcNAc-1-phosphotransferase deficiency (GNPTG gamma subunit), with
later onset and survival into adulthood.
disease_term:
preferred_term: GNPTG-mucolipidosis
term:
id: MONDO:0009652
label: GNPTG-mucolipidosis
distinguishing_features:
- GNPTG defects with attenuated, slowly progressive connective-tissue disease, versus the severe fatal infantile GNPTAB form.
evidence:
- reference: PMID:30882951
reference_title: "The lysosomal storage disorders mucolipidosis type II, type III alpha/beta, and type III gamma: Update on GNPTAB and GNPTG mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "MLIII alpha/beta and MLIII gamma are\nattenuated forms of the disease"
explanation: "GNPTG-related ML III is an attenuated form, distinct from the severe infantile GNPTAB ML II."
treatments:
- name: Supportive Care
description: >-
No disease-modifying therapy is established; management is supportive (orthopedic,
cardiorespiratory, nutritional). Hematopoietic stem cell transplantation has been
investigated with limited benefit.
treatment_term:
preferred_term: Supportive Care
term:
id: NCIT:C15747
label: Supportive Care
- name: Hematopoietic Stem Cell Transplantation
description: >-
Hematopoietic stem cell transplantation has been investigated in ML II with limited and
inconsistent benefit; it does not halt the skeletal or cardiac disease.
treatment_term:
preferred_term: hematopoietic cell transplantation
term:
id: NCIT:C15431
label: Hematopoietic Cell Transplantation
definitions:
- name: Clinical case definition of mucolipidosis II (I-cell disease)
definition_type: CASE_DEFINITION
description: >-
Mucolipidosis type II is the severe GlcNAc-1-phosphotransferase deficiency caused by
biallelic GNPTAB variants, defined by a mannose-6-phosphate targeting defect with severe
multisystem lysosomal storage, dysostosis multiplex, coarse facies, and death in early
childhood.
scope: Disease-level case definition for GNPTAB-related mucolipidosis II.
evidence:
- reference: PMID:20301728
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "biallelic pathogenic (or likely pathogenic) variants in GNPTAB"
explanation: "Anchors the case definition in biallelic GNPTAB-related mucolipidosis II."
References & Deep Research
References
1Deep Research
1Mucolipidosis Type II (MLII; I‑cell disease) — Disease Characteristics Research Report
Executive summary (current understanding)
Mucolipidosis type II (MLII) is an ultra-rare, severe, autosomal recessive lysosomal storage disorder (LSD) caused by biallelic pathogenic variants in GNPTAB, resulting in deficiency of N‑acetylglucosamine‑1‑phosphotransferase (GlcNAc‑1‑phosphotransferase) and failure to generate mannose‑6‑phosphate (M6P) targeting signals on many lysosomal hydrolases. Consequently, lysosomal enzymes are missorted and hypersecreted, leading to multisystem storage pathology with prenatal/neonatal onset, progressive skeletal disease (dysostosis multiplex), growth failure, cardiorespiratory complications, and profound developmental impairment; survival is typically limited to childhood in classic MLII (dogterom2021mucolipidosistypeii pages 1-2, he2023outcomesafterhsct pages 1-2).
Recent 2023–2024 literature emphasizes (i) improved quantitative longitudinal neurodevelopmental characterization (suggesting sustained but very slow skill gain rather than clear progressive neurodegeneration in one cohort) and (ii) advances in biochemical screening/diagnosis using multiplex MS/MS enzyme activity patterns in dried blood spots (DBS) as a route to earlier detection (ammer2023cnsmanifestationsin pages 1-2, hong2023multiplextandemmass pages 1-2).
1. Disease information
1.1 Definition and overview
MLII (I‑cell disease) is a GNPTAB-related mucolipidosis in which loss of GlcNAc‑1‑phosphotransferase activity prevents M6P tagging of lysosomal enzymes in the Golgi, causing their secretion rather than lysosomal delivery, with secondary intracellular accumulation of undegraded macromolecules (he2023outcomesafterhsct pages 1-2, dogterom2021mucolipidosistypeii pages 1-2).
1.2 Key identifiers and synonyms
A normalization table for identifiers captured in the retrieved evidence is provided below.
| Field | Value | Evidence |
|---|---|---|
| Disease name | Mucolipidosis type II | (he2023outcomesafterhsct pages 1-2, dogterom2021mucolipidosistypeii pages 1-2) |
| Preferred synonym | I-cell disease | (he2023outcomesafterhsct pages 1-2, badenetti2024investigatingneuronalpathogenesisa pages 24-28) |
| Other synonyms in retrieved sources | Inclusion-cell disease; MLII | (badenetti2024investigatingneuronalpathogenesisa pages 24-28, khan2020mucolipidosesoverviewpast pages 1-3) |
| OMIM ID (MLII) | 252500 | (badenetti2024investigatingneuronalpathogenesisa pages 24-28) |
| OMIM ID (related MLIII alpha/beta) | 252600 | (dogterom2021mucolipidosistypeii pages 1-2) |
| OMIM ID (related MLIII gamma) | 252605 | (dogterom2021mucolipidosistypeii pages 1-2) |
| MONDO ID | not found in retrieved sources | (he2023outcomesafterhsct pages 1-2) |
| Orphanet ID | not found in retrieved sources | (he2023outcomesafterhsct pages 1-2) |
| ICD-10/ICD-11 | not found in retrieved sources | (he2023outcomesafterhsct pages 1-2) |
| MeSH | not found in retrieved sources | (he2023outcomesafterhsct pages 1-2) |
| Primary causal gene(s) | GNPTAB (encodes the alpha/beta precursor subunits of GlcNAc-1-phosphotransferase) | (he2023outcomesafterhsct pages 1-2, dogterom2021mucolipidosistypeii pages 1-2, khan2020mucolipidosesoverviewpast pages 1-3) |
| Related gene(s) in allelic/related mucolipidosis | GNPTG (MLIII gamma); GNPTAB also causes MLIII alpha/beta | (dogterom2021mucolipidosistypeii pages 1-2, feng2024clinicalandmolecular pages 1-2) |
| Core molecular defect | Deficiency of N-acetylglucosamine-1-phosphotransferase with failure of mannose-6-phosphate tagging and missorting/hypersecretion of lysosomal enzymes | (he2023outcomesafterhsct pages 1-2, dogterom2021mucolipidosistypeii pages 1-2, khan2020mucolipidosesoverviewpast pages 1-3) |
| Inheritance | Autosomal recessive | (badenetti2024investigatingneuronalpathogenesisa pages 24-28, dogterom2021mucolipidosistypeii pages 1-2, khan2020mucolipidosesoverviewpast pages 1-3) |
| Key nomenclature note | MLII is the severe end of the GNPTAB-related mucolipidosis spectrum; MLIII alpha/beta is the attenuated related form | (feng2024clinicalandmolecular pages 1-2, dogterom2021mucolipidosistypeii pages 1-2) |
Table: This table summarizes the core disease nomenclature and identifiers for mucolipidosis type II (I-cell disease), including related OMIM entries and causal genes. It is useful as a normalization reference for a disease knowledge base when some ontology identifiers were not available in the retrieved evidence.
