GM1 Gangliosidosis Type 2
GM1 gangliosidosis type 2 (late-infantile/juvenile GM1 gangliosidosis) is the intermediate-severity form of GLB1-related lysosomal beta-galactosidase deficiency, with partial residual enzyme activity. GM1 ganglioside accumulates in neurons, producing later-onset and more slowly progressive neurodegeneration than the infantile form, with developmental regression, seizures, and ataxia, and less prominent visceral and skeletal involvement.
Ask OpenScientist
Ask a research question about GM1 Gangliosidosis Type 2. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Mappings
Definitions
1Show evidence (1 reference)
Inheritance
1Pathophysiology
2Show evidence (1 reference)
Show evidence (1 reference)
Pathograph
Phenotypes
12Cardiovascular 1
Show evidence (1 reference)
Digestive 1
Show evidence (1 reference)
Eye 2
Show evidence (1 reference)
Show evidence (1 reference)
Musculoskeletal 2
Show evidence (1 reference)
Show evidence (1 reference)
Nervous System 5
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Other 1
Show evidence (1 reference)
Genetic Associations
1Show evidence (1 reference)
Medical Actions
1Show evidence (1 reference)
Differential Diagnoses
1Conditions with similar clinical presentations that must be differentiated from GM1 Gangliosidosis Type 2:
- Earlier onset, near-absent enzyme activity, and death by ~3 years, versus the slower late-infantile/juvenile course.
Show evidence (1 reference)
Clinical Trials
2Show evidence (1 reference)
Show evidence (1 reference)
Source YAML
click to showname: GM1 Gangliosidosis Type 2
creation_date: "2026-06-13T00:00:00Z"
description: >-
GM1 gangliosidosis type 2 (late-infantile/juvenile GM1 gangliosidosis) is the
intermediate-severity form of GLB1-related lysosomal beta-galactosidase deficiency,
with partial residual enzyme activity. GM1 ganglioside accumulates in neurons, producing
later-onset and more slowly progressive neurodegeneration than the infantile form, with
developmental regression, seizures, and ataxia, and less prominent visceral and skeletal
involvement.
category: Mendelian
disease_term:
preferred_term: GM1 gangliosidosis type 2
term:
id: MONDO:0009261
label: GM1 gangliosidosis type 2
mappings:
mondo_mappings:
- term:
id: MONDO:0009261
label: GM1 gangliosidosis type 2
mapping_predicate: skos:exactMatch
mapping_source: MONDO
mapping_justification: Primary MONDO disease identifier for this late-infantile/juvenile GM1 gangliosidosis entry.
external_assertions:
- name: Orphanet GM1 gangliosidosis type 2 record
source: Orphanet
assertion_type: structured_disease_record
external_id: ORPHA:79256
url: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=79256
description: >
Orphanet's ORPHA:79256 structured record for GM1 gangliosidosis type 2
includes the exact MONDO and OMIM cross-references, autosomal recessive
inheritance, GLB1 disease-gene assertion, definition, and epidemiology used
in this entry.
evidence:
- reference: ORPHA:79256
reference_title: GM1 gangliosidosis type 2 (Orphanet structured-database record)
supports: SUPPORT
evidence_source: OTHER
snippet: "MONDO:0009261 | Exact"
explanation: Orphanet maps ORPHA:79256 exactly to the MONDO identifier used by this entry.
- reference: ORPHA:79256
reference_title: GM1 gangliosidosis type 2 (Orphanet structured-database record)
supports: SUPPORT
evidence_source: OTHER
snippet: "OMIM:230600 | Exact"
explanation: Orphanet lists OMIM:230600 as an exact external cross-reference.
synonyms:
- Late-infantile GM1 gangliosidosis
- Juvenile GM1 gangliosidosis
- Intermediate GM1 gangliosidosis
- GM1 gangliosidosis, type II
references:
- reference: PMID:24156116
title: "GLB1-Related Disorders."
tags:
- GeneReviews
- reference: PMID:38313286
title: "GM1 Gangliosidosis Type II: Results of a 10-Year Prospective Study."
parents:
- sphingolipidosis
pathophysiology:
- name: Partial Beta-Galactosidase Deficiency and GM1 Ganglioside Accumulation
conforms_to: "lysosomal_substrate_accumulation#Lysosomal Hydrolase or Cofactor Deficiency"
description: >-
Biallelic GLB1 variants reduce lysosomal beta-galactosidase activity. Residual activity
higher than in the infantile form yields a later-onset, more slowly progressive neuronal
GM1 ganglioside accumulation.
gene:
preferred_term: GLB1
term:
id: hgnc:4298
label: GLB1
biological_processes:
- preferred_term: ganglioside catabolic process
modifier: DECREASED
term:
id: GO:0006689
label: ganglioside catabolic process
evidence:
- reference: PMID:33859490
reference_title: "GM1 Gangliosidosis: Mechanisms and Management."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Mutation of the GLB1 gene, which codes for β-gal, prevents cleavage of the terminal β-1,4-linked galactose residue from GM1 ganglioside."
explanation: "GLB1 deficiency blocks GM1 ganglioside degradation; partial activity gives the intermediate phenotype."
downstream:
- target: Neuronal GM1 Storage and Neurodegeneration
description: Reduced enzyme activity allows neuronal GM1 ganglioside accumulation.
causal_link_type: DIRECT
evidence:
- reference: PMID:34539759
reference_title: "GM1 Gangliosidosis-A Mini-Review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Absent or reduced β-galactosidase activity leads to the accumulation of β-linked galactose-containing glycoconjugates including the glycosphingolipid (GSL) GM1-ganglioside in neuronal tissue."
explanation: Reduced beta-galactosidase activity directly causes neuronal GM1 ganglioside accumulation.
- target: Decreased beta-galactosidase activity
description: GLB1 variants reduce measured beta-galactosidase enzymatic activity.
causal_link_type: DIRECT
evidence:
- reference: PMID:30187681
reference_title: Protein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "an enzyme assay showed reduced β-galactosidase-1 activity"
explanation: The type II case report documents reduced beta-galactosidase-1 activity.
- name: Neuronal GM1 Storage and Neurodegeneration
conforms_to: "lysosomal_substrate_accumulation#Lysosomal Substrate Accumulation"
description: >-
GM1 ganglioside accumulates in neuronal lysosomes, impairing cell physiology and
precipitating progressive neurodegeneration at a slower pace than the infantile form.
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
cellular_components:
- preferred_term: lysosome
term:
id: GO:0005764
label: lysosome
evidence:
- reference: PMID:34539759
reference_title: "GM1 Gangliosidosis-A Mini-Review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Absent or reduced β-galactosidase activity leads to the accumulation of β-linked galactose-containing glycoconjugates including the glycosphingolipid (GSL) GM1-ganglioside in neuronal tissue."
explanation: "Reduced beta-galactosidase activity causes neuronal GM1 ganglioside accumulation."
downstream:
- target: Developmental regression
description: Neuronal GM1 storage produces progressive psychomotor regression.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:31937438
reference_title: "The natural history of Type 1 infantile GM1 gangliosidosis: A literature-based meta-analysis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Type 2 patients exhibit similar symptoms to Type 1 patients, including psychomotor regression and eye and bone abnormalities, but have an attenuated progression"
explanation: Literature review supports psychomotor regression in type II GM1 gangliosidosis.
- target: Seizures
description: Neuronal storage and neurodegeneration are associated with seizures.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:30187681
reference_title: Protein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "include progressive motor abnormalities, muscle weakness, seizures, strabismus, corneal clouding, and diffuse brain atrophy"
explanation: Type II clinical summary lists seizures among the neurologic manifestations.
- target: Dysphagia / feeding difficulties
description: Progressive neurologic decline causes feeding difficulty and aspiration risk.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:24156116
reference_title: "GLB1-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "progressive feeding \ndifficulties leading to aspiration risk"
explanation: GeneReviews links type II GM1 progression to feeding difficulties and aspiration risk.
- target: Speech decline / dysarthria
description: CNS storage disease produces progressive motor and speech decline.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:24156116
reference_title: "GLB1-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "progressive cognitive, motor, and speech\ndecline as measured by psychometric\ntesting"
explanation: GeneReviews supports progressive speech decline in type II GM1 gangliosidosis.
- target: Corneal clouding
description: Storage disease can include ocular corneal clouding.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:24156116
reference_title: "GLB1-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "There may be mild corneal clouding, hepatosplenomegaly, and/or\ncardiomyopathy"
explanation: GeneReviews supports mild corneal clouding in type II GM1 gangliosidosis.
- target: Cerebral atrophy
description: Progressive neuronal storage disease produces brain atrophy on serial MRI.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:38313286
reference_title: "GM1 Gangliosidosis Type II: Results of a 10-Year Prospective Study."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Serial MRIs demonstrated progressive brain atrophy"
explanation: Prospective type II cohort supports progressive brain atrophy.
- target: Muscle weakness
description: Progressive neuromotor disease includes muscle weakness.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:30187681
reference_title: Protein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "include progressive motor abnormalities, muscle weakness, seizures, strabismus, corneal clouding, and diffuse brain atrophy"
explanation: Type II clinical summary lists muscle weakness among the motor manifestations.
- target: Strabismus
description: Type II GM1 neurologic and ocular involvement includes strabismus.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:30187681
reference_title: Protein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "include progressive motor abnormalities, muscle weakness, seizures, strabismus, corneal clouding, and diffuse brain atrophy"
explanation: Type II clinical summary lists strabismus among the clinical manifestations.
phenotypes:
- category: Biochemical
name: Decreased beta-galactosidase activity
description: Reduced lysosomal beta-galactosidase activity is the diagnostic enzymatic defect.
phenotype_term:
preferred_term: Decreased beta-galactosidase activity
term:
id: HP:0008166
label: Decreased beta-galactosidase activity
evidence:
- reference: PMID:30187681
reference_title: Protein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "an enzyme assay showed reduced β-galactosidase-1 activity"
explanation: The type II case report documents reduced beta-galactosidase-1 activity.
- name: Developmental regression
description: Loss of acquired developmental milestones after a period of normal or near-normal early development.
phenotype_term:
preferred_term: Developmental regression
term:
id: HP:0002376
label: Developmental regression
evidence:
- reference: PMID:31937438
reference_title: "The natural history of Type 1 infantile GM1 gangliosidosis: A literature-based meta-analysis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Type 2 patients exhibit similar symptoms to Type 1 patients, including psychomotor regression and eye and bone abnormalities, but have an attenuated progression"
explanation: "Type 2 GM1 gangliosidosis features psychomotor regression with an attenuated progression compared with Type 1."
- name: Seizures
description: >-
Seizures occur in late-infantile and juvenile type II GM1 gangliosidosis (D'Souza 2024
prospective cohort; per-subtype frequencies are reported in the full-text tables, not the
abstract).
phenotype_term:
preferred_term: Seizure
term:
id: HP:0001250
label: Seizure
evidence:
- reference: PMID:24156116
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "seizure medication dosages that result in excessive sedation"
explanation: GeneReviews surveillance guidance addresses seizure management in GLB1-related disease, indicating seizures occur and require treatment; per-subtype type II frequencies are in the full text.
- reference: PMID:30187681
reference_title: Protein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "include progressive motor abnormalities, muscle weakness, seizures, strabismus, corneal clouding, and diffuse brain atrophy"
explanation: Type II clinical summary lists seizures among the manifestations.
