Tay–Sachs Disease, AB Variant (GM2 Activator Deficiency): Comprehensive Disease Characteristics Report
Executive summary
Tay–Sachs disease AB variant (also called GM2 gangliosidosis AB variant) is an ultra-rare, autosomal-recessive lysosomal storage disorder caused by biallelic pathogenic variants in GM2A, which encodes the GM2 activator protein (GM2AP). GM2AP is required for β-hexosaminidase A (HexA) to hydrolyze GM2 ganglioside; thus, AB-variant patients can have a Tay–Sachs-like phenotype despite normal HexA/HexB enzyme activities on routine assays. Recent (2023) preclinical work demonstrates proof-of-concept AAV9-GM2A gene replacement and establishes a more severe, translationally relevant Gm2a−/−Neu3−/− mouse model by removing a mouse-specific compensatory GM2 catabolic pathway. (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 1-3, hall2017gm2activatordeficiency pages 1-3, deschenes2023characterizationofa pages 8-10, vyas2023efficacyofadenoassociated pages 1-2)
Key resource field summary (for knowledge base curation)
Table (click to expand)
| Knowledge-base field | Summary |
|---|---|
| Disease definition / synonyms / identifiers | Ultra-rare autosomal-recessive GM2 gangliosidosis caused by deficiency of the GM2 activator protein (GM2AP), clinically often indistinguishable from Tay-Sachs disease. Common names: Tay-Sachs disease AB variant, GM2 gangliosidosis AB variant, GM2 activator deficiency, GM2 activator protein deficiency. OMIM disease entry reported as 272750; GM2A gene OMIM *613109; MONDO Tay-Sachs disease AB variant = MONDO_0010099; broader GM2 gangliosidosis = MONDO_0017720. Fewer than 30 cases were reported in the literature by 2022; older reviews counted 9-10 molecularly confirmed cases worldwide (sheth2016gm2gangliosidosisab pages 1-3, ganne2022gm2gangliosidosisab pages 1-5, hall2017gm2activatordeficiency pages 1-3, OpenTargets Search: GM2 gangliosidosis,Tay-Sachs disease-GM2A,HEXA,HEXB). |
| Causal gene and inheritance | Caused by biallelic GM2A variants (gene encodes ganglioside GM2 activator, a non-enzymatic lysosomal cofactor required to present GM2 to Hex A for hydrolysis). Inheritance is autosomal recessive. Mechanistically distinct from classic Tay-Sachs (HEXA) and Sandhoff (HEXB) despite overlapping phenotype (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 3-4, sheth2016gm2gangliosidosisab pages 1-3, gualdronfrias2019taysachsdisease pages 3-4). |
| Hallmark diagnostic pattern | Key clue: GM2 gangliosidosis phenotype with normal Hex A and Hex B enzyme activity in leukocytes/blood. Recommended workup includes GM2A sequencing, with copy-number analysis because exon-level deletions can be missed by routine sequencing; supportive/confirmatory tools include plasma GM2 LC-MS/MS, fibroblast GM2 studies, EM/IF, and RNA/cDNA analyses in unresolved cases (hall2017gm2activatordeficiency pages 1-3, ganne2022gm2gangliosidosisab pages 5-8, ganne2022gm2gangliosidosisab pages 8-14, sheth2016gm2gangliosidosisab pages 3-4). |
| Main phenotypes by onset: infantile | Usually onset in the first year (often ~3-12 months). Features include developmental delay/regression, hypotonia, hyperacusis/exaggerated startle, seizures, poor visual attention/nystagmus, bilateral cherry-red maculae, and progressive neurodegeneration. MRI may show putaminal hyperintensity, thalamic hypointensity, delayed/unmyelinated white matter. Reported outcome: severe disability and early death around 4-5 years, sometimes earlier from respiratory complications (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 3-4, sheth2016gm2gangliosidosisab pages 1-3, deschenes2023characterizationofa pages 1-2, noites2025gm2gangliosidosisab pages 2-3). |
| Main phenotypes by onset: juvenile | Reported onset roughly 2-10 years with ataxia, psychomotor deterioration/regression, spasticity, seizures and progressive neurologic decline; historically death before adulthood in described juvenile GM2 gangliosidosis summaries. AB-variant-specific juvenile cases are rare and sparsely characterized (ganne2022gm2gangliosidosisab pages 1-5). |