Note on missing ontology IDs: In this tool run, MONDO, Orphanet, ICD‑10/ICD‑11, and MeSH identifiers were not present in the retrieved full texts and therefore cannot be asserted with citation-compliant evidence here (artifact-00).
1.3 Evidence source type
The evidence base here includes aggregated, disease-level systematic review data (historic literature through Aug 2019), recent retrospective and cohort studies, single-patient clinical reports, and observational clinicaltrials.gov protocols (dogterom2021mucolipidosistypeii pages 3-4, ammer2023cnsmanifestationsin pages 1-2, NCT01891422 chunk 1).
2. Etiology
2.1 Disease causal factors
- Primary cause (genetic): Biallelic pathogenic variants in GNPTAB (autosomal recessive) causing deficiency/absence of GlcNAc‑1‑phosphotransferase and failure of M6P-dependent lysosomal enzyme trafficking (dogterom2021mucolipidosistypeii pages 1-2, he2023outcomesafterhsct pages 1-2).
- Related allelic disorders: Reduced residual GlcNAc‑1‑phosphotransferase activity yields attenuated phenotypes classified as mucolipidosis type III α/β; GNPTG variants primarily cause MLIII γ (dogterom2021mucolipidosistypeii pages 1-2, feng2024clinicalandmolecular pages 1-2).
2.2 Risk factors / protective factors / GxE
For a Mendelian disease with early onset, non-genetic risk factors and protective factors are not well established in the retrieved evidence. Disease risk is driven by inheritance of pathogenic GNPTAB alleles (dogterom2021mucolipidosistypeii pages 1-2). Gene–environment interactions were not identified in the retrieved MLII-specific sources.
3. Phenotypes (clinical features; HPO suggestions)
A phenotype-to-HPO mapping table with frequencies (when available) is provided below.
| Phenotype | Phenotype type | Typical onset | Progression / severity | Frequency / quantitative data | Suggested HPO term(s) | Evidence |
|---|---|---|---|---|---|---|
| Coarse/dysmorphic facial features | Physical manifestation | Prenatal-neonatal or first months of life | Early, prominent, severe in classic MLII | Presenting feature in 47.4% of MLII cases in systematic review | Coarse facial features (HP:0000280); Dysmorphism | (dogterom2021mucolipidosistypeii pages 3-4, he2023outcomesafterhsct pages 1-2) |
| Developmental delay / global developmental impairment | Neurodevelopmental sign | Infancy | Progressive impairment in function, but longitudinal data suggest continued slow skill gain rather than true neurocognitive regression | Presenting feature in 24.2%; mean developmental quotient 36.7% (SD 20.4) at last assessment; gain of 0.28 age-equivalent score points/month (95% CI 0.17-0.38) | Global developmental delay (HP:0001263); Intellectual disability (HP:0001249) | (dogterom2021mucolipidosistypeii pages 3-4, ammer2023cnsmanifestationsin pages 1-2) |
| Skeletal abnormalities / dysostosis multiplex | Physical manifestation / radiographic abnormality | Prenatal-infancy | Progressive, severe multisystem skeletal disease | Presenting feature in 20.0%; severe orthopaedic disease and dysostosis multiplex reported in all patients in CNS cohort | Dysostosis multiplex; Abnormality of the skeletal system (HP:0000924) | (dogterom2021mucolipidosistypeii pages 3-4, ammer2023cnsmanifestationsin pages 4-6, he2023outcomesafterhsct pages 1-2) |
| Growth retardation / short stature | Physical manifestation | Infancy | Progressive growth failure | Presenting feature in 12.6%; severe growth abnormalities described | Growth delay (HP:0001510); Short stature (HP:0004322) | (dogterom2021mucolipidosistypeii pages 3-4, he2023outcomesafterhsct pages 1-2, feng2024clinicalandmolecular pages 1-2) |
| Restricted joint range of motion / joint stiffness | Clinical sign | Infancy to early childhood | Progressive; common musculoskeletal hallmark | Presenting feature in 10.7%; main manifestation in recent Chinese cohort | Joint stiffness (HP:0001387); Decreased range of motion of joint | (dogterom2021mucolipidosistypeii pages 3-4, feng2024clinicalandmolecular pages 1-2) |
| Abnormal skull shape / craniosynostosis-related skull changes | Physical manifestation | Infancy | May progress; part of dysostosis multiplex spectrum | Presenting feature in 10.7% | Abnormal skull morphology (HP:0000928); Craniosynostosis (HP:0001363) | (dogterom2021mucolipidosistypeii pages 3-4, khan2020mucolipidosesoverviewpast pages 1-3) |
| Cardiac involvement | Organ/system involvement | Prenatal-neonatal or infancy | Serious contributor to mortality | Frequently described clinically; cardiac failure accounted for 15/93 reported deaths and combined cardiopulmonary causes were common | Abnormality of the cardiovascular system (HP:0001626); Heart failure (HP:0001635) | (dogterom2021mucolipidosistypeii pages 3-4, he2023outcomesafterhsct pages 1-2) |