- name: Ataxia
description: >-
Cerebellar ataxia is part of the progressive neuromotor decline in type II GM1
gangliosidosis (D'Souza 2024 cohort; full-text frequency data).
phenotype_term:
preferred_term: Ataxia
term:
id: HP:0001251
label: Ataxia
- name: Dysphagia / feeding difficulties
description: >-
Progressive feeding difficulties leading to aspiration risk are part of the typical
type II GM1 gangliosidosis course as the neurological disease advances.
phenotype_term:
preferred_term: Dysphagia
term:
id: HP:0002015
label: Dysphagia
evidence:
- reference: PMID:24156116
reference_title: "GLB1-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "progressive feeding \ndifficulties leading to aspiration risk"
explanation: GeneReviews explicitly describes progressive feeding difficulties leading to aspiration risk as part of the typical type II course.
- name: Speech decline / dysarthria
description: Progressive speech decline (dysarthria) accompanies the CNS dysfunction.
phenotype_term:
preferred_term: Dysarthria
term:
id: HP:0001260
label: Dysarthria
evidence:
- reference: PMID:24156116
reference_title: "GLB1-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "progressive cognitive, motor, and speech\ndecline as measured by psychometric\ntesting"
explanation: CNS dysfunction in type II manifests as progressive cognitive, motor, and speech decline.
- name: Skeletal disease (kyphosis)
description: Progressive skeletal disease including kyphosis occurs in some individuals.
phenotype_term:
preferred_term: Kyphosis
term:
id: HP:0002808
label: Kyphosis
evidence:
- reference: PMID:24156116
reference_title: "GLB1-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "progressive skeletal disease in some individuals (including kyphosis"
explanation: Progressive skeletal disease including kyphosis is part of the type II phenotype.
- name: Corneal clouding
description: Mild corneal clouding may be present.
phenotype_term:
preferred_term: Corneal opacity
term:
id: HP:0007957
label: Corneal opacity
evidence:
- reference: PMID:24156116
reference_title: "GLB1-Related Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "There may be mild corneal clouding, hepatosplenomegaly, and/or\ncardiomyopathy"
explanation: Mild corneal clouding is a recognized feature of type II GM1 gangliosidosis.
- name: Cardiomyopathy
description: Cardiomyopathy and thickened cardiac valves can develop, especially in juvenile onset.
phenotype_term:
preferred_term: Cardiomyopathy
term:
id: HP:0001638
label: Cardiomyopathy
evidence:
- reference: PMID:38313286
reference_title: "GM1 Gangliosidosis Type II: Results of a 10-Year Prospective Study."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Some\nolder children with juvenile onset developed thickened aortic and/or mitral\nvalves with regurgitation"
explanation: Juvenile-onset type II patients develop thickened cardiac valves with regurgitation.
- name: Cerebral atrophy
description: Progressive brain atrophy on serial neuroimaging.
phenotype_term:
preferred_term: Cerebral atrophy
term:
id: HP:0002059
label: Cerebral atrophy
evidence:
- reference: PMID:38313286
reference_title: "GM1 Gangliosidosis Type II: Results of a 10-Year Prospective Study."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Serial MRIs demonstrated progressive brain atrophy"
explanation: Serial MRIs in the type II cohort showed progressive brain atrophy.
- name: Muscle weakness
description: Muscle weakness occurs as part of the progressive motor involvement in type II GM1 gangliosidosis.
phenotype_term:
preferred_term: Muscle weakness
term:
id: HP:0001324
label: Muscle weakness
evidence:
- reference: PMID:30187681
reference_title: Protein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "include progressive motor abnormalities, muscle weakness, seizures, strabismus, corneal clouding, and diffuse brain atrophy"
explanation: Type II clinical summary lists muscle weakness among the manifestations.
- name: Strabismus
description: Strabismus can occur with type II GM1 gangliosidosis ocular and neurologic involvement.
phenotype_term:
preferred_term: Strabismus
term:
id: HP:0000486
label: Strabismus
evidence:
- reference: PMID:30187681
reference_title: Protein modeling and clinical description of a novel in-frame GLB1 deletion causing GM1 gangliosidosis type II.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "include progressive motor abnormalities, muscle weakness, seizures, strabismus, corneal clouding, and diffuse brain atrophy"
explanation: Type II clinical summary lists strabismus among the manifestations.
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
genetic:
- name: GLB1
association: Biallelic GLB1 variants with partial residual beta-galactosidase activity
relationship_type: CAUSATIVE
variant_origin: GERMLINE
gene_term:
preferred_term: GLB1
term:
id: hgnc:4298
label: GLB1
evidence:
- reference: PMID:34539759
reference_title: "GM1 Gangliosidosis-A Mini-Review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "GM1 gangliosidosis is a progressive, neurosomatic, lysosomal storage disorder caused by mutations in the GLB1 gene encoding the enzyme β-galactosidase."
explanation: "GLB1 mutations cause GM1 gangliosidosis; partial activity yields the late-infantile/juvenile form."
progression:
- phase: Late-infantile/juvenile intermediate course
notes: >-
Type II is intermediate in severity, with later onset and slower progression than the
infantile form; the three GM1 forms are distinguished by age of onset.
evidence:
- reference: PMID:34539759
reference_title: "GM1 Gangliosidosis-A Mini-Review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Type I (infantile), Type II (late-infantile and juvenile), and Type III (adult)], based on the age of onset of clinical symptoms"
explanation: "Type II is the late-infantile/juvenile form defined by intermediate age of onset."
diagnosis:
- name: Beta-galactosidase enzyme assay
diagnosis_term:
preferred_term: clinical laboratory procedure
term:
id: MAXO:0000006
label: clinical laboratory procedure
description: Demonstration of reduced lysosomal beta-galactosidase activity in leukocytes or fibroblasts.
markers: Reduced (partial residual) beta-galactosidase activity.
evidence:
- reference: PMID:34539759
reference_title: "GM1 Gangliosidosis-A Mini-Review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "caused by mutations in the GLB1 gene encoding the enzyme β-galactosidase"
explanation: "Beta-galactosidase activity testing underlies diagnosis."
- name: GLB1 molecular genetic testing
diagnosis_term:
preferred_term: genetic testing
term:
id: MAXO:0000127
label: genetic testing
description: Confirmatory biallelic GLB1 sequencing.
evidence:
- reference: PMID:34539759
reference_title: "GM1 Gangliosidosis-A Mini-Review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "caused by mutations in the GLB1 gene encoding the enzyme β-galactosidase"
explanation: "GLB1 sequencing provides molecular confirmation."
differential_diagnoses:
- name: GM1 gangliosidosis type 1
description: >-
The severe infantile form with near-absent enzyme activity and rapidly fatal course.
disease_term:
preferred_term: GM1 gangliosidosis type 1
term:
id: MONDO:0009260
label: GM1 gangliosidosis type 1
distinguishing_features:
- Earlier onset, near-absent enzyme activity, and death by ~3 years, versus the slower late-infantile/juvenile course.
evidence:
- reference: PMID:31937438
reference_title: "The natural history of Type 1 infantile GM1 gangliosidosis: A literature-based meta-analysis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Type 2 patients exhibit similar symptoms to Type 1 patients, including psychomotor regression and eye and bone abnormalities, but have an attenuated progression"
explanation: "Type 1 differs from Type 2 by earlier onset and faster progression; Type 2 has an attenuated course."
treatments:
- name: Supportive Care
description: >-
No FDA-approved disease-modifying therapy exists; management is supportive, including
seizure control and rehabilitation.
treatment_term:
preferred_term: Supportive Care
term:
id: NCIT:C15747
label: Supportive Care
evidence:
- reference: PMID:33859490
reference_title: "GM1 Gangliosidosis: Mechanisms and Management."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Beyond palliative and supportive care, no FDA-approved treatments exist for GM1 patients."
explanation: "Care is supportive in the absence of approved disease-modifying therapy."
clinical_trials:
- name: NCT03952637
phase: PHASE_I
status: RECRUITING
description: >-
Intravenous AAV9 gene transfer expressing human beta-galactosidase (GLB1) in Type I and
Type II GM1 gangliosidosis.
evidence:
- reference: clinicaltrials:NCT03952637
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A gene therapy may help the body make this enzyme"
explanation: AAV9-GLB1 gene therapy aims to restore beta-galactosidase in GM1 gangliosidosis, including type II.
- name: NCT04713475
phase: PHASE_I
status: ACTIVE_NOT_RECRUITING
description: >-
PBGM01 (AAVhu68-GLB1) delivered into the cisterna magna in pediatric Type 1 and Type 2a
GM1 gangliosidosis.
evidence:
- reference: clinicaltrials:NCT04713475
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "PBGM01 is a gene therapy for GM1 gangliosidosis intended to deliver a functional copy of the GLB1 gene"
explanation: PBGM01 delivers a functional GLB1 gene to the CNS in type 1 and type 2a GM1 gangliosidosis.
definitions:
- name: Clinical case definition of late-infantile/juvenile GM1 gangliosidosis
definition_type: CASE_DEFINITION
description: >-
Type 2 GM1 gangliosidosis is the intermediate form of GLB1-related beta-galactosidase
deficiency, defined by biallelic GLB1 variants with partial residual activity, neuronal
GM1 ganglioside accumulation, and a late-infantile/juvenile-onset neurodegenerative
course slower than the infantile form.
scope: Disease-level case definition for the late-infantile/juvenile GM1 gangliosidosis subtype.
evidence:
- reference: PMID:34539759
reference_title: "GM1 Gangliosidosis-A Mini-Review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "GM1 gangliosidosis is a progressive, neurosomatic, lysosomal storage disorder caused by mutations in the GLB1 gene encoding the enzyme β-galactosidase."
explanation: "Anchors the case definition in GLB1/beta-galactosidase deficiency."