| Main phenotypes by onset: late / adult | First late-onset AB-variant case reported in 2022. Phenotype may include gait disorder beginning around age 10, lower motor neuron involvement, spinocerebellar ataxia, muscular atrophy, mild cognitive/executive deficits, psychiatric features, and subtle cerebellar vermis atrophy with normal Hex A/B activity. Prognosis appears better than infantile disease but data are very limited (ganne2022gm2gangliosidosisab pages 5-8, ganne2022gm2gangliosidosisab pages 1-5). |
| Representative pathogenic variants reported | Reported variants include c.472G>T (p.Glu158Ter / p.E158X), homozygous exon 2 deletion, c.79A>T (p.Lys27Ter), c.415C>T (p.Pro139Ser; evaluated as likely hypomorphic / VUS in trans with truncating allele), and earlier literature variants such as c.160G>T (p.Glu54Ter), c.164C>T (p.Pro55Leu), c.412T>C, c.506G>C (p.Arg169Pro), c.522T>G (p.Leu174Arg) plus frameshift/deletion alleles. Population data are sparse; some variants were absent from gnomAD/ExAC/TOPMed and one missense allele had a single prior Latin American report (sheth2016gm2gangliosidosisab pages 3-4, hall2017gm2activatordeficiency pages 1-3, ganne2022gm2gangliosidosisab pages 5-8, sheth2016gm2gangliosidosisab pages 1-3, ganne2022gm2gangliosidosisab pages 14-17). |
| Epidemiology / population notes | Rarest GM2 gangliosidosis subtype. Literature-based counts: 9 cases / 7 mutations in older review, 10 molecularly proven cases by 2016 review table, and <30 cases by 2022 review. Reported ancestries/populations include Indian, Saudi, Spanish, US Black, Laotian/Hmong, and later Portuguese; consanguinity is reported in some infantile cases but no robust carrier-frequency estimate exists for AB variant (sheth2016gm2gangliosidosisab pages 1-3, ganne2022gm2gangliosidosisab pages 1-5, gowda2022clinicalandlaboratory pages 1-2, noites2025gm2gangliosidosisab pages 1-2). |
| 2023-2024 translational development: AAV9-GM2A preclinical | 2023 AB-variant-specific gene therapy proof-of-concept: systemic ssAAV9-GM2A in Gm2a-/- mice at 1 × 10^14 vg/kg (reported also as 1 × 10^11 vg/mouse) given at postnatal day 1 or 6 weeks produced long-term vector persistence in brain/liver, reduced CNS GM2 accumulation, and improved rotarod performance especially in 6-week-treated animals; long-term biochemical correction was partial, suggesting need for higher dose/optimization. Separate 2023 intrathecal scAAV9.hGM2A study showed dose-responsive biochemical correction with 0.5, 1.0, 2.0 × 10^11 vg/mouse, broad CNS distribution, persistence up to 104 weeks, and no severe adverse events (deschenes2023biochemicalcorrectionof pages 1-2, deschenes2023biochemicalcorrectionof pages 2-4, vyas2023efficacyofadenoassociated pages 7-9, deschenes2023biochemicalcorrectionof pages 14-15, vyas2023efficacyofadenoassociated pages 11-13, vyas2023efficacyofadenoassociated pages 1-2, vyas2023efficacyofadenoassociated pages 2-4, vyas2023efficacyofadenoassociated pages 9-10, vyas2023efficacyofadenoassociated media 2eb6c65b). |
| 2023-2024 translational development: severe double-KO model | 2023 Gm2a-/-Neu3-/- double knockout mouse established as a more severe and translationally relevant AB-variant model. Rationale: single Gm2a-/- mice are mild because murine NEU3 provides an alternative GM2 catabolic route. Double KO causes marked CNS GM2 accumulation (~3-4-fold vs Gm2a-/-), ataxia, reduced mobility/coordination, weight loss, vacuolization, onset of deficits by ~12-16 weeks, and shortened lifespan (~27 weeks vs ~92 weeks for Gm2a-/-), better approximating severe human disease for preclinical testing (deschenes2023characterizationofa pages 8-10, vyas2023efficacyofadenoassociated pages 9-10, deschenes2023characterizationofa pages 1-2, deschenes2023characterizationofa pages 2-3, deschenes2023characterizationofa pages 3-4). |
| Relevant clinical trials for GM2 gangliosidoses: TSHA-101 | NCT04798235; TSHA-101 bicistronic AAV9-HEXA/HEXB gene therapy; intrathecal, one-time; target infantile-onset GM2 gangliosidosis; ACTIVE_NOT_RECRUITING; start date 2021-03-12; phase 1/2; planned enrollment 3. Primary focus: safety/tolerability; secondary outcomes include survival, Hex A activity, motor/neurologic measures (NCT04798235 chunk 1). |