| Respiratory involvement / recurrent infections / respiratory failure | Organ/system involvement | Prenatal-neonatal or infancy | Progressive; major life-limiting complication | Respiratory failure 24/93 and pneumonia 32/93 among reported causes of death | Recurrent respiratory infections (HP:0002205); Respiratory insufficiency (HP:0002093) | (dogterom2021mucolipidosistypeii pages 3-4, he2023outcomesafterhsct pages 1-2, khan2020mucolipidosesoverviewpast pages 1-3) |
| Hip dysplasia | Musculoskeletal sign | Childhood (often evident early) | Progressive orthopedic burden | 9/11 patients (81%) in CNS cohort | Hip dysplasia (HP:0001385) | (ammer2023cnsmanifestationsin pages 4-6) |
| Sensorineural hearing impairment | Sensory abnormality | Childhood | Variable, often chronic | Confirmed in 6/11 patients (55%) | Sensorineural hearing impairment (HP:0000407) | (ammer2023cnsmanifestationsin pages 4-6) |
| Middle ear disease | ENT manifestation | Childhood | Recurrent/chronic | Present in 10/11 patients (91%) | Otitis media; Middle ear abnormality | (ammer2023cnsmanifestationsin pages 4-6) |
| Ophthalmologic abnormalities | Sensory/ocular manifestation | Childhood | Variable | Reported in 7/11 patients (63%) | Abnormality of the eye (HP:0000478) | (ammer2023cnsmanifestationsin pages 4-6) |
| Carpal tunnel syndrome | Neuromuscular / orthopedic sign | Childhood to later course | Can require intervention | 2/11 patients (18%) in CNS cohort | Carpal tunnel syndrome (HP:0100543) | (ammer2023cnsmanifestationsin pages 4-6) |
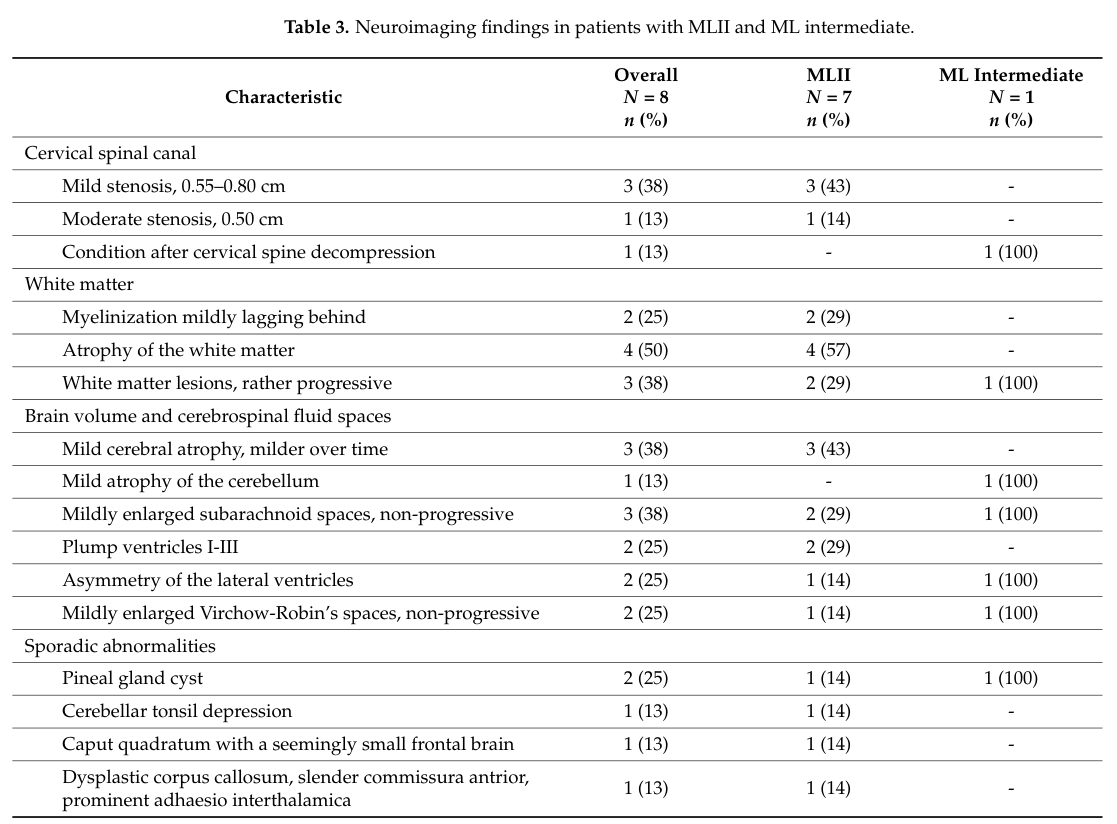
| Cervical spinal canal stenosis | Neuroimaging / structural abnormality | Childhood | Important complication; may require surveillance/intervention | 63% on neuroimaging | Cervical spinal canal stenosis; Spinal canal stenosis (HP:0003369) | (ammer2023cnsmanifestationsin pages 1-2, ammer2023cnsmanifestationsin media 4b113608) |
| Mild brain atrophy | Neuroimaging abnormality | Childhood | Reported as mild and non-progressive in cohort | 50% in figure/table summary from imaging context | Cerebral atrophy (HP:0002059) | (ammer2023cnsmanifestationsin media 4b113608) |
| White matter lesions / abnormalities | Neuroimaging abnormality | Childhood | Non-progressive / nonspecific in available cohort | Reported, frequency not numerically extracted in text summary | Abnormality of cerebral white matter (HP:0002500) | (ammer2023cnsmanifestationsin pages 1-2, ammer2023cnsmanifestationsin media 4b113608) |
| Hypotonia / reduced muscle tone | Neurologic sign | Infancy | Contributes to delayed motor development | Described in case-based clinical reports and HSCT follow-up | Hypotonia (HP:0001252) | (he2023outcomesafterhsct pages 1-2) |
| Gingival hyperplasia | Craniofacial/oral sign | Infancy | Characteristic, progressive | Frequently cited as characteristic feature of MLII | Gingival overgrowth (HP:0000212) | (ammer2021ishematopoieticstem pages 1-2, khan2020mucolipidosesoverviewpast pages 1-3) |
Table: This table summarizes the major clinical and imaging phenotypes reported for mucolipidosis type II, including onset, progression, and approximate frequencies where available. It is useful for populating phenotype fields in a disease knowledge base and mapping features to HPO terms.
3.1 Recent quantitative CNS phenotype characterization (2023)
A 2023 retrospective longitudinal analysis of 11 MLII patients reported profound impairment but continued developmental gains: mean developmental quotient (DQ) at last assessment 36.7% (SD 20.4) and an average gain of 0.28 age-equivalent score points/month (95% CI 0.17–0.38) (ammer2023cnsmanifestationsin pages 1-2). The abstract concludes: “MLII is associated with profound developmental impairment, but not with neurodegeneration and neurocognitive decline.” (Journal of Clinical Medicine; published June 2023; URL in citation metadata) (ammer2023cnsmanifestationsin pages 1-2).
3.2 Visual evidence (neurodevelopment and MRI frequencies)
Figure/table crops from the 2023 CNS cohort summarize longitudinal neurocognitive trajectories and the frequencies of MRI abnormalities (including cervical spinal stenosis and mild brain atrophy) (ammer2023cnsmanifestationsin media 4b113608, ammer2023cnsmanifestationsin media 18508077).
4. Genetic / molecular information
4.1 Causal genes
- GNPTAB (major causal gene for MLII; encodes α/β subunits of GlcNAc‑1‑phosphotransferase) (he2023outcomesafterhsct pages 1-2, dogterom2021mucolipidosistypeii pages 1-2).
- GNPTG is relevant primarily to MLIII γ and for differential diagnosis within mucolipidosis subtypes (dogterom2021mucolipidosistypeii pages 1-2).