References & Deep Research
References
2Deep Research
1GM1 Gangliosidosis Type 2 (late-infantile/juvenile): Disease Characteristics Research Report
Target disease: GM1 gangliosidosis type 2 (Type II; late-infantile and juvenile)
Category: Mendelian (autosomal recessive lysosomal storage disorder)
Core ontology IDs: MONDO:0009261 (type 2) and MONDO:0018149 (general GM1 gangliosidosis) (OpenTargets Search: GM1 gangliosidosis)
| Category | Item | Value | Source type | URL | Evidence |
|---|---|---|---|---|---|
| Disease identifier | MONDO (general) | MONDO:0018149 — GM1 gangliosidosis | OpenTargets | https://platform.opentargets.org/disease/MONDO_0018149 | (OpenTargets Search: GM1 gangliosidosis) |
| Disease identifier | MONDO (type II) | MONDO:0009261 — GM1 gangliosidosis type 2 | OpenTargets | https://platform.opentargets.org/disease/MONDO_0009261 | (OpenTargets Search: GM1 gangliosidosis) |
| Disease identifier | ICD-10 | E75.1 | Paper | https://doi.org/10.3389/fgene.2024.1344051 | (zagaynova2024casereportpreimplantation pages 1-2) |
| Disease identifier | OMIM (type I / type II / type III series) | 230500 / 230600 / 230650 | Paper | https://doi.org/10.3389/fgene.2024.1344051 | (zagaynova2024casereportpreimplantation pages 1-2) |
| Disease identifier | OMIM (type II specific) | 230600 | Paper | https://doi.org/10.1007/8904_2015_451 | (ferreira2020theskeletalphenotype pages 1-6) |
| Classification | MeSH term | Gangliosidosis, GM1 | Clinical trial metadata | https://clinicaltrials.gov/study/NCT04041102 | (NCT04041102 chunk 2, NCT04041102 chunk 3) |
| Classification | Broader category | Lysosomal storage disease; sphingolipidosis; nervous system lysosomal storage disease | Clinical trial metadata | https://clinicaltrials.gov/study/NCT04041102 | (NCT04041102 chunk 2, NCT04041102 chunk 3) |
| Causal gene | HGNC gene symbol | GLB1 | OpenTargets / papers | https://platform.opentargets.org/target/ENSG00000170266 | (OpenTargets Search: GM1 gangliosidosis, rha2021gm1gangliosidosismechanisms pages 1-2) |
| Gene product | Enzyme | Lysosomal acid β-galactosidase; beta-galactosidase | Papers | https://doi.org/10.2147/TACG.S206076 | (rha2021gm1gangliosidosismechanisms pages 1-2, rha2021gm1gangliosidosismechanisms pages 2-3) |
| Synonym | Common disease name | GM1 gangliosidosis type II | Papers | https://doi.org/10.1016/j.gim.2024.101144 | (d’souza2024gm1gangliosidosistype pages 1-3) |
| Synonym | Common disease name | Intermediate GM1 gangliosidosis | Paper | https://doi.org/10.1016/j.bone.2019.115142 | (ferreira2020theskeletalphenotype pages 1-6) |
| Synonym | Common disease name | Juvenile GM1 gangliosidosis | Papers / trial | https://doi.org/10.1186/s12881-017-0417-4 | (karimzadeh2017casereportsof pages 1-2, NCT04041102 chunk 2) |
| Synonym | Common disease name | Late-infantile GM1 gangliosidosis | Paper | https://doi.org/10.1016/j.jpeds.2019.08.016 | (arashkaps2019theclinicaland pages 1-2) |
| Synonym | Combined subtype label | Late-infantile/juvenile GM1 gangliosidosis | Paper | https://doi.org/10.3389/fgene.2024.1344051 | (zagaynova2024casereportpreimplantation pages 1-2) |
| Synonym | Alternative subtype notation | Type 2a (late-infantile) / Type 2b (juvenile) | Papers | https://doi.org/10.1016/j.jpeds.2019.08.016 | (arashkaps2019theclinicaland pages 1-2, lang2020thenaturalhistory pages 1-2) |
| Distinguishing description | Spectrum concept | GM1 gangliosidosis is a clinical continuum; type II comprises the late-infantile and juvenile forms | Papers | https://doi.org/10.3389/fgene.2021.734878 | (nicoli2021gm1gangliosidosis—aminireview pages 1-2, arashkaps2019theclinicaland pages 1-2) |
Table: This table compiles the main disease identifiers, molecular anchors, and commonly used names for GM1 gangliosidosis type II. It is useful as a normalization aid for mapping literature, ontology terms, and trial records to the same disease concept.
1. Disease Information
1.1 Concise overview
GM1 gangliosidosis is a progressive, neurosomatic lysosomal storage disorder caused by pathogenic variants in GLB1, resulting in deficiency of lysosomal acid β-galactosidase (β-gal) and accumulation of GM1 ganglioside and related glycoconjugates. Clinical severity spans a continuum but is commonly categorized by age of onset into infantile (type I), late-infantile/juvenile (type II), and adult/chronic (type III). Type II is generally less rapidly progressive than type I but remains neurodegenerative and life-limiting. (rha2021gm1gangliosidosismechanisms pages 1-2, nicoli2021gm1gangliosidosis—aminireview pages 1-2)
1.2 Key identifiers and synonyms
- MONDO: type II GM1 gangliosidosis = MONDO:0009261; GM1 gangliosidosis (general) = MONDO:0018149 (OpenTargets Search: GM1 gangliosidosis).
- ICD-10: E75.1 (reported in a 2024 clinical genetics case report) (zagaynova2024casereportpreimplantation pages 1-2).
- OMIM: Type I 230500; Type II 230600; Type III 230650 (zagaynova2024casereportpreimplantation pages 1-2).
- MeSH term: “Gangliosidosis, GM1” (as used in ClinicalTrials.gov condition-browse metadata) (NCT04041102 chunk 2, NCT04041102 chunk 3).
Common synonyms used in the literature include “late-infantile GM1 gangliosidosis,” “juvenile GM1 gangliosidosis,” “intermediate GM1 gangliosidosis,” and subtype labels type IIa (late infantile) / type IIb (juvenile) (arashkaps2019theclinicaland pages 1-2, lang2020thenaturalhistory pages 1-2, ferreira2020theskeletalphenotype pages 1-6).
Orphanet (ORPHA) ID and ICD-11 code: not found in the retrieved full-text evidence; would require direct lookup in Orphanet/WHO ICD-11 resources (gap).
1.3 Evidence source type
This report is derived from: (i) aggregated disease-level reviews and cohort studies, (ii) prospective natural history observational cohorts, (iii) clinical-trial registry records, and (iv) translational biomarker and model-organism studies.
2. Etiology
2.1 Disease causal factors
Primary cause: biallelic pathogenic variants in GLB1 encoding β-galactosidase → deficient lysosomal β-gal activity → lysosomal storage of GM1 ganglioside and other β-linked galactose-containing substrates (including keratan sulfate–related glycoconjugates) (arashkaps2019theclinicaland pages 1-2, rha2021gm1gangliosidosismechanisms pages 1-2).
Inheritance: autosomal recessive (karimzadeh2017casereportsof pages 1-2, rha2021gm1gangliosidosismechanisms pages 1-2).
2.2 Risk factors
For a Mendelian disorder, the dominant “risk factors” are genetic:
* Carrier status in parents (autosomal recessive) and consanguinity (illustrated in juvenile type II case series from consanguineous families) (karimzadeh2017casereportsof pages 1-2).
Founder effects / high carrier frequency* in specific populations (Roma and Cypriot village example; see §9) (rha2021gm1gangliosidosismechanisms pages 1-2).
2.3 Protective factors and gene–environment interactions
No robust protective genetic variants or gene–environment interactions were identified in the retrieved evidence. Some genotype–phenotype association concepts are noted (e.g., type II/III often retaining residual β-gal activity), but these reflect allelic severity rather than protective modifiers (lang2020thenaturalhistory pages 1-2, lang2020thenaturalhistory pages 2-3).
3. Phenotypes (Type II: late-infantile and juvenile)
Large, systematically phenotyped type II cohorts now exist. A key 2024 10-year prospective observational study enrolled 41 individuals with type II GM1 (late infantile ~17; juvenile ~24), providing multi-system assessments including MRI/MRS and adaptive behavior measures (d’souza2024gm1gangliosidosistype pages 1-3).
The table below consolidates key phenotypes and suggested HPO terms.
| Phenotype (plain language) | Suggested HPO term(s) | Typical onset/subtype | Frequency / statistics if available | Progression notes | Key source (PMID if known, else DOI) | Evidence citation ids |
|---|---|---|---|---|---|---|
| Developmental plateau/regression and loss of acquired milestones | HP:0002376 Developmental regression; HP:0012758 Neurodevelopmental delay | Late-infantile typically after normal early milestones to ~12 months; juvenile often after initially normal development at 3–5 years | Late-infantile patients “usually exhibited delay or non-acquisition of major milestones”; juvenile patients “usually attained these milestones on time” before later decline | Progressive; late-infantile loses skills earlier and faster, juvenile shows slower decline | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144; Ferreira et al., 2020, DOI:10.1016/j.bone.2019.115142 | (d’souza2024gm1gangliosidosistype pages 6-9, ferreira2020theskeletalphenotype pages 1-6) |
| Impaired walking / loss of ambulation | HP:0002355 Difficulty walking; HP:0002505 Ataxia; HP:0001288 Gait disturbance | Late-infantile: 12–18 months onward; juvenile: often first recognized at 3–5 years | Late-infantile: by age 2 most are non-ambulatory; juvenile: many remain ambulatory for years but most are wheelchair-bound by mid-teens | Worsens over time; within-person mobility decline documented longitudinally | Ferreira et al., 2020, DOI:10.1016/j.bone.2019.115142; D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (ferreira2020theskeletalphenotype pages 1-6, d’souza2024gm1gangliosidosistype pages 9-12) |
| Ataxia / coordination problems | HP:0001251 Ataxia | Common presenting feature, especially juvenile/type 2b | No cohort percentage reported in extracted text; repeatedly described as an early manifestation | Progressive and often followed by dystonia/spasticity | Karimzadeh et al., 2017, DOI:10.1186/s12881-017-0417-4; D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (karimzadeh2017casereportsof pages 1-2, d’souza2024gm1gangliosidosistype pages 3-6) |
| Dystonia / abnormal movements | HP:0001332 Dystonia | More typical in juvenile and later type II course, but can occur in both late-infantile and juvenile | No cohort percentage reported in extracted text | Progressive movement disorder; often follows gait/ataxia symptoms | Karimzadeh et al., 2017, DOI:10.1186/s12881-017-0417-4; Arash-Kaps et al., 2019, DOI:10.1016/j.jpeds.2019.08.016 | (karimzadeh2017casereportsof pages 1-2, arashkaps2019theclinicaland pages 1-2) |
| Spasticity / hyperreflexia | HP:0001257 Spasticity; HP:0001347 Hyperreflexia | Variable, often later in type II | No percentage in extracted text | Progressive upper motor neuron features in a subset | Rha et al., 2021, DOI:10.2147/TACG.S206076 | (rha2021gm1gangliosidosismechanisms pages 2-3) |
| Speech disorder / dysarthria / loss of speech | HP:0001260 Dysarthria; HP:0002167 Delayed speech and language development; HP:0001344 Progressive neurologic deterioration | Late-infantile: limited expressive language and progressive anarthria; juvenile: often normal speech first, early “stuttering” then dysarthria | Juvenile speech scores decline with age (rho = -0.60); language rho = -0.53 | Progressive communication loss, more severe/earlier in late-infantile | Ferreira et al., 2020, DOI:10.1016/j.bone.2019.115142; D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (ferreira2020theskeletalphenotype pages 1-6, d’souza2024gm1gangliosidosistype pages 12-15) |
| Swallowing difficulty / dysphagia | HP:0002015 Dysphagia | Seen in both; often clinically important in later childhood | VFSS performed in 82% late-infantile (14/17) and 96% juvenile (23/24); juvenile dietary restriction score worsened with age (rho = -0.68) | Generally progressive feeding/swallow impairment; aspiration risk variable | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (d’souza2024gm1gangliosidosistype pages 6-9, d’souza2024gm1gangliosidosistype pages 9-12) |
| Seizures / epileptiform EEG abnormalities | HP:0001250 Seizure; HP:0002353 EEG abnormality | Variable; can emerge during childhood in both subtypes | At enrollment, seizure treatment in 65% (11/17) late-infantile and 29% (7/24) juvenile; epileptiform EEG activity in 40% (4/10) of abnormal late-infantile EEGs | Progressive neurologic disease; EEG abnormalities common, especially late-infantile | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (d’souza2024gm1gangliosidosistype pages 12-15, d’souza2024gm1gangliosidosistype pages 37-41) |