| Relevant clinical trials for GM2 gangliosidoses: AXO-AAV-GM2 interventional | NCT04669535; AXO-AAV-GM2 dual-vector gene therapy for Tay-Sachs/Sandhoff; delivery described as bilateral intraparenchymal thalamic plus intracisternal/intrathecal; pediatric target population; TERMINATED in trial search output; start year 2021; phase 1; enrollment reported as 9 in trial search output (review chapter listed 18 planned). No posted efficacy results in the retrieved context (NCT04669535 chunk 2). |
| Relevant clinical trials for GM2 gangliosidoses: AXO-AAV-GM2 long-term follow-up | NCT06614569; long-term follow-up of previously treated AXO-AAV-GM2 subjects; parent intervention route bilateral intraparenchymal thalamic and intracisternal/intrathecal; ACTIVE_NOT_RECRUITING; actual start 2024-09-17; estimated enrollment 7; main purpose is delayed safety plus longitudinal neurocognitive/motor follow-up for up to 5 years (NCT06614569 chunk 1). |
| Relevant clinical trials for GM2 gangliosidoses: natural history | NCT00668187; A Natural History Study of the Gangliosidoses; observational, no intervention; includes Tay-Sachs/Sandhoff/GM1; RECRUITING; start 2010-12; estimated enrollment 52. Important for defining progression and outcome measures for future AB-variant and broader GM2 trials (NCT00668187 chunk 1). |
Table: This table compiles the core knowledge-base fields for Tay-Sachs disease AB variant (GM2A deficiency), including disease definition, genetics, phenotype, diagnostics, and 2023-2024 translational advances. It also summarizes relevant GM2 gangliosidosis clinical trials to support curation and therapeutic landscape review.
1. Disease information
1.1 What is the disease?
GM2 gangliosidosis AB variant is a GM2 gangliosidosis caused by deficiency of the non-enzymatic cofactor GM2 activator protein. Clinically, it is often described as indistinguishable from Tay–Sachs disease, because GM2 accumulates in neurons in both conditions, leading to progressive neurodegeneration. (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 1-3)
Current understanding: AB variant is best conceptualized as a third genetic cause of the Tay–Sachs/Sandhoff clinical spectrum: HEXA (Tay–Sachs), HEXB (Sandhoff), and GM2A (AB variant). (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 1-3)
1.2 Key identifiers
- OMIM (disease): 272750 (reported in a case-report review of AB variant). (sheth2016gm2gangliosidosisab pages 1-3)
- OMIM (gene GM2A): *613109 (reported in a 2022 AB-variant review). (ganne2022gm2gangliosidosisab pages 1-5)
- MONDO:
- Tay–Sachs disease AB variant: MONDO_0010099 (OpenTargets disease label). (OpenTargets Search: GM2 gangliosidosis,Tay-Sachs disease-GM2A,HEXA,HEXB)
- GM2 gangliosidosis (broader parent concept): MONDO_0017720. (OpenTargets Search: GM2 gangliosidosis,Tay-Sachs disease-GM2A,HEXA,HEXB)
Not found in retrieved sources for AB variant: Orphanet ID, ICD-10/ICD-11 codes, and MeSH unique ID were not explicitly provided in the retrieved full-text evidence. (sheth2016gm2gangliosidosisab pages 1-3, ganne2022gm2gangliosidosisab pages 1-5)
1.3 Synonyms / alternative names
- GM2 gangliosidosis AB variant (preferred in many papers) (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 1-3)
- GM2 activator deficiency / GM2 activator protein deficiency (hall2017gm2activatordeficiency pages 1-3)
- GM2AB / ABGM2 (in some recent literature) (deschenes2023characterizationofa pages 1-2, vyas2023efficacyofadenoassociated pages 1-2)
1.4 Evidence provenance (individual vs aggregated)
Most AB-variant knowledge is derived from individual case reports and small case series/reviews (reflecting extreme rarity), plus animal-model and in vitro studies for mechanistic and therapeutic development. (sheth2016gm2gangliosidosisab pages 1-3, ganne2022gm2gangliosidosisab pages 1-5, deschenes2023characterizationofa pages 8-10, vyas2023efficacyofadenoassociated pages 7-9)
2. Etiology
2.1 Disease causal factors
Primary cause: biallelic loss-of-function (or severely hypomorphic) variants in GM2A leading to GM2AP deficiency and failure of GM2 hydrolysis in lysosomes. (sheth2016gm2gangliosidosisab pages 1-3, sheth2016gm2gangliosidosisab pages 3-4, hall2017gm2activatordeficiency pages 1-3)