4.2 Variant classes and genotype–phenotype
Severe GNPTAB loss-of-function (e.g., frameshift/nonsense) correlates with MLII, while partial activity is associated with MLIII α/β (dogterom2021mucolipidosistypeii pages 1-2). Recent cohort work continues expanding variant spectra: a 2024 Chinese series reported detection of GNPTAB mutations in 87.5% of alleles and identified several novel variants, noting plasma arylsulfatase A and hexosaminidase A were significantly elevated with normal urinary GAGs (BMC Pediatrics; 2024; https://doi.org/10.1186/s12887-024-05223-x) (feng2024clinicalandmolecular pages 1-2).
A 2023 HSCT case report described novel compound heterozygous GNPTAB variants (c.673C>T; c.1090C>T) in China (Frontiers in Pediatrics; July 2023; https://doi.org/10.3389/fped.2023.1199489) (he2023outcomesafterhsct pages 1-2).
4.3 Functional consequence
Loss of M6P tagging leads to hypersecretion of lysosomal hydrolases and multi-substrate lysosomal accumulation (e.g., GAGs and lipids), a hallmark reflected clinically by elevated circulating lysosomal enzyme activities (he2023outcomesafterhsct pages 1-2, feng2024clinicalandmolecular pages 1-2).
5. Environmental information
No MLII-specific environmental, lifestyle, or infectious causal factors were identified in the retrieved evidence; MLII is primarily explained by GNPTAB-mediated enzyme trafficking failure (dogterom2021mucolipidosistypeii pages 1-2).
6. Mechanism / pathophysiology
6.1 Core pathway (M6P-dependent lysosomal enzyme trafficking)
GlcNAc‑1‑phosphotransferase is required for the first step in M6P tagging of lysosomal enzymes; failure of this step leads to impaired lysosomal targeting and extracellular hypersecretion of hydrolases, with intracellular deficiency of many lysosomal enzymes (khan2020mucolipidosesoverviewpast pages 1-3, he2023outcomesafterhsct pages 1-2).
Suggested pathways/ontologies: - GO Biological Process (suggested): lysosomal enzyme targeting; protein glycosylation; Golgi vesicle transport; lysosomal organization. - GO Cellular Component (suggested): Golgi apparatus; lysosome.
6.2 Downstream cellular consequences (recent mechanistic synthesis; 2024)
Recent mechanistic synthesis in Gnptab-deficient models links defective M6P sorting to: - NPC2 missorting and reduced lysosomal NPC2, contributing to cholesterol trafficking defects and cholesterol accumulation (badenetti2024investigatingneuronalpathogenesisa pages 24-28). - Autophagy impairment with accumulation of p62 and LC3-II, increased lysosome number (LAMP1/2 elevation), and defective autolysosomes; mitochondrial dysfunction in MLII fibroblasts could be rescued by inhibiting autophagosome generation, implicating impaired autophagic flux in pathogenesis (badenetti2024investigatingneuronalpathogenesisa pages 24-28).
Suggested GO terms (process): autophagy; cholesterol transport; lysosome organization.
Suggested CL terms (cell types): fibroblast; cardiomyocyte; neuron (based on cited model systems) (badenetti2024investigatingneuronalpathogenesisa pages 24-28).
7. Anatomical structures affected
MLII is multisystemic; heavily affected systems include: - Skeletal system (dysostosis multiplex, joint restriction, hip dysplasia) (ammer2023cnsmanifestationsin pages 4-6, dogterom2021mucolipidosistypeii pages 3-4). - Respiratory system (recurrent infections/respiratory failure; major cause of death) (dogterom2021mucolipidosistypeii pages 3-4). - Cardiovascular system (cardiac failure and combined cardiopulmonary causes of death) (dogterom2021mucolipidosistypeii pages 3-4). - Central nervous system (severe developmental impairment; cervical spinal stenosis frequent on imaging) (ammer2023cnsmanifestationsin pages 1-2, ammer2023cnsmanifestationsin media 4b113608).
UBERON suggestions (term names): bone; cartilage; heart; lung; brain; cervical spinal cord.
8. Temporal development (onset and progression)
- Onset: Typically prenatal/neonatal/early infancy. Systematic review data reported median age at first symptoms 0.0 years (IQR 0.0–0.3) for MLII (dogterom2021mucolipidosistypeii pages 3-4).
- Diagnosis: Median age at diagnosis 0.7 years (IQR 0.2–1.4) in pooled literature (dogterom2021mucolipidosistypeii pages 3-4).
- Progression/course: Progressive multisystem disease; leading fatal complications are pulmonary and cardiac (dogterom2021mucolipidosistypeii pages 3-4).
9. Inheritance and population
9.1 Inheritance
Autosomal recessive inheritance is consistently reported for MLII (dogterom2021mucolipidosistypeii pages 1-2).
9.2 Epidemiology and survival statistics
A systematic review of 843 published MLII/MLIII cases provides the most consolidated quantitative evidence in the retrieved corpus: - Median survival (Kaplan–Meier) for MLII: 5.0 years (95% CI 3.8–6.2) (dogterom2021mucolipidosistypeii pages 3-4). - Median age of death: 1.8 years (IQR 0.2–4.1) (dogterom2021mucolipidosistypeii pages 3-4). - Reported causes of death among MLII cases included pneumonia (32/93), respiratory failure (24/93), and cardiac failure (15/93), consistent with cardiopulmonary dominance (dogterom2021mucolipidosistypeii pages 3-4).
The same review reported estimated combined incidence of mucolipidosis (MLII/III overall) of 0.22–2.70 per 100,000 live births (dogterom2021mucolipidosistypeii pages 1-2).
10. Diagnostics
A structured diagnostic workflow summary is provided below.