| Cerebral and cerebellar atrophy on MRI | HP:0002059 Cerebral atrophy; HP:0001272 Cerebellar atrophy | Both subtypes, more severe/rapid in late-infantile | Late-infantile: cerebellar atrophy 11/14 (79%), cerebral cortical atrophy 11/14 (79%); juvenile: cerebral cortical atrophy 17/21 (81%), cerebellar atrophy 6/21 (29%) | Progressive; serial MRI shows faster progression in late-infantile | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (d’souza2024gm1gangliosidosistype pages 12-15, d’souza2024gm1gangliosidosistype pages 1-3) |
| White matter / myelination abnormalities | HP:0002500 Abnormal cerebral white matter morphology; HP:0012447 Delayed myelination | Both; especially notable in late-infantile | Late-infantile: all 8 assessed for myelination abnormal; juvenile white matter injury 13/19 (68%) | Progressive imaging abnormality; used as a biomarker in natural history and trials | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (d’souza2024gm1gangliosidosistype pages 12-15) |
| Visual impairment with strabismus / nystagmus | HP:0000505 Visual impairment; HP:0000486 Strabismus; HP:0000639 Nystagmus | More severe in late-infantile; can occur in juvenile | Late-infantile: strabismus 17/17 (100%), nystagmus 9/17 (53%), cortical visual impairment 12/17 (71%); juvenile average visual acuity better and less cortical visual impairment | Progressive visual dysfunction; unlike type I, cherry-red spots typically absent | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144; Rha et al., 2021, DOI:10.2147/TACG.S206076 | (d’souza2024gm1gangliosidosistype pages 6-9, rha2021gm1gangliosidosismechanisms pages 2-3) |
| Hearing usually normal or near-normal | HP:0000365 Hearing impairment (typically absent); HP:0010788 Abnormal auditory brainstem response | Both subtypes | Peripheral hearing normal in 88% (15/17) late-infantile and 92% (22/24) juvenile; ABR normal in 53% late-infantile and 64% juvenile | Auditory phenotype relatively preserved versus infantile GM1, though ABR abnormalities may occur | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (d’souza2024gm1gangliosidosistype pages 6-9, d’souza2024gm1gangliosidosistype pages 9-12) |
| Skeletal dysplasia / low bone mineral density / odontoid hypoplasia | HP:0002652 Skeletal dysplasia; HP:0000939 Osteopenia; HP:0003307 Odontoid hypoplasia; HP:0100923 Avascular necrosis of the femoral head | Both; odontoid hypoplasia especially late-infantile, hip AVN in some juvenile patients | Lumbar spine, femoral neck, total hip BMD Z-scores about -2.1, -2.2, -1.8; all late-infantile patients had odontoid hypoplasia in the cited cohort | Chronic orthopedic burden; contributes to disability and wheelchair dependence | Ferreira et al., 2020, DOI:10.1016/j.bone.2019.115142 | (ferreira2020theskeletalphenotype pages 1-6) |
| Cardiac valvular thickening/regurgitation | HP:0001654 Cardiac valvular defect; HP:0001644 Dilated ascending aorta not established here; HP:0006682 Mitral regurgitation; HP:0001653 Aortic regurgitation | More often older juvenile patients | Juvenile ECHO abnormalities in 3/13 with aortic leaflet thickening/regurgitation; one late-infantile patient had mitral valve prolapse | Appears later and is not universal | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (d’souza2024gm1gangliosidosistype pages 1-3, d’souza2024gm1gangliosidosistype pages 9-12) |
| Low β-galactosidase activity (laboratory abnormality) | HP:0012379 Abnormal enzyme/coenzyme activity (suggested broad term) | Present in both subtypes from diagnosis | Serum β-gal 0–5% of pediatric controls (median 0.028); CSF 2–8% (median 0.05) of controls | Stable underlying biochemical defect; central to diagnosis and trial pharmacodynamics | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (d’souza2024gm1gangliosidosistype pages 9-12) |
| Elevated AST / mild liver chemistry abnormality | HP:0031964 Elevated circulating aspartate aminotransferase concentration | Both, more common in late-infantile | AST elevated in 76% late-infantile and 29% juvenile | Usually mild; part of multisystem monitoring rather than dominant clinical phenotype | D’Souza et al., 2024, DOI:10.1016/j.gim.2024.101144 | (d’souza2024gm1gangliosidosistype pages 9-12) |
Table: This table summarizes the main clinical, imaging, and laboratory phenotypes reported for GM1 gangliosidosis type II, split where possible between late-infantile and juvenile presentations. It is useful for mapping disease features to HPO terms and for distinguishing common progression patterns and frequencies from recent natural-history studies.
3.1 Phenotype highlights with quantitative data (recent)
- Diagnostic delay: median time from symptom onset to diagnosis in probands was 1.53 years (late infantile) vs 5.5 years (juvenile) in the 2024 prospective cohort—consistent with milder/atypical presentations in juvenile type II (d’souza2024gm1gangliosidosistype pages 6-9). In a skeletal-focused cohort, average time from symptom onset to diagnosis was 1.9 years (late infantile) vs 6.3 years (juvenile) (ferreira2020theskeletalphenotype pages 1-6).
- Preserved hearing compared with type I: peripheral hearing was normal in 88% (15/17) late-infantile and 92% (22/24) juvenile participants; abnormal auditory brainstem responses occurred in a subset (d’souza2024gm1gangliosidosistype pages 9-12).
- Neuroimaging biomarkers: MRI showed high prevalence of atrophy and white matter abnormalities with subtype differences (e.g., late infantile cerebellar atrophy 79% vs juvenile 29%) and age correlations for some atrophy ratings (ρage up to 0.79) (d’souza2024gm1gangliosidosistype pages 12-15).
- MRS biomarkers: both groups had elevated myo-inositol and decreased N-acetylaspartate (NAA) with age correlations (juvenile NAA ρ=-0.74; late infantile NAA ρ=-0.59) (d’souza2024gm1gangliosidosistype pages 12-15).
3.2 Quality of life (QoL) impact
Direct patient QoL instruments were not extracted from the type II natural history texts; however, caregiving burden data from the U.S. indicate high functional dependence in juvenile/late-onset GM1/GM2: caregivers reported frequent mobility aid use and speech difficulties, plus substantial psychological and financial burden (qualitative; not subtype-specific) (rodriguez2025burdenofcaregiving pages 1-2).
4. Genetic / Molecular Information
4.1 Causal gene
- GLB1 (galactosidase beta 1) is the primary causal gene for GM1 gangliosidosis and is strongly supported in disease–target resources (OpenTargets) and clinical literature (OpenTargets Search: GM1 gangliosidosis, rha2021gm1gangliosidosismechanisms pages 1-2).
4.2 Pathogenic variants and genotype–phenotype themes
- Allelic heterogeneity is extensive: a review notes “more than 200 pathogenic GLB1 mutations” and very large numbers of possible allele combinations, complicating molecular interpretation (rha2021gm1gangliosidosismechanisms pages 2-3).
- Type II/III patients frequently show residual enzyme activity and are often compound heterozygotes (e.g., one null and one missense allele), consistent with attenuated severity compared with type I (lang2020thenaturalhistory pages 1-2, lang2020thenaturalhistory pages 2-3).
- In the 2024 prospective type II cohort, classification of 37 distinct variants resulted in upgrades and new pathogenic/likely pathogenic submissions, underscoring ongoing variant reclassification work in clinically followed cohorts (d’souza2024gm1gangliosidosistype pages 1-3).
4.3 Modifier genes / epigenetics / chromosomal abnormalities
No validated modifier genes, epigenetic drivers, or chromosomal abnormalities specific to type II GM1 were identified in the retrieved evidence.
5. Environmental Information
GM1 type II is fundamentally genetic; no consistent environmental or lifestyle contributors were identified in the retrieved literature.
6. Mechanism / Pathophysiology
6.1 Causal chain (current understanding)
- GLB1 loss-of-function → deficient lysosomal β-galactosidase activity (rha2021gm1gangliosidosismechanisms pages 1-2).
- Substrate accumulation (GM1 ganglioside and other β-gal substrates) in lysosomes and internal membranes (rha2021gm1gangliosidosismechanisms pages 1-2).
- ER stress and Ca2+ dysregulation: GM1 enrichment in the ER increases ER Ca2+ efflux and activates the unfolded protein response (UPR). GM1-enriched microdomains increase ER–mitochondria contact sites (MAMs), promoting abnormal Ca2+ transfer into mitochondria and mitochondrial Ca2+ overload, triggering apoptosis (nicoli2021gm1gangliosidosis—aminireview pages 4-5, rha2021gm1gangliosidosismechanisms pages 3-5).
- Synaptic dysfunction / neurotransmission changes: model data indicate Ca2+ influx/efflux abnormalities and impaired neurotransmitter uptake associated with stored lipids, contributing to circuit dysfunction and neurodegeneration (rha2021gm1gangliosidosismechanisms pages 5-6).
- Downstream tissue injury: progressive neuronal dysfunction and loss, with varying degrees of gliosis/neuroinflammation depending on stage/model (nicoli2021gm1gangliosidosis—aminireview pages 4-5, eikelberg2020axonopathyandreduction pages 1-3).
6.2 Recent developments (2023–2024): single-nucleus transcriptomics
A 2024 single-nucleus RNA-seq study in a GM1 mouse model highlighted cell-type–specific changes and pathway enrichments consistent with neurodegeneration and synaptic dysfunction. The authors reported that affected pathways included oxidative phosphorylation and neuroactive ligand–receptor interactions, and suggested neurotransmitter/circuit disruption may be more prominent than early inflammatory activation at the studied stage (16 weeks), with no detected microglial/astrocyte activation or innate immunity gene upregulation at that time point (Sep 2024; https://doi.org/10.3390/ijms25179712) (liu2024insightsintothe pages 1-2, liu2024insightsintothe pages 13-15).
6.3 Suggested ontology terms
- GO biological processes (examples): lysosomal catabolic process; unfolded protein response; regulation of intracellular calcium ion homeostasis; mitochondrion-mediated apoptotic signaling; synaptic transmission. Supported mechanistic themes include UPR-mediated apoptosis and synaptic dysfunction (rha2021gm1gangliosidosismechanisms pages 3-5, liu2024insightsintothe pages 13-15).
- Cell types (CL, examples): neuron; astrocyte; microglial cell; oligodendrocyte; oligodendrocyte precursor cell. Cell-type-specific transcriptomic changes were reported across neurons and glia (liu2024insightsintothe pages 1-2, liu2024insightsintothe pages 13-15).
7. Anatomical Structures Affected
7.1 Organ/system level
- Central nervous system (primary): progressive neurodegeneration, brain atrophy, white matter abnormalities (d’souza2024gm1gangliosidosistype pages 12-15).
- Eye/visual system: strabismus, nystagmus, cortical/neurologic visual impairment in a substantial fraction of late-infantile patients (d’souza2024gm1gangliosidosistype pages 6-9).
- Skeletal system: dysostosis multiplex spectrum, odontoid hypoplasia, low bone mineral density, avascular necrosis in some juvenile patients (ferreira2020theskeletalphenotype pages 1-6).
- Cardiac valves (subset, more in older juveniles): aortic/mitral valve thickening with regurgitation (d’souza2024gm1gangliosidosistype pages 1-3, d’souza2024gm1gangliosidosistype pages 9-12).
7.2 Tissue/cell level and subcellular localization
- Cellular compartment: lysosome (primary storage) and ER/MAM involvement (mechanistic) (nicoli2021gm1gangliosidosis—aminireview pages 4-5, rha2021gm1gangliosidosismechanisms pages 3-5).
- Affected CNS cell populations: neurons and glia (microglia/astrocytes/oligodendrocyte lineage) show transcriptional alterations in model systems (liu2024insightsintothe pages 1-2, liu2024insightsintothe pages 13-15).
8. Temporal Development (Type II)
8.1 Onset
Type II is commonly subdivided by onset age:
* Late-infantile (type IIa): onset roughly 1–3 years (and more narrowly 7 months–2 years in some summaries) (arashkaps2019theclinicaland pages 1-2, lang2020thenaturalhistory pages 1-2).