| Diagnostic component | Specimen / modality | Key MLII finding | Practical notes / thresholds | Evidence |
|---|---|---|---|---|
| Core biochemical screen | Plasma / serum lysosomal enzyme assay | Markedly increased circulating lysosomal hydrolase activities due to missorting/hypersecretion; reported markers include arylsulfatase A, hexosaminidase A, and β-glucuronidase | Feng 2024 notes enzyme activities 10–20× above normal support diagnosis; urinary glycosaminoglycans may be normal, so normal urine GAGs do not exclude MLII | (feng2024clinicalandmolecular pages 1-2, he2023outcomesafterhsct pages 1-2) |
| Example plasma enzyme references | Plasma enzyme panel | Arylsulfatase A and hexosaminidase A are highlighted as significantly elevated in MLII/III α/β cohorts | Reference ranges reported by Feng 2024: ASA 50–140 nmol/mg·17 h; HexA 29.8–63.8 nmol/mg·h; interpret against local laboratory standards | (feng2024clinicalandmolecular pages 1-2) |
| Dried blood spot (DBS) multiplex enzymology | DBS tandem MS/MS panel | MLII/III profile can show significantly elevated acid sphingomyelinase (ASM), iduronate-2-sulfatase (IDS), and alpha-N-acetylglucosaminidase (NAGLU); some enzymes may appear reduced in the same panel | Hong 2023 studied 15 MLII/III patients versus >500 newborn DBS; authors suggest adding an elevated-activity cutoff may enable MLII/III detection as a secondary newborn-screening finding | (hong2023multiplextandemmass pages 1-2, hong2023multiplextandemmass pages 4-5) |
| Emerging biomarker study | Plasma MS-based biomarker discovery | BioML trial seeks a new mass-spectrometry plasma biomarker for ML I/II/III/IV, explicitly including MLII | Secondary aims include robustness, specificity, and long-term stability over 12- and 24-month time frames; observational study was withdrawn, so not clinical standard yet | (NCT02298673 chunk 1, NCT02298673 chunk 2) |
| Molecular confirmation | Germline DNA testing (single gene, panel, exome/genome) | Biallelic pathogenic GNPTAB variants confirm GNPTAB-related MLII / MLIII α/β | Targeted GNPTAB sequencing is confirmatory after biochemical suspicion; severe loss-of-function variants are strongly associated with MLII | (he2023outcomesafterhsct pages 1-2, dogterom2021mucolipidosistypeii pages 1-2) |
| Differential molecular context | Related gene testing when phenotype overlaps attenuated forms | GNPTG testing is relevant mainly for MLIII gamma rather than classic MLII | Helps separate MLII from related mucolipidosis subtypes in broader lysosomal/glycoproteinosis panels | (dogterom2021mucolipidosistypeii pages 1-2) |
| Skeletal imaging | Plain radiographs / bone survey | Characteristic dysostosis multiplex and progressive bone disease; presenting bone abnormalities are common | Useful early when coarse facies, growth failure, joint restriction, or abnormal skull shape raise suspicion; Dogterom reports bone abnormalities among common presenting findings | (dogterom2021mucolipidosistypeii pages 3-4, khan2020mucolipidosesoverviewpast pages 1-3) |
| Neuroimaging | Brain and cervical spine MRI | MRI abnormalities are often nonspecific; cervical spinal stenosis 63% in one 11-patient cohort, with mild brain atrophy and white-matter lesions reported | Ammer 2023 found profound developmental impairment but no clear progressive neurodegeneration; cervical imaging is especially relevant because stenosis may require surveillance/intervention | (ammer2023cnsmanifestationsin pages 1-2, ammer2023cnsmanifestationsin media 4b113608) |
| Neurodevelopmental assessment | Standardized developmental testing (e.g., BSID-III, VABS-II) | Severe developmental impairment is typical; mean developmental quotient at last assessment was 36.7% (SD 20.4) in Ammer 2023 | Not diagnostic alone, but valuable for baseline characterization, prognosis, and longitudinal follow-up | (ammer2023cnsmanifestationsin pages 1-2) |
| Natural-history / registry-based evaluation | Blood, urine, fibroblasts, clinical records, serial exams | Longitudinal glycoproteinosis studies include MLII to define progression, genotype-phenotype relationships, and current supportive care patterns | NCT01891422 collects blood, urine, fibroblasts, neuropsychology, and skeletal imaging data; useful for research and future endpoint development rather than routine diagnosis | (NCT01891422 chunk 1, dogterom2021mucolipidosistypeii pages 1-2) |
| Diagnostic synthesis | Integrated clinicobiochemical-genetic workup | Diagnosis is strongest when early severe phenotype + markedly elevated plasma lysosomal enzymes + biallelic GNPTAB variants are present | MLII often presents prenatally/neonatally/infancy with coarse facial features, skeletal disease, growth failure, and developmental delay; biochemical plus genetic confirmation is recommended | (he2023outcomesafterhsct pages 1-2, feng2024clinicalandmolecular pages 1-2, dogterom2021mucolipidosistypeii pages 1-2) |
Table: This table summarizes the main diagnostic modalities used for mucolipidosis type II, from plasma enzyme testing and DBS MS/MS screening to GNPTAB confirmation and imaging. It is useful as a concise, evidence-linked workflow for populating diagnostic fields in a disease knowledge base.
10.1 Key diagnostic concepts and definitions
- “Hypersecretion” signature: In MLII, lysosomal enzyme activities in plasma can be markedly increased due to missorting; one recent cohort used 10–20× above normal as a supportive diagnostic criterion, while urinary GAGs may remain normal (feng2024clinicalandmolecular pages 1-2).
10.2 Recent development: multiplex MS/MS enzyme activity assay (2023)
Hong et al. (Molecular Genetics and Metabolism Reports; June 2023; https://doi.org/10.1016/j.ymgmr.2023.100978) reported a multiplex tandem MS DBS assay measuring activities of 12 lysosomal enzymes and found acid sphingomyelinase (ASM), iduronate-2-sulfatase (IDS), and alpha-N-acetylglucosaminidase (NAGLU) were significantly elevated in MLII/III compared with newborn DBS (hong2023multiplextandemmass pages 1-2, hong2023multiplextandemmass pages 4-5). They suggest incorporating an elevated-activity cutoff could permit MLII/III detection as a secondary newborn-screening finding (hong2023multiplextandemmass pages 4-5).
10.3 Emerging biomarker research programs (real-world implementation)
- NCT01891422 (Completed; Greenwood Genetic Center; 2009 entry): Longitudinal observational study including MLII, designed to define natural history, document supportive therapies, and collect biospecimens (blood, urine, fibroblasts) alongside neuropsychological and skeletal imaging endpoints (NCT01891422 chunk 1).
- NCT02298673 (Withdrawn; CENTOGENE; 2018 entry): Proposed development/validation of an MS-based plasma biomarker for ML I–IV, including MLII, with robustness/specificity/stability testing up to 24 months (NCT02298673 chunk 2).
11. Outcome / prognosis
MLII carries a poor prognosis with early childhood mortality in classic disease; systematic review-derived median survival is 5.0 years and cardiopulmonary complications dominate reported causes of death (dogterom2021mucolipidosistypeii pages 3-4, dogterom2021mucolipidosistypeii pages 1-2).
A 2023 longitudinal CNS cohort adds nuance: profound developmental impairment is typical, but the cohort-level pattern did not support progressive neurocognitive decline over the observed period, with non-progressive/nonspecific MRI findings except frequent cervical spinal stenosis (ammer2023cnsmanifestationsin pages 1-2, ammer2023cnsmanifestationsin media 4b113608).
12. Treatment
A structured treatment/management summary (including MAXO term suggestions) is provided below.
| Management domain | Intervention / implementation | Evidence and real-world notes | Limitations / caveats | Suggested MAXO term(s) | Citation |
|---|---|---|---|---|---|
| Supportive multidisciplinary care | Ongoing multispecialty management including cardiopulmonary monitoring, orthopedic care, developmental services, audiology/ophthalmology follow-up, nutrition, and symptom-directed procedures | MLII is a progressive multisystem disorder with pulmonary and cardiac complications as leading causes of death; published natural-history cohorts document frequent use of life-extending/supportive procedures such as tracheostomy, gastric tube placement, craniosynostosis surgery, cervical spine surgery, and cardiac interventions. Longitudinal natural-history study NCT01891422 was designed in part to document disease progression, supportive therapies, surgeries, and subspecialty needs in glycoproteinoses including MLII. | Supportive care improves safety and quality of life but is not disease-modifying; burden remains high and skeletal disease often progresses despite intervention. | supportive care; respiratory management; cardiac monitoring; physical therapy; occupational therapy; nutritional support; surgical management | (dogterom2021mucolipidosistypeii pages 1-2, dogterom2021mucolipidosistypeii pages 4-5, NCT01891422 chunk 1) |
| Hematopoietic stem cell transplantation (HSCT) | Allogeneic HSCT / umbilical cord blood transplantation in selected severe cases | He et al. 2023 reported HSCT at 12 months from a 9/10 HLA-matched unrelated donor with neutrophil and platelet engraftment on days 10 and 11 and short-term improvement in muscle tone, gross/fine motor skills, and developmental testing. Earlier MLII HSCT experience reviewed by Ammer et al. shows donor cells can provide M6P-tagged enzymes for cross-correction and may preserve some cardiac function and ambulation. | Evidence base is extremely small; no cure established. Benefits remain uncertain, neurodevelopmental impairment and skeletal progression may persist, and post-transplant mortality/serious complications occur. In one detailed follow-up case, disease markers improved but the child died at 6.6 years from pneumonia. | hematopoietic stem cell transplantation; allogeneic stem cell transplantation; cord blood transplantation | (he2023outcomesafterhsct pages 1-2, ammer2021ishematopoieticstem pages 1-2, dogterom2021mucolipidosistypeii pages 4-5) |
| HSCT expert interpretation | HSCT as a potential but unproven disease-modifying approach | Case-based and review evidence suggests HSCT may prolong life or improve quality of life in some patients, consistent with broader LSD transplant experience summarized in neurological LSD therapy reviews. | Current evidence is insufficient for routine recommendation as standard of care in MLII; exact benefit remains unclear and requires longer-term prospective study. | clinical monitoring after transplantation; multidisciplinary transplant evaluation | (he2023outcomesafterhsct pages 1-2, ammer2021ishematopoieticstem pages 1-2) |
| Natural history / care infrastructure | Longitudinal observational follow-up and registry-style characterization | NCT01891422 follows patients with glycoproteinoses including MLII using annual history, neuropsychology, growth data, skeletal imaging, surgery tracking, and biospecimen collection to define progression and current care patterns. This is a real-world implementation that supports clinical counseling and endpoint development. | Observational only; does not test efficacy of therapy directly. | disease progression monitoring; longitudinal phenotyping; care coordination | (NCT01891422 chunk 1) |
| Biomarker development | Plasma MS-based biomarker discovery for earlier/sensitive diagnosis | BioML (NCT02298673) explicitly included MLII and aimed to develop an MS-based plasma biomarker, with secondary goals of testing robustness, specificity, and long-term stability over 12–24 months. This represents a translational research direction that could support screening and treatment monitoring. | Trial status was withdrawn; no validated MLII-specific plasma biomarker established from this record. | biomarker testing; blood biomarker analysis; mass spectrometry assay | (NCT02298673 chunk 1, NCT02298673 chunk 2) |
| Biochemical diagnostic innovation with management relevance | DBS multiplex MS/MS enzyme activity profiling | Hong et al. 2023 showed DBS multiplex tandem MS can detect MLII/III-associated enzyme patterns, with significant elevation of ASM, IDS, and NAGLU in affected patients versus newborn controls; authors suggest screening algorithms could detect MLII/III as a secondary finding. Earlier diagnosis could enable earlier supportive planning and transplant consideration. | Not a treatment; assay performance is influenced by storage conditions and non-age-matched samples, and the authors caution against using their values as reference ranges. | newborn screening; enzyme activity testing; dried blood spot testing | (hong2023multiplextandemmass pages 1-2, hong2023multiplextandemmass pages 4-5) |
| Experimental / preclinical gene therapy | Gene replacement or CNS-directed gene therapy concepts for lysosomal disease | Reviews of neurological LSD therapeutics highlight rapid development of AAV and hematopoietic stem cell gene therapy approaches across LSDs, establishing a relevant platform for MLII even though no MLII clinical gene therapy trial was identified in the retrieved evidence. Disease-specific review material cited in MLII sources points to gene therapy as a likely future option. | No MLII human gene therapy trial or approved product identified in retrieved sources; evidence for MLII remains preclinical/prospective. | gene therapy; viral vector gene therapy; ex vivo gene therapy | (dogterom2021mucolipidosistypeii pages 1-2, khan2020mucolipidosesoverviewpast pages 18-20) |
| Experimental substrate reduction / small-molecule approaches | Candidate substrate reduction, chaperone, or immunomodulatory strategies in cell models | Retrieved ML-focused preclinical literature notes reduced heparan sulfate in patient fibroblasts with agents such as miglustat, genistein, and thalidomide, supporting exploratory non-transplant therapeutic directions for mucolipidoses. | Evidence is in vitro and mainly MLIII-focused in retrieved sources; no clinical efficacy data for MLII. | substrate reduction therapy; small molecule therapy | (khan2020mucolipidosesoverviewpast pages 18-20) |
| Overall current standard | No established curative therapy; management remains largely supportive with selective experimental use of HSCT | Systematic review and case reports consistently state that no curative or clearly disease-modifying therapy is established for MLII at present; treatment in practice is dominated by supportive multidisciplinary care, with HSCT attempted in rare cases and biomarker/gene-therapy research ongoing. | Prognosis remains poor in severe MLII despite advances in diagnosis and supportive care. | palliative care; supportive care; multidisciplinary care pathway | (he2023outcomesafterhsct pages 1-2, dogterom2021mucolipidosistypeii pages 1-2) |
Table: This table summarizes current real-world management of mucolipidosis type II, emphasizing supportive multidisciplinary care, the limited but important HSCT experience, and active translational research directions such as biomarker development and preclinical gene therapy.
12.1 Current standard of care and real-world management
There is no established curative or clearly disease-modifying therapy; management is largely supportive and multidisciplinary. In the systematic review, multiple “potential life-extending procedures” were reported in MLII, including HSCT/BMT/cord blood transplant, tracheostomy, gastrostomy, craniosynostosis surgery, cervical spine surgery, and cardiac interventions (dogterom2021mucolipidosistypeii pages 4-5).