Juvenile (type IIb): onset roughly 3–10 years* (often described as 3–5 years in clinical cohorts) (arashkaps2019theclinicaland pages 1-2, ferreira2020theskeletalphenotype pages 1-6).
8.2 Progression and staging proxies
- Late-infantile type II often reaches ~12 months with typical milestones, then plateaus and loses skills between 12–18 months, with many becoming non-ambulatory by age 2 in one cohort (ferreira2020theskeletalphenotype pages 1-6).
- Juvenile type II often begins with subtle gait/speech changes (“stuttering” → dysarthria), later progressing to wheelchair dependence in many by mid-teens (ferreira2020theskeletalphenotype pages 1-6).
Quantitative longitudinal staging is increasingly supported by MRI/MRS and adaptive behavior scales (Vineland) in prospective cohorts (d’souza2024gm1gangliosidosistype pages 12-15).
9. Inheritance and Population
9.1 Epidemiology
Across multiple sources, GM1 gangliosidosis incidence is commonly cited as ~1 in 100,000–200,000 live births (rha2021gm1gangliosidosismechanisms pages 1-2, arashkaps2019theclinicaland pages 1-2). Higher regional incidence has been reported, e.g. southern Brazil ~1:17,000 live births (kannebley2015clinicalfindingsand pages 1-2) and Malta ~1:3,700 live births (rha2021gm1gangliosidosismechanisms pages 1-2). These figures are for GM1 overall rather than type II specifically.
9.2 Population genetics (founder effects / carrier frequency)
Carrier-frequency enrichment has been reported in some populations, including Roma (~1 in 50 overall; up to 1 in 10 in the Rudari sub-isolate) and a Cypriot village where ~1 in 12 residents are carriers (rha2021gm1gangliosidosismechanisms pages 1-2). Such data support targeted genetic counseling and cascade screening in high-risk groups.
10. Diagnostics
10.1 Core diagnostic workflow
- Biochemical testing: assay of β-galactosidase activity remains a common first-line test (rha2021gm1gangliosidosismechanisms pages 2-3). In the 2024 type II cohort, serum β-gal was 0–5% of controls (median 0.028) and CSF β-gal 2–8% of controls (median 0.05), illustrating the severe enzymatic deficit even in type II (d’souza2024gm1gangliosidosistype pages 9-12).
- Molecular confirmation: GLB1 sequencing (single gene, panels, exome/genome) to identify biallelic pathogenic variants (rha2021gm1gangliosidosismechanisms pages 2-3).
10.2 Biomarkers and monitoring (major recent advance: 2023)
A 2023 eBioMedicine biomarker paper developed/validated LC-MS/MS assays for glycan biomarkers and reported H3N2b as a high-dynamic-range pharmacodynamic biomarker: * Abstract-supported core claim (from retrieved evidence text): two pentasaccharides were “>18-fold elevated” in patient plasma/CSF/urine, and H3N2b was “negatively correlated with β-galactosidase activity” and decreased after AAV gene therapy in a cat model and in a treated patient (Jun 2023; https://doi.org/10.1016/j.ebiom.2023.104627) (kell2023apentasaccharidefor pages 1-2, kell2023apentasaccharidefor pages 2-3).
10.3 Imaging and electrophysiology
MRI/MRS abnormalities are common and quantifiable in type II GM1 and can serve as trial endpoints (e.g., atrophy, white matter injury, elevated myo-inositol, reduced NAA) (d’souza2024gm1gangliosidosistype pages 12-15). EEG abnormalities were frequent, particularly in late-infantile type II (d’souza2024gm1gangliosidosistype pages 12-15).
10.4 Differential diagnosis
Because juvenile type II may lack classic infantile signs (e.g., cherry-red maculae, hepatosplenomegaly), it can be mistaken for other ataxic or primarily neurologic disorders; clinicians are advised to consider GM1 in progressive neurodegeneration with spastic-dystonic movement disorders even without visceral findings (arashkaps2019theclinicaland pages 1-2, karimzadeh2017casereportsof pages 1-2).
11. Outcome / Prognosis
Type II GM1 is progressive and life-limiting. A 2021 review summarizes type II as typically presenting between ~7 months and 5 years and leading to death “in mid-childhood to early adulthood,” with variability related to residual β-gal activity (rha2021gm1gangliosidosismechanisms pages 1-2). Detailed type II survival distributions were not extracted from the retrieved type II natural history text excerpts (gap).
12. Treatment
12.1 Current real-world management
No globally approved disease-modifying therapy was identified in the retrieved evidence for GM1 (including type II); care is largely supportive and multidisciplinary (rha2021gm1gangliosidosismechanisms pages 1-2, d’souza2024gm1gangliosidosistype pages 1-3).
12.2 Advanced therapeutics and clinical trials (emphasis 2023–2024)
| Modality | Agent / program | Mechanism | Route | Key trial(s) | Phase / status | Key endpoints | Published results / evidence | Notes |
|---|---|---|---|---|---|---|---|---|
| Supportive care | Multidisciplinary symptomatic management | Seizure control, nutrition/swallow support, PT/OT/ST, orthopedic and cardiac monitoring, respiratory care | Standard clinical care | Natural-history studies used as comparator rather than interventional trial | Standard of care; no disease-modifying approval | Function, safety, quality of life, complication prevention | 10-year prospective type II cohort documented common use of antiseizure therapy, swallow studies, mobility assessment, and multisystem monitoring; no approved disease-modifying therapy as of 2024 (2024, DOI:10.1016/j.gim.2024.101144; 2021, DOI:10.2147/TACG.S206076) (d’souza2024gm1gangliosidosistype pages 37-41, d’souza2024gm1gangliosidosistype pages 1-3, rha2021gm1gangliosidosismechanisms pages 1-2) | Remains the real-world baseline for type II GM1; important for tertiary prevention and trial contextualization |
| Substrate reduction therapy (SRT) | Miglustat | Inhibits glycosphingolipid synthesis to reduce upstream substrate load | Oral | Historical case series/small studies; not a current pivotal GM1 type II registration trial in retrieved context | Off-label / exploratory; mixed evidence | Neurologic function, symptom stabilization | Review summarizing small Italian experience reported gradual neurologic improvements in very small cohorts; separate infantile gangliosidoses U.S. miglustat study was terminated (2024, DOI:10.3389/fnins.2024.1392683) (foster2024therapeuticdevelopmentsfor pages 5-6, foster2024therapeuticdevelopmentsfor media 704a7e6f, foster2024therapeuticdevelopmentsfor media 3fd6f6a9) | Evidence base is weak and heterogeneous; not established as standard disease-modifying therapy for type II GM1 |
| Substrate reduction therapy (SRT) | Venglustat | Brain-penetrant glucosylceramide synthase inhibitor intended to reduce glycosphingolipid biosynthesis | Oral | NCT04221451 | Phase 3; TERMINATED | Efficacy, pharmacodynamics, PK, safety | Trial record retrieved as a multinational randomized placebo-controlled study; review literature notes venglustat as an investigational small molecule for gangliosidoses/related disorders, but no GM1 type II efficacy results were available in retrieved 2023-2024 sources (ClinicalTrials.gov; 2025 biomarker review mentions investigational role) (foster2024therapeuticdevelopmentsfor pages 4-5, OpenTargets Search: GM1 gangliosidosis) | Important to mention because natural-history paper cites ongoing small-molecule substrate inhibitor development, but retrieved registry title is late-onset GM2-focused and status is terminated; relevance to GM1 type II appears indirect/in basket-program context rather than proven benefit |
| Gene therapy | Intravenous AAV9-GLB1 (NHGRI/Sio program; often described as AAV9/GLB1) | Gene replacement delivering human GLB1 to increase lysosomal β-galactosidase and reduce GM1 storage | Single IV infusion | NCT03952637 | Phase 1/2; RECRUITING | Primary: safety; secondary/exploratory: biomarkers, neurologic development, motor function, brain volume/myelination, immune tolerance | Trial design published in registry; review notes dosing cohorts up to 1.5E13, 4.5E13, 7.5E13 vg/kg for type II and 3-year follow-up (2024 review). Early clinical results became available later: first 9 type II participants showed increased CSF β-gal, decreased CSF GM1, imaging improvement signals, and relative stabilization in some Vineland domains, with one vector-attributed SAE and transient transaminase elevations (2025 preprint, DOI:10.1101/2025.07.28.25332074) (NCT03952637 chunk 1, foster2024therapeuticdevelopmentsfor pages 4-5, lewis2025aav9genetherapy pages 1-4) | Most directly relevant current systemic gene-therapy program for GM1 type II; 2023-2024 context supports active development though peer-reviewed efficacy data were not yet mature in 2024 |
| Gene therapy | PBGM01 (AAVhu68-GLB1; Imagine-1) | CNS-directed GLB1 gene replacement using AAVhu68 | Single injection into cisterna magna | NCT04713475 | Phase 1/2; ACTIVE_NOT_RECRUITING | Primary: safety / treatment-related AEs/SAEs and developmental milestone change; secondary: Vineland-II, β-gal activity, GM1 substrate levels, NfL, MRI, QoL, ventilator-free survival | Registry details include dose-escalation and expansion cohorts for Type 1 and Type 2a patients; 2-year efficacy assessment with 3-year safety extension. Reviews in 2023-2024 identify PBGM01 as one of the leading active GM1 gene-therapy programs, but no peer-reviewed efficacy data were available in the retrieved 2024 literature (ClinicalTrials.gov; 2024 review DOI:10.3389/fnins.2024.1392683) (NCT04713475 chunk 1, foster2024therapeuticdevelopmentsfor pages 5-6, shaimardanova2023genetherapyof pages 4-6) | Key CNS-targeted alternative to IV AAV9; specifically includes late-onset infantile/type 2a patients |
| Gene therapy | LYS-GM101 (AAVrh10-GLB1; Lysogene) | CNS-directed GLB1 gene replacement | Intracisternal / CNS-directed administration | NCT04273269 | Phase 1/2; TERMINATED | Safety, dose finding, feasibility | 2023 gene-therapy review lists this as one of three clinical AAV programs entering trials; no positive clinical efficacy data retrieved, and the registry status is terminated (shaimardanova2023genetherapyof pages 12-13, shaimardanova2023genetherapyof pages 4-6) | Relevant as part of the competitive clinical landscape, but not a current active front-runner based on retrieved status |
| Biomarker-enabled monitoring | H3N2b pentasaccharide biomarker | Natural β-gal substrate glycan used as pharmacodynamic biomarker for treatment response | Measured in urine, plasma/serum, CSF | Embedded in gene-therapy translational studies; used alongside AAV programs | Preclinical-to-clinical translational biomarker; not a therapy itself | Reduction in H3N2b as evidence of biochemical response; assay development and validation | 2023 eBioMedicine study showed H3N2b was >18-fold elevated in patient plasma/CSF/urine, negatively correlated with β-gal activity, and fell after AAV gene therapy in cats and in a treated patient; proposed as a non-invasive pharmacodynamic biomarker (2023, DOI:10.1016/j.ebiom.2023.104627) (kell2023apentasaccharidefor pages 1-2, kell2023apentasaccharidefor pages 2-3, kell2023apentasaccharidefor pages 16-16) | Especially important for type II trials because slow disease progression makes conventional clinical endpoints difficult; useful adjunct to CSF GM1, β-gal, MRI/MRS |
| Experimental / preclinical | Pharmacological chaperones (e.g., iminosugar compound 12) | Stabilize residual mutant β-galactosidase to enhance lysosomal trafficking/activity | Oral/small-molecule concept; preclinical | No GM1 type II clinical trial in retrieved context | Preclinical | Enzyme activity rescue in variant-specific cells | 2022 medicinal chemistry study reported ~40% β-gal activity enhancement in patient leukocytes with p.Ile51Asn/p.Arg201His using a candidate chaperone (2022, DOI:10.3390/molecules27134008) (srivastava2026novelgalactosidasebeta1variant pages 1-3) | Precision-medicine potential for residual-activity type II genotypes, but far from clinical implementation |
| Experimental / preclinical | Base editing / gene editing of GLB1 | Correct pathogenic GLB1 SNVs in patient-derived cells | Ex vivo / in vitro | No registered human interventional GM1 type II gene-editing trial retrieved | Preclinical | Editing efficiency, β-gal restoration, off-target profile | 2023 CRISPR Journal study reported that 41% of pathogenic GLB1 SNVs may be targetable by adenine base editors and demonstrated restoration of therapeutic β-gal levels in patient fibroblasts (2023, DOI:10.1089/crispr.2022.0045) (shaimardanova2023genetherapyof pages 12-13) | Important emerging direction but not yet clinically implemented for type II GM1 |
| Experimental / future | Prenatal AAV9-GLB1 gene transfer | Very-early gene replacement before advanced neurodegeneration | Prenatal IV fetal administration | NCT07479953 | Phase 1; NOT_YET_RECRUITING | Feasibility, safety, prenatal delivery outcomes | Trial registry indicates fetal enrollment at 28-35+6 weeks with genetically confirmed type I or II GM1 and extensive prenatal genetic confirmation requirements (ClinicalTrials.gov) (NCT07479953 chunk 1) | Highly experimental; conceptually important because earlier intervention is widely viewed as critical in neuronopathic lysosomal disease |
Table: This table summarizes the main current and emerging therapeutic approaches and clinical trials relevant to GM1 gangliosidosis type II, with emphasis on 2023-2024 developments. It highlights active gene-therapy programs, SRT efforts, and the H3N2b biomarker that is increasingly important for monitoring treatment response in slow-progressing disease.
Key active programs for type II include:
* Intravenous AAV9-GLB1 gene transfer (NCT03952637; recruiting): primary objective safety; exploratory endpoints include biomarkers, neurodevelopment/motor outcomes, and MRI measures (ClinicalTrials.gov record 2019; https://clinicaltrials.gov/study/NCT03952637) (NCT03952637 chunk 1).
Cisterna magna AAVhu68-GLB1 (PBGM01)* (NCT04713475; active not recruiting): endpoints include safety, Bayley developmental milestones, Vineland, β-gal and substrate levels (including GM1), NfL, MRI, QoL, and ventilator-free survival vs natural history (ClinicalTrials.gov record 2021; https://clinicaltrials.gov/study/NCT04713475) (NCT04713475 chunk 1).
Biomarker-enabled implementation: H3N2b is increasingly used as a pharmacodynamic marker in translational pipelines, aligning with the need for sensitive endpoints in slowly progressive type II disease (kell2023apentasaccharidefor pages 1-2).
12.3 MAXO term suggestions (examples)
- Gene therapy (in vivo AAV-mediated gene transfer)
- Substrate reduction therapy
- Symptomatic treatment; physical therapy; occupational therapy; speech therapy; anticonvulsant therapy
(These are suggested for knowledge base mapping; specific MAXO IDs not retrieved in evidence.)
13. Prevention
Primary prevention is feasible only through reproductive/genetic strategies:
* Carrier testing and genetic counseling in families and high-risk populations (rha2021gm1gangliosidosismechanisms pages 1-2).
Prenatal diagnosis (e.g., amniocentesis or CVS) is described as an option when parents are carriers (rha2021gm1gangliosidosismechanisms pages 2-3).
Preimplantation genetic testing (PGT-M) can prevent affected births in carrier couples. A 2024 case report describes ART with PGT-M using direct mutation testing plus STR haplotyping around GLB1, followed by prenatal confirmation and birth of an unaffected infant (Feb 2024; https://doi.org/10.3389/fgene.2024.1344051) (zagaynova2024casereportpreimplantation pages 1-2).
Secondary prevention (early detection) is conceptually enabled by newborn screening approaches that combine enzyme and biomarker assays on dried blood spots, but the retrieved evidence does not document routine population-scale NBS implementation for GM1 type II (kell2023apentasaccharidefor pages 16-16, rha2021gm1gangliosidosismechanisms pages 2-3).
14. Other Species / Natural Disease
Naturally occurring GM1-like disease has been described in multiple species including cats, dogs, cattle, sheep, and others, and these have been used for biomarker and therapy studies (eikelberg2020axonopathyandreduction pages 1-3, rha2021gm1gangliosidosismechanisms pages 2-3). Large-animal models are particularly important for CNS-directed delivery and biomarker development relevant to human trials (rha2021gm1gangliosidosismechanisms pages 21-21).
15. Model Organisms
Commonly used models span mouse, large-animal, and human in vitro systems:
* Mouse (Glb1−/− knockouts and engineered models): develop CNS lesions and later motor deficits (ataxia/tremor), with neuronal storage pathology and gliosis; used for mechanistic and gene-therapy testing (eikelberg2020axonopathyandreduction pages 1-3, rha2021gm1gangliosidosismechanisms pages 2-3).
Feline models (naturally occurring): closely recapitulate late-infantile/juvenile neurologic disease and have supported AAV gene-therapy studies and biomarker development (rha2021gm1gangliosidosismechanisms pages 2-3, foster2024therapeuticdevelopmentsfor pages 4-5).
Human GLB1-knockout cerebral organoids: derived from isogenic GLB1 knockout iPSCs with <5% β-gal activity; show progressive GM1 accumulation and contain CNS-relevant structures and cell types, enabling human-tissue testing of AAV9-GLB1 (Dec 2019; https://doi.org/10.1016/j.ymgmr.2019.100513) (latour2019humanglb1knockout pages 1-2).
Visual evidence (treatment landscape)
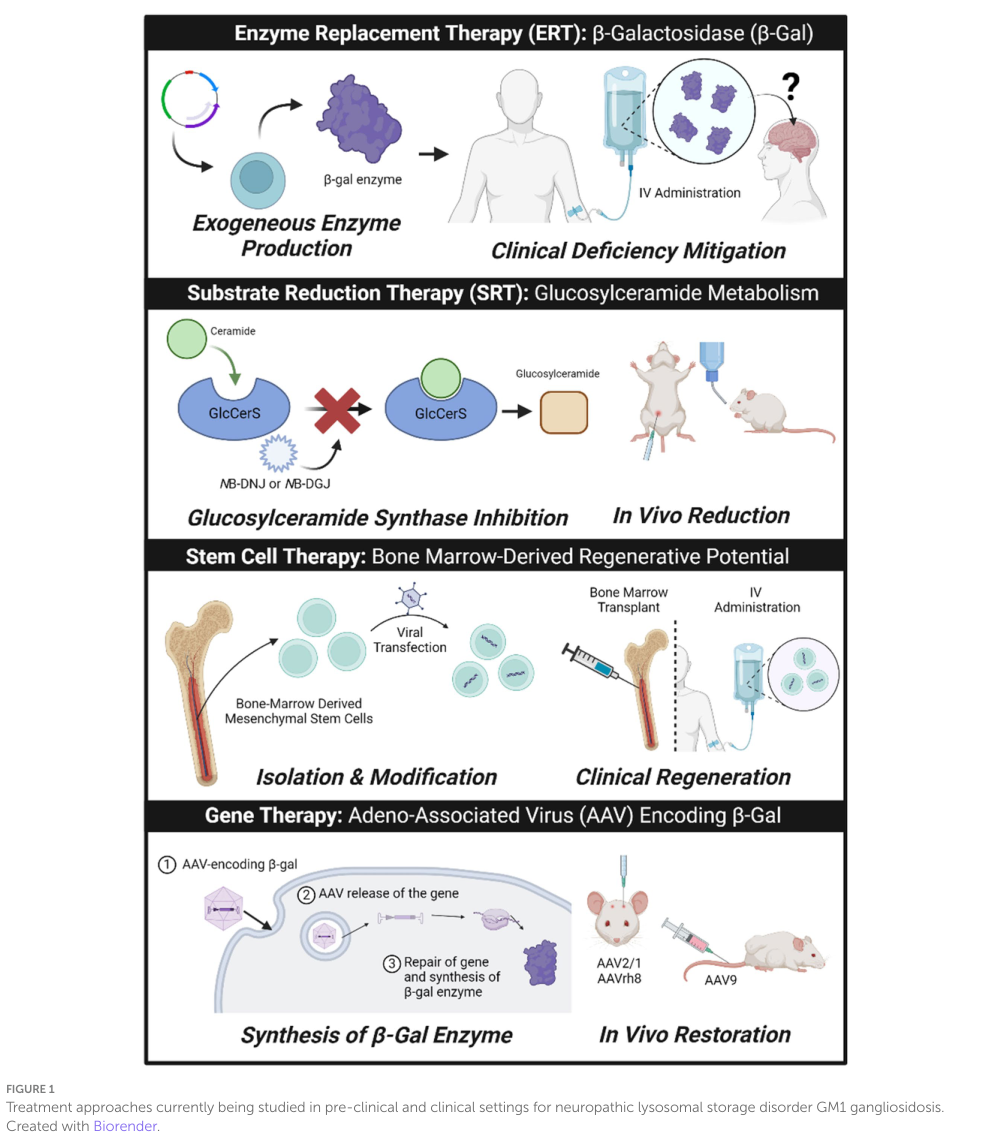
A 2024 Frontiers in Neuroscience review includes a schematic of major therapeutic modalities (ERT, SRT, stem cell therapy, gene therapy) and a table summarizing human clinical trials for GM1 gangliosidosis; these provide a concise visual overview for knowledge base curation and trial tracking (foster2024therapeuticdevelopmentsfor media 704a7e6f, foster2024therapeuticdevelopmentsfor media 3fd6f6a9).
Notes on evidence gaps and reliability
- Several requested identifier types (Orphanet/ICD-11/MeSH tree numbers) were not directly retrievable from the available full-text evidence in this run; they should be added via direct database queries.
- Some emerging clinical gene-therapy outcomes were retrieved as a 2025 preprint (medRxiv) rather than peer-reviewed 2023–2024 publications; therefore, 2023–2024 treatment “latest research” is best represented here by trial activity plus biomarker development (H3N2b) and mechanistic multi-omics work (snRNA-seq) (lewis2025aav9genetherapy pages 1-4, kell2023apentasaccharidefor pages 1-2, liu2024insightsintothe pages 1-2).
References
-
(OpenTargets Search: GM1 gangliosidosis): Open Targets Query (GM1 gangliosidosis, 9 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(zagaynova2024casereportpreimplantation pages 1-2): Valeria A. Zagaynova, Yulia A. Nasykhova, Ziravard N. Tonyan, Maria M. Danilova, Natalya M. Dvoynova, Tatyana E. Lazareva, Tatyana E. Ivashchenko, Elena S. Shabanova, Inna O. Krikheli, Elena A. Lesik, Olesya N. Bespalova, Igor Yu. Kogan, and Andrey S. Glotov. Case report: preimplantation genetic testing for infantile gm1 gangliosidosis. Frontiers in Genetics, Feb 2024. URL: https://doi.org/10.3389/fgene.2024.1344051, doi:10.3389/fgene.2024.1344051. This article has 0 citations and is from a peer-reviewed journal.