12.2 HSCT (limited human evidence)
A 2023 case report explicitly states: “No cure for MLII exists.” and summarizes typical severity and outcomes; it reports an HSCT performed at 12 months with early engraftment and short-term motor improvements (Frontiers in Pediatrics; July 2023; https://doi.org/10.3389/fped.2023.1199489) (he2023outcomesafterhsct pages 1-2). The authors conclude: “Our data show that HSCT is a potential way to prolong the life of patients and improve their quality of life… the exact benefit remains unclear in MLII patients.” (he2023outcomesafterhsct pages 1-2).
12.3 Experimental / translational directions
No MLII-specific clinical gene therapy or ERT trials were identified in the retrieved evidence set; however, systematic review authors highlight gene therapy as a likely future direction for ML (dogterom2021mucolipidosistypeii pages 1-2). Translational efforts currently visible in retrieved sources include (i) biomarker discovery protocols and (ii) multiplex MS/MS approaches that could enable earlier detection and better endpoint definition (NCT02298673 chunk 2, hong2023multiplextandemmass pages 4-5).
13. Prevention
As a Mendelian disorder, primary prevention is primarily genetic: - Genetic counseling for autosomal recessive inheritance and family planning (suggested MAXO: genetic counseling). - Prenatal diagnosis / carrier testing are implied in the genetic-testing framework but not detailed in retrieved sources.
Newborn screening is not standard for MLII in the retrieved sources, but the DBS multiplex enzymology work provides a plausible route for secondary detection in LSD screening programs (hong2023multiplextandemmass pages 4-5).
14. Other species / natural disease
A naturally occurring MLII-like disease has been described in domestic cats with a GNPTAB nonsense variant and clinical/biochemical similarities to human MLII, positioning feline MLII as a large-animal model for pathophysiology and therapeutic exploration (BMC Veterinary Research; Dec 2018; https://doi.org/10.1186/s12917-018-1728-1) (wang2018agnptabnonsense pages 8-8).
15. Model organisms
Multiple model systems are used to study GNPTAB deficiency: - Mouse models: gene-trap and knock-in Gnptab models recapitulating key aspects of MLII, including elevated circulating lysosomal enzymes, skeletal pathology, and neurodegeneration in severe models (khan2020mucolipidosesoverviewpast pages 7-9). - Zebrafish: subunit-deficient zebrafish models are cited as informative for enzyme-specific mannose phosphorylation differences (wang2018agnptabnonsense pages 8-8). - Human cellular models: patient-derived fibroblasts (and other cell types) show characteristic storage and trafficking defects and are widely used to study downstream consequences such as cholesterol accumulation and impaired autophagy (badenetti2024investigatingneuronalpathogenesisa pages 24-28).
Key evidence statistics (quick reference)
- Median age at diagnosis: 0.7 years (dogterom2021mucolipidosistypeii pages 3-4).
- Median survival: 5.0 years (95% CI 3.8–6.2) (dogterom2021mucolipidosistypeii pages 3-4).
- Causes of death (examples): pneumonia 32/93, respiratory failure 24/93, cardiac failure 15/93 (dogterom2021mucolipidosistypeii pages 3-4).
- Developmental quotient (DQ) in one longitudinal cohort: mean 36.7% (SD 20.4) (ammer2023cnsmanifestationsin pages 1-2).
- Cervical spinal stenosis on MRI in one cohort: 63% (ammer2023cnsmanifestationsin pages 1-2, ammer2023cnsmanifestationsin media 4b113608).
Evidence gaps and constraints (for knowledge base curation)
- Ontology identifiers (MONDO/Orphanet/ICD/MeSH) were not present in retrieved full texts during this run; they should be added from dedicated ontology resources (artifact-00).
- Validated longitudinal biomarkers for treatment monitoring remain limited; clinicaltrials.gov shows biomarker discovery intent but one biomarker study was withdrawn (NCT02298673 chunk 2).
- Clinical gene therapy/ERT trials for MLII were not identified in the retrieved evidence set; statements here are limited to prospects and platform-level developments rather than MLII-specific trial outcomes (dogterom2021mucolipidosistypeii pages 1-2).
References
-
(dogterom2021mucolipidosistypeii pages 1-2): Emma J. Dogterom, Margreet A.E.M. Wagenmakers, Martina Wilke, Serwet Demirdas, Nicole M. Muschol, Sandra Pohl, Jan C. van der Meijden, Dimitris Rizopoulos, Ans T. van der Ploeg, and Esmée Oussoren. Mucolipidosis type ii and type iii: a systematic review of 843 published cases. Genetics in Medicine, 23:2047-2056, Nov 2021. URL: https://doi.org/10.1038/s41436-021-01244-4, doi:10.1038/s41436-021-01244-4. This article has 44 citations and is from a highest quality peer-reviewed journal.
-
(he2023outcomesafterhsct pages 1-2): Si-jia He, Dong-jun Li, Wen-qiong Lv, Wen-hao Tang, Shu-wen Sun, Yi-ping Zhu, Ying Liu, Jin Wu, and Xiao-xi Lu. Outcomes after hsct for mucolipidosis ii (i-cell disease) caused by novel compound heterozygous gnptab mutations. Frontiers in Pediatrics, Jul 2023. URL: https://doi.org/10.3389/fped.2023.1199489, doi:10.3389/fped.2023.1199489. This article has 4 citations.
-
(ammer2023cnsmanifestationsin pages 1-2): Luise Sophie Ammer, Karolin Täuber, Anna Perez, Thorsten Dohrmann, Jonas Denecke, René Santer, Ulrike Blümlein, Ann-Kathrin Ozga, Sandra Pohl, and Nicole Maria Muschol. Cns manifestations in mucolipidosis type ii—a retrospective analysis of longitudinal data on neurocognitive development and neuroimaging in eleven patients. Journal of Clinical Medicine, 12:4114, Jun 2023. URL: https://doi.org/10.3390/jcm12124114, doi:10.3390/jcm12124114. This article has 8 citations.
-
(hong2023multiplextandemmass pages 1-2): Xinying Hong, Laura Pollard, Miao He, Michael H. Gelb, and Timothy C. Wood. Multiplex tandem mass spectrometry enzymatic activity assay for the screening and diagnosis of mucolipidosis type ii and iii. Jun 2023. URL: https://doi.org/10.1016/j.ymgmr.2023.100978, doi:10.1016/j.ymgmr.2023.100978. This article has 6 citations.
-
(badenetti2024investigatingneuronalpathogenesisa pages 24-28): L Badenetti. Investigating neuronal pathogenesis in mucopolysaccharidosis type ii and mucolipidosis type ii disease models. Unknown journal, 2024.
-
(khan2020mucolipidosesoverviewpast pages 1-3): Shaukat A. Khan and Saori C. Tomatsu. Mucolipidoses overview: past, present, and future. International Journal of Molecular Sciences, 21:6812, Sep 2020. URL: https://doi.org/10.3390/ijms21186812, doi:10.3390/ijms21186812. This article has 78 citations.