-
(ferreira2020theskeletalphenotype pages 1-6): Carlos R. Ferreira, Debra S. Regier, Robin Yoon, Kristen S. Pan, Jean M. Johnston, Sandra Yang, Jürgen W. Spranger, and Cynthia J. Tifft. The skeletal phenotype of intermediate gm1 gangliosidosis: clinical, radiographic and densitometric features, and implications for clinical monitoring and intervention. Feb 2020. URL: https://doi.org/10.1016/j.bone.2019.115142, doi:10.1016/j.bone.2019.115142. This article has 21 citations and is from a domain leading peer-reviewed journal.
-
(NCT04041102 chunk 2): Natural History Study of Infantile and Juvenile GM1 Gangliosidosis (GM1) Patients. University of Pennsylvania. 2020. ClinicalTrials.gov Identifier: NCT04041102
-
(NCT04041102 chunk 3): Natural History Study of Infantile and Juvenile GM1 Gangliosidosis (GM1) Patients. University of Pennsylvania. 2020. ClinicalTrials.gov Identifier: NCT04041102
-
(rha2021gm1gangliosidosismechanisms pages 1-2): Allisandra K. Rha, Anne S. Maguire, and Douglas R. Martin. Gm1 gangliosidosis: mechanisms and management. The Application of Clinical Genetics, 14:209-233, Apr 2021. URL: https://doi.org/10.2147/tacg.s206076, doi:10.2147/tacg.s206076. This article has 96 citations.
-
(rha2021gm1gangliosidosismechanisms pages 2-3): Allisandra K. Rha, Anne S. Maguire, and Douglas R. Martin. Gm1 gangliosidosis: mechanisms and management. The Application of Clinical Genetics, 14:209-233, Apr 2021. URL: https://doi.org/10.2147/tacg.s206076, doi:10.2147/tacg.s206076. This article has 96 citations.
-
(d’souza2024gm1gangliosidosistype pages 1-3): Precilla D’Souza, Cristan Farmer, Jean M. Johnston, Sangwoo T. Han, David Adams, Adam L. Hartman, Wadih Zein, Laryssa A. Huryn, Beth Solomon, Kelly King, Christopher P. Jordan, Jennifer G. Myles, Elena-Raluca Nicoli, C. Rothermel, Yoliann Mojica Algarin, Reyna L Huang, Rachel Quimby, Mosufa Zainab, Sarah Bowden, Anna Crowell, A. Buckley, Carmen Brewer, Debra S Regier, Brian P. Brooks, M. Acosta, Eva H Baker, Gilbert Vezina, Audrey Thurm, and C. Tifft. Gm1 gangliosidosis type ii: results of a 10-year prospective study. Genetics in medicine : official journal of the American College of Medical Genetics, 26:101144-101144, Apr 2024. URL: https://doi.org/10.1016/j.gim.2024.101144, doi:10.1016/j.gim.2024.101144. This article has 23 citations.
-
(karimzadeh2017casereportsof pages 1-2): Parvaneh Karimzadeh, Samaneh Naderi, Farzaneh Modarresi, Hassan Dastsooz, Hamid Nemati, Tayebeh Farokhashtiani, Bibi Shahin Shamsian, Soroor Inaloo, and Mohammad Ali Faghihi. Case reports of juvenile gm1 gangliosidosisis type ii caused by mutation in glb1 gene. BMC Medical Genetics, Jul 2017. URL: https://doi.org/10.1186/s12881-017-0417-4, doi:10.1186/s12881-017-0417-4. This article has 28 citations and is from a peer-reviewed journal.
-
(arashkaps2019theclinicaland pages 1-2): Laila Arash-Kaps, Katalin Komlosi, Marlene Seegräber, Stefan Diederich, Eduard Paschke, Yasmina Amraoui, Skadi Beblo, Andrea Dieckmann, Martin Smitka, and Julia B. Hennermann. The clinical and molecular spectrum of gm1 gangliosidosis. The Journal of Pediatrics, 215:152-157.e3, Dec 2019. URL: https://doi.org/10.1016/j.jpeds.2019.08.016, doi:10.1016/j.jpeds.2019.08.016. This article has 49 citations.
-
(lang2020thenaturalhistory pages 1-2): Frederick M. Lang, Paul Korner, Mark Harnett, Ajith Karunakara, and Cynthia J. Tifft. The natural history of type 1 infantile gm1 gangliosidosis: a literature-based meta-analysis. Mar 2020. URL: https://doi.org/10.1016/j.ymgme.2019.12.012, doi:10.1016/j.ymgme.2019.12.012. This article has 54 citations and is from a peer-reviewed journal.
-
(nicoli2021gm1gangliosidosis—aminireview pages 1-2): Elena-Raluca Nicoli, Ida Annunziata, Alessandra d’Azzo, Frances M. Platt, Cynthia J. Tifft, and Karolina M. Stepien. Gm1 gangliosidosis—a mini-review. Frontiers in Genetics, Sep 2021. URL: https://doi.org/10.3389/fgene.2021.734878, doi:10.3389/fgene.2021.734878. This article has 107 citations and is from a peer-reviewed journal.
-
(lang2020thenaturalhistory pages 2-3): Frederick M. Lang, Paul Korner, Mark Harnett, Ajith Karunakara, and Cynthia J. Tifft. The natural history of type 1 infantile gm1 gangliosidosis: a literature-based meta-analysis. Mar 2020. URL: https://doi.org/10.1016/j.ymgme.2019.12.012, doi:10.1016/j.ymgme.2019.12.012. This article has 54 citations and is from a peer-reviewed journal.
-
(d’souza2024gm1gangliosidosistype pages 6-9): Precilla D’Souza, Cristan Farmer, Jean M. Johnston, Sangwoo T. Han, David Adams, Adam L. Hartman, Wadih Zein, Laryssa A. Huryn, Beth Solomon, Kelly King, Christopher P. Jordan, Jennifer G. Myles, Elena-Raluca Nicoli, C. Rothermel, Yoliann Mojica Algarin, Reyna L Huang, Rachel Quimby, Mosufa Zainab, Sarah Bowden, Anna Crowell, A. Buckley, Carmen Brewer, Debra S Regier, Brian P. Brooks, M. Acosta, Eva H Baker, Gilbert Vezina, Audrey Thurm, and C. Tifft. Gm1 gangliosidosis type ii: results of a 10-year prospective study. Genetics in medicine : official journal of the American College of Medical Genetics, 26:101144-101144, Apr 2024. URL: https://doi.org/10.1016/j.gim.2024.101144, doi:10.1016/j.gim.2024.101144. This article has 23 citations.
-
(d’souza2024gm1gangliosidosistype pages 9-12): Precilla D’Souza, Cristan Farmer, Jean M. Johnston, Sangwoo T. Han, David Adams, Adam L. Hartman, Wadih Zein, Laryssa A. Huryn, Beth Solomon, Kelly King, Christopher P. Jordan, Jennifer G. Myles, Elena-Raluca Nicoli, C. Rothermel, Yoliann Mojica Algarin, Reyna L Huang, Rachel Quimby, Mosufa Zainab, Sarah Bowden, Anna Crowell, A. Buckley, Carmen Brewer, Debra S Regier, Brian P. Brooks, M. Acosta, Eva H Baker, Gilbert Vezina, Audrey Thurm, and C. Tifft. Gm1 gangliosidosis type ii: results of a 10-year prospective study. Genetics in medicine : official journal of the American College of Medical Genetics, 26:101144-101144, Apr 2024. URL: https://doi.org/10.1016/j.gim.2024.101144, doi:10.1016/j.gim.2024.101144. This article has 23 citations.
-
(d’souza2024gm1gangliosidosistype pages 3-6): Precilla D’Souza, Cristan Farmer, Jean M. Johnston, Sangwoo T. Han, David Adams, Adam L. Hartman, Wadih Zein, Laryssa A. Huryn, Beth Solomon, Kelly King, Christopher P. Jordan, Jennifer G. Myles, Elena-Raluca Nicoli, C. Rothermel, Yoliann Mojica Algarin, Reyna L Huang, Rachel Quimby, Mosufa Zainab, Sarah Bowden, Anna Crowell, A. Buckley, Carmen Brewer, Debra S Regier, Brian P. Brooks, M. Acosta, Eva H Baker, Gilbert Vezina, Audrey Thurm, and C. Tifft. Gm1 gangliosidosis type ii: results of a 10-year prospective study. Genetics in medicine : official journal of the American College of Medical Genetics, 26:101144-101144, Apr 2024. URL: https://doi.org/10.1016/j.gim.2024.101144, doi:10.1016/j.gim.2024.101144. This article has 23 citations.
-
(d’souza2024gm1gangliosidosistype pages 12-15): Precilla D’Souza, Cristan Farmer, Jean M. Johnston, Sangwoo T. Han, David Adams, Adam L. Hartman, Wadih Zein, Laryssa A. Huryn, Beth Solomon, Kelly King, Christopher P. Jordan, Jennifer G. Myles, Elena-Raluca Nicoli, C. Rothermel, Yoliann Mojica Algarin, Reyna L Huang, Rachel Quimby, Mosufa Zainab, Sarah Bowden, Anna Crowell, A. Buckley, Carmen Brewer, Debra S Regier, Brian P. Brooks, M. Acosta, Eva H Baker, Gilbert Vezina, Audrey Thurm, and C. Tifft. Gm1 gangliosidosis type ii: results of a 10-year prospective study. Genetics in medicine : official journal of the American College of Medical Genetics, 26:101144-101144, Apr 2024. URL: https://doi.org/10.1016/j.gim.2024.101144, doi:10.1016/j.gim.2024.101144. This article has 23 citations.
-
(d’souza2024gm1gangliosidosistype pages 37-41): Precilla D’Souza, Cristan Farmer, Jean M. Johnston, Sangwoo T. Han, David Adams, Adam L. Hartman, Wadih Zein, Laryssa A. Huryn, Beth Solomon, Kelly King, Christopher P. Jordan, Jennifer G. Myles, Elena-Raluca Nicoli, C. Rothermel, Yoliann Mojica Algarin, Reyna L Huang, Rachel Quimby, Mosufa Zainab, Sarah Bowden, Anna Crowell, A. Buckley, Carmen Brewer, Debra S Regier, Brian P. Brooks, M. Acosta, Eva H Baker, Gilbert Vezina, Audrey Thurm, and C. Tifft. Gm1 gangliosidosis type ii: results of a 10-year prospective study. Genetics in medicine : official journal of the American College of Medical Genetics, 26:101144-101144, Apr 2024. URL: https://doi.org/10.1016/j.gim.2024.101144, doi:10.1016/j.gim.2024.101144. This article has 23 citations.
-
(rodriguez2025burdenofcaregiving pages 1-2): Maria Belen Rodriguez, Ruth Pulikottil-Jacob, Karli Heuer, Nancy Gabriela Perez, Christine Waggoner, Diana Jussila, Chad Gwaltney, Robert Krupnick, and Daisy Ng-Mak. Burden of caregiving of individuals with gm1 and gm2 gangliosidoses in the united states: a qualitative study. Orphanet Journal of Rare Diseases, Nov 2025. URL: https://doi.org/10.1186/s13023-025-04030-6, doi:10.1186/s13023-025-04030-6. This article has 0 citations and is from a peer-reviewed journal.
-
(nicoli2021gm1gangliosidosis—aminireview pages 4-5): Elena-Raluca Nicoli, Ida Annunziata, Alessandra d’Azzo, Frances M. Platt, Cynthia J. Tifft, and Karolina M. Stepien. Gm1 gangliosidosis—a mini-review. Frontiers in Genetics, Sep 2021. URL: https://doi.org/10.3389/fgene.2021.734878, doi:10.3389/fgene.2021.734878. This article has 107 citations and is from a peer-reviewed journal.
-
(rha2021gm1gangliosidosismechanisms pages 3-5): Allisandra K. Rha, Anne S. Maguire, and Douglas R. Martin. Gm1 gangliosidosis: mechanisms and management. The Application of Clinical Genetics, 14:209-233, Apr 2021. URL: https://doi.org/10.2147/tacg.s206076, doi:10.2147/tacg.s206076. This article has 96 citations.
-
(rha2021gm1gangliosidosismechanisms pages 5-6): Allisandra K. Rha, Anne S. Maguire, and Douglas R. Martin. Gm1 gangliosidosis: mechanisms and management. The Application of Clinical Genetics, 14:209-233, Apr 2021. URL: https://doi.org/10.2147/tacg.s206076, doi:10.2147/tacg.s206076. This article has 96 citations.
-
(eikelberg2020axonopathyandreduction pages 1-3): Deborah Eikelberg, Annika Lehmbecker, Graham Brogden, Witchaya Tongtako, Kerstin Hahn, Andre Habierski, Julia B. Hennermann, Hassan Y. Naim, Felix Felmy, Wolfgang Baumgärtner, and Ingo Gerhauser. Axonopathy and reduction of membrane resistance: key features in a new murine model of human gm1-gangliosidosis. Journal of Clinical Medicine, 9:1004, Apr 2020. URL: https://doi.org/10.3390/jcm9041004, doi:10.3390/jcm9041004. This article has 24 citations.
-
(liu2024insightsintothe pages 1-2): Sichi Liu, Ting Xie, and Yonglan Huang. Insights into the pathobiology of gm1 gangliosidosis from single-nucleus transcriptomic analysis of cns cells in a mouse model. International Journal of Molecular Sciences, 25:9712, Sep 2024. URL: https://doi.org/10.3390/ijms25179712, doi:10.3390/ijms25179712. This article has 4 citations.
-
(liu2024insightsintothe pages 13-15): Sichi Liu, Ting Xie, and Yonglan Huang. Insights into the pathobiology of gm1 gangliosidosis from single-nucleus transcriptomic analysis of cns cells in a mouse model. International Journal of Molecular Sciences, 25:9712, Sep 2024. URL: https://doi.org/10.3390/ijms25179712, doi:10.3390/ijms25179712. This article has 4 citations.
-
(kannebley2015clinicalfindingsand pages 1-2): João Stein Kannebley, Laura Silveira-Moriyama, Laís Orrico Donnabella Bastos, and Carlos Eduardo Steiner. Clinical findings and natural history in ten unrelated families with juvenile and adult gm1 gangliosidosis. JIMD reports, 24:115-22, Jan 2015. URL: https://doi.org/10.1007/8904_2015_451, doi:10.1007/8904_2015_451. This article has 30 citations and is from a peer-reviewed journal.
-
(kell2023apentasaccharidefor pages 1-2): Pamela Kell, Rohini Sidhu, Mingxing Qian, Sonali Mishra, Elena-Raluca Nicoli, Precilla D'Souza, Cynthia J. Tifft, Amanda L. Gross, Heather L. Gray-Edwards, Douglas R. Martin, Miguel Sena- Esteves, Dennis J. Dietzen, Manmilan Singh, Jingqin Luo, Jean E. Schaffer, Daniel S. Ory, and Xuntian Jiang. A pentasaccharide for monitoring pharmacodynamic response to gene therapy in gm1 gangliosidosis. Jun 2023. URL: https://doi.org/10.1016/j.ebiom.2023.104627, doi:10.1016/j.ebiom.2023.104627. This article has 19 citations and is from a peer-reviewed journal.
-
(kell2023apentasaccharidefor pages 2-3): Pamela Kell, Rohini Sidhu, Mingxing Qian, Sonali Mishra, Elena-Raluca Nicoli, Precilla D'Souza, Cynthia J. Tifft, Amanda L. Gross, Heather L. Gray-Edwards, Douglas R. Martin, Miguel Sena- Esteves, Dennis J. Dietzen, Manmilan Singh, Jingqin Luo, Jean E. Schaffer, Daniel S. Ory, and Xuntian Jiang. A pentasaccharide for monitoring pharmacodynamic response to gene therapy in gm1 gangliosidosis. Jun 2023. URL: https://doi.org/10.1016/j.ebiom.2023.104627, doi:10.1016/j.ebiom.2023.104627. This article has 19 citations and is from a peer-reviewed journal.
-
(foster2024therapeuticdevelopmentsfor pages 5-6): Dorian Foster, Lucian Williams, Noah Arnold, and Jessica Larsen. Therapeutic developments for neurodegenerative gm1 gangliosidosis. Frontiers in Neuroscience, Apr 2024. URL: https://doi.org/10.3389/fnins.2024.1392683, doi:10.3389/fnins.2024.1392683. This article has 21 citations and is from a peer-reviewed journal.
-
(foster2024therapeuticdevelopmentsfor media 704a7e6f): Dorian Foster, Lucian Williams, Noah Arnold, and Jessica Larsen. Therapeutic developments for neurodegenerative gm1 gangliosidosis. Frontiers in Neuroscience, Apr 2024. URL: https://doi.org/10.3389/fnins.2024.1392683, doi:10.3389/fnins.2024.1392683. This article has 21 citations and is from a peer-reviewed journal.
-
(foster2024therapeuticdevelopmentsfor media 3fd6f6a9): Dorian Foster, Lucian Williams, Noah Arnold, and Jessica Larsen. Therapeutic developments for neurodegenerative gm1 gangliosidosis. Frontiers in Neuroscience, Apr 2024. URL: https://doi.org/10.3389/fnins.2024.1392683, doi:10.3389/fnins.2024.1392683. This article has 21 citations and is from a peer-reviewed journal.
-
(foster2024therapeuticdevelopmentsfor pages 4-5): Dorian Foster, Lucian Williams, Noah Arnold, and Jessica Larsen. Therapeutic developments for neurodegenerative gm1 gangliosidosis. Frontiers in Neuroscience, Apr 2024. URL: https://doi.org/10.3389/fnins.2024.1392683, doi:10.3389/fnins.2024.1392683. This article has 21 citations and is from a peer-reviewed journal.
-
(NCT03952637 chunk 1): A Phase 1/2 Study of Intravenous Gene Transfer With an AAV9 Vector Expressing Human Beta-galactosidase in Type I and Type II GM1 Gangliosidosis. National Human Genome Research Institute (NHGRI). 2019. ClinicalTrials.gov Identifier: NCT03952637
-
(lewis2025aav9genetherapy pages 1-4): Connor J. Lewis, Precilla D’Souza, Jean M. Johnston, Maria T. Acosta, Cristan Farmer, Eva H. Baker, Anna Crowell, Yoliann Mojica, Sumaiya Rahman, Lisa Joseph, Adam Hartman, Gilbert Vézina, Zenaide Quezado, Muhammad H. Yousef, Amelia Luckett, Zeynep Vardar, Mohammad Salman Shazeeb, Manuela Corti, Meghan Blackwood, Kirsten Coleman, Audrey Thurm, Erika De Boever, William A. Gahl, Barry J. Byrne, Terence R. Flotte, Xuntian Jiang, Amanda L. Gross, Allison M. Keeler, Heather Gray-Edwards, Douglas R. Martin, Miguel Sena-Esteves, and Cynthia J. Tifft. Aav9 gene therapy in gm1 gangliosidosis type ii: a phase 1/2 trial. medRxiv, Jul 2025. URL: https://doi.org/10.1101/2025.07.28.25332074, doi:10.1101/2025.07.28.25332074. This article has 3 citations.
-
(NCT04713475 chunk 1): Study of Safety, Tolerability and Efficacy of PBGM01 in Pediatric Participants With GM1 Gangliosidosis. Gemma Biotherapeutics. 2021. ClinicalTrials.gov Identifier: NCT04713475
-
(shaimardanova2023genetherapyof pages 4-6): Alisa A. Shaimardanova, Valeriya V. Solovyeva, Shaza S. Issa, and Albert A. Rizvanov. Gene therapy of sphingolipid metabolic disorders. International Journal of Molecular Sciences, 24:3627, Feb 2023. URL: https://doi.org/10.3390/ijms24043627, doi:10.3390/ijms24043627. This article has 34 citations.
-
(shaimardanova2023genetherapyof pages 12-13): Alisa A. Shaimardanova, Valeriya V. Solovyeva, Shaza S. Issa, and Albert A. Rizvanov. Gene therapy of sphingolipid metabolic disorders. International Journal of Molecular Sciences, 24:3627, Feb 2023. URL: https://doi.org/10.3390/ijms24043627, doi:10.3390/ijms24043627. This article has 34 citations.
-
(kell2023apentasaccharidefor pages 16-16): Pamela Kell, Rohini Sidhu, Mingxing Qian, Sonali Mishra, Elena-Raluca Nicoli, Precilla D'Souza, Cynthia J. Tifft, Amanda L. Gross, Heather L. Gray-Edwards, Douglas R. Martin, Miguel Sena- Esteves, Dennis J. Dietzen, Manmilan Singh, Jingqin Luo, Jean E. Schaffer, Daniel S. Ory, and Xuntian Jiang. A pentasaccharide for monitoring pharmacodynamic response to gene therapy in gm1 gangliosidosis. Jun 2023. URL: https://doi.org/10.1016/j.ebiom.2023.104627, doi:10.1016/j.ebiom.2023.104627. This article has 19 citations and is from a peer-reviewed journal.
-
(srivastava2026novelgalactosidasebeta1variant pages 1-3): Preeti Srivastava, Abhishek Kumar, Shikhar Deep Jain, Ratan Kumar, Shikha Swaroop, and Tapas Sarangi. Novel galactosidase-beta-1 variant in infantile gm1 gangliosidosis: a case report. Cureus, 18 1:e102121, Jan 2026. URL: https://doi.org/10.7759/cureus.102121, doi:10.7759/cureus.102121. This article has 0 citations.
-
(NCT07479953 chunk 1): Tippi Mackenzie. Prenatal Intravenous Gene Transfer With an AAV9 Vector Expressing Human Beta-galactosidase in Type I and Type II GM1 Gangliosidosis Clinical Trial. Tippi Mackenzie. 2026. ClinicalTrials.gov Identifier: NCT07479953
-
(rha2021gm1gangliosidosismechanisms pages 21-21): Allisandra K. Rha, Anne S. Maguire, and Douglas R. Martin. Gm1 gangliosidosis: mechanisms and management. The Application of Clinical Genetics, 14:209-233, Apr 2021. URL: https://doi.org/10.2147/tacg.s206076, doi:10.2147/tacg.s206076. This article has 96 citations.
-
(latour2019humanglb1knockout pages 1-2): Yvonne L. Latour, Robin Yoon, Sarah E. Thomas, Christina Grant, Cuiling Li, Miguel Sena-Esteves, Maria L. Allende, Richard L. Proia, and Cynthia J. Tifft. Human glb1 knockout cerebral organoids: a model system for testing aav9-mediated glb1 gene therapy for reducing gm1 ganglioside storage in gm1 gangliosidosis. Molecular Genetics and Metabolism Reports, 21:100513, Dec 2019. URL: https://doi.org/10.1016/j.ymgmr.2019.100513, doi:10.1016/j.ymgmr.2019.100513. This article has 64 citations.