-
(feng2024clinicalandmolecular pages 1-2): Yuyu Feng, Yonglan Huang, Xiaoyuan Zhao, Huiying Sheng, Xueying Su, Xi Yin, Liu Li, and Wen Zhang. Clinical and molecular characteristics of 20 chinese probands with mucolipidosis type ii and iii alpha/beta. BMC Pediatrics, Dec 2024. URL: https://doi.org/10.1186/s12887-024-05223-x, doi:10.1186/s12887-024-05223-x. This article has 2 citations and is from a peer-reviewed journal.
-
(dogterom2021mucolipidosistypeii pages 3-4): Emma J. Dogterom, Margreet A.E.M. Wagenmakers, Martina Wilke, Serwet Demirdas, Nicole M. Muschol, Sandra Pohl, Jan C. van der Meijden, Dimitris Rizopoulos, Ans T. van der Ploeg, and Esmée Oussoren. Mucolipidosis type ii and type iii: a systematic review of 843 published cases. Genetics in Medicine, 23:2047-2056, Nov 2021. URL: https://doi.org/10.1038/s41436-021-01244-4, doi:10.1038/s41436-021-01244-4. This article has 44 citations and is from a highest quality peer-reviewed journal.
-
(NCT01891422 chunk 1): Longitudinal Studies of the Glycoproteinoses. Greenwood Genetic Center. 2009. ClinicalTrials.gov Identifier: NCT01891422
-
(ammer2023cnsmanifestationsin pages 4-6): Luise Sophie Ammer, Karolin Täuber, Anna Perez, Thorsten Dohrmann, Jonas Denecke, René Santer, Ulrike Blümlein, Ann-Kathrin Ozga, Sandra Pohl, and Nicole Maria Muschol. Cns manifestations in mucolipidosis type ii—a retrospective analysis of longitudinal data on neurocognitive development and neuroimaging in eleven patients. Journal of Clinical Medicine, 12:4114, Jun 2023. URL: https://doi.org/10.3390/jcm12124114, doi:10.3390/jcm12124114. This article has 8 citations.
-
(ammer2023cnsmanifestationsin media 4b113608): Luise Sophie Ammer, Karolin Täuber, Anna Perez, Thorsten Dohrmann, Jonas Denecke, René Santer, Ulrike Blümlein, Ann-Kathrin Ozga, Sandra Pohl, and Nicole Maria Muschol. Cns manifestations in mucolipidosis type ii—a retrospective analysis of longitudinal data on neurocognitive development and neuroimaging in eleven patients. Journal of Clinical Medicine, 12:4114, Jun 2023. URL: https://doi.org/10.3390/jcm12124114, doi:10.3390/jcm12124114. This article has 8 citations.
-
(ammer2021ishematopoieticstem pages 1-2): Luise Sophie Ammer, Sandra Pohl, Sandra Rafaela Breyer, Charlotte Aries, Jonas Denecke, Anna Perez, Martin Petzoldt, Johanna Schrum, Ingo Müller, and Nicole Maria Muschol. Is hematopoietic stem cell transplantation a therapeutic option for mucolipidosis type ii? Mar 2021. URL: https://doi.org/10.1016/j.ymgmr.2020.100704, doi:10.1016/j.ymgmr.2020.100704. This article has 9 citations.
-
(ammer2023cnsmanifestationsin media 18508077): Luise Sophie Ammer, Karolin Täuber, Anna Perez, Thorsten Dohrmann, Jonas Denecke, René Santer, Ulrike Blümlein, Ann-Kathrin Ozga, Sandra Pohl, and Nicole Maria Muschol. Cns manifestations in mucolipidosis type ii—a retrospective analysis of longitudinal data on neurocognitive development and neuroimaging in eleven patients. Journal of Clinical Medicine, 12:4114, Jun 2023. URL: https://doi.org/10.3390/jcm12124114, doi:10.3390/jcm12124114. This article has 8 citations.
-
(hong2023multiplextandemmass pages 4-5): Xinying Hong, Laura Pollard, Miao He, Michael H. Gelb, and Timothy C. Wood. Multiplex tandem mass spectrometry enzymatic activity assay for the screening and diagnosis of mucolipidosis type ii and iii. Jun 2023. URL: https://doi.org/10.1016/j.ymgmr.2023.100978, doi:10.1016/j.ymgmr.2023.100978. This article has 6 citations.
-
(NCT02298673 chunk 1): Biomarker for Mucolipidosis Disorder Type I, II, III, IV (BioML). CENTOGENE GmbH Rostock. 2018. ClinicalTrials.gov Identifier: NCT02298673
-
(NCT02298673 chunk 2): Biomarker for Mucolipidosis Disorder Type I, II, III, IV (BioML). CENTOGENE GmbH Rostock. 2018. ClinicalTrials.gov Identifier: NCT02298673
-
(dogterom2021mucolipidosistypeii pages 4-5): Emma J. Dogterom, Margreet A.E.M. Wagenmakers, Martina Wilke, Serwet Demirdas, Nicole M. Muschol, Sandra Pohl, Jan C. van der Meijden, Dimitris Rizopoulos, Ans T. van der Ploeg, and Esmée Oussoren. Mucolipidosis type ii and type iii: a systematic review of 843 published cases. Genetics in Medicine, 23:2047-2056, Nov 2021. URL: https://doi.org/10.1038/s41436-021-01244-4, doi:10.1038/s41436-021-01244-4. This article has 44 citations and is from a highest quality peer-reviewed journal.
-
(khan2020mucolipidosesoverviewpast pages 18-20): Shaukat A. Khan and Saori C. Tomatsu. Mucolipidoses overview: past, present, and future. International Journal of Molecular Sciences, 21:6812, Sep 2020. URL: https://doi.org/10.3390/ijms21186812, doi:10.3390/ijms21186812. This article has 78 citations.
-
(wang2018agnptabnonsense pages 8-8): Ping Wang, Hamutal Mazrier, Jessica Caverly Rae, Karthik Raj, and Urs Giger. A gnptab nonsense variant is associated with feline mucolipidosis ii (i-cell disease). BMC Veterinary Research, Dec 2018. URL: https://doi.org/10.1186/s12917-018-1728-1, doi:10.1186/s12917-018-1728-1. This article has 10 citations and is from a peer-reviewed journal.
-
(khan2020mucolipidosesoverviewpast pages 7-9): Shaukat A. Khan and Saori C. Tomatsu. Mucolipidoses overview: past, present, and future. International Journal of Molecular Sciences, 21:6812, Sep 2020. URL: https://doi.org/10.3390/ijms21186812, doi:10.3390/ijms21186812. This article has 78 citations.