2.2 Risk factors
Genetic risk factors
- Autosomal-recessive inheritance: affected individuals typically have biallelic GM2A variants, with parents being heterozygous carriers in reported families. (sheth2016gm2gangliosidosisab pages 3-4, sheth2016gm2gangliosidosisab pages 1-3)
- Consanguinity: reported in some cases and cohorts (e.g., an Indian AB-variant case born to consanguineous parents; and a southern India gangliosidosis cohort with high consanguinity overall). (sheth2016gm2gangliosidosisab pages 1-3, gowda2022clinicalandlaboratory pages 1-2)
Environmental risk factors
No environmental or lifestyle risk factors were identified in the retrieved evidence; AB variant is a monogenic lysosomal disorder. (sheth2016gm2gangliosidosisab pages 1-3)
2.3 Protective factors
No protective genetic/environmental factors were identified in the retrieved evidence. In mice, an alternate GM2 catabolic pathway mediated by NEU3 partially compensates for GM2A deficiency (a species-specific modifier), but this is not established as a human protective factor. (deschenes2023characterizationofa pages 1-2, vyas2023efficacyofadenoassociated pages 9-10)
2.4 Gene–environment interactions
Not reported in the retrieved evidence. (sheth2016gm2gangliosidosisab pages 1-3)
3. Phenotypes
3.1 Core phenotype spectrum (human)
AB variant is frequently described as phenotypically similar to Tay–Sachs disease, with severe neurodegeneration in infantile forms and more heterogeneous manifestations in later-onset disease. (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 1-3)
Infantile-onset AB variant (symptoms/signs)
Commonly reported phenotypes include: * Global developmental delay/regression (HPO suggestion: HP:0001263 Global developmental delay; HP:0002376 Developmental regression) (sheth2016gm2gangliosidosisab pages 1-3) * Hypotonia (HP:0001252) (sheth2016gm2gangliosidosisab pages 1-3) * Hyperacusis / exaggerated startle (HP:0000347 Hyperacusis; HP:0002343 Startle response) (sheth2016gm2gangliosidosisab pages 1-3, noites2025gm2gangliosidosisab pages 2-3) * Seizures (HP:0001250) (ganne2022gm2gangliosidosisab pages 1-5, noites2025gm2gangliosidosisab pages 2-3) * Nystagmus / visual impairment (HP:0000639 Nystagmus; HP:0000505 Visual impairment) (sheth2016gm2gangliosidosisab pages 1-3) * Cherry-red spot of the macula (HP:0001103) (sheth2016gm2gangliosidosisab pages 1-3)
Neuroimaging features (infantile): basal ganglia and thalamic signal abnormalities and delayed myelination have been reported (e.g., putaminal hyperintensity, thalamic hypointensity, unmyelinated white matter). (sheth2016gm2gangliosidosisab pages 1-3)
Temporal course: infantile AB variant typically presents in the first year and progresses to severe disability and early death (premature death around 4–5 years is cited in review summaries; individual infantile cases may die earlier due to complications). (ganne2022gm2gangliosidosisab pages 1-5, noites2025gm2gangliosidosisab pages 2-3)
Juvenile-onset AB variant
Summary descriptions (limited AB-variant-specific case detail in retrieved evidence): * Onset: ~2–10 years (ganne2022gm2gangliosidosisab pages 1-5) * Phenotypes: ataxia, psychomotor deterioration, spasticity, seizures (HPO suggestions: HP:0001251 Ataxia; HP:0001257 Spasticity) (ganne2022gm2gangliosidosisab pages 1-5) * Outcome: progression and death before adulthood is described in GM2 gangliosidosis subtype summaries (AB-variant-specific longitudinal datasets remain sparse). (ganne2022gm2gangliosidosisab pages 1-5)
Late-onset/adult AB variant
A 2022 report described a first late-onset AB-variant case with: * Gait disorder beginning ~age 10 and progressive course (ganne2022gm2gangliosidosisab pages 5-8) * Lower motor neuronopathy and spinocerebellar ataxia (HPO: HP:0001272 Cerebellar ataxia; HP:0000739 Abnormality of the corticospinal tract can be considered depending on exam; lower motor neuron involvement is captured by HP:0003433 Lower motor neuron dysfunction) (ganne2022gm2gangliosidosisab pages 5-8, ganne2022gm2gangliosidosisab pages 1-5) * Mild cognitive/executive deficits (HP:0001263 / HP:0002143 Abnormal executive function) (ganne2022gm2gangliosidosisab pages 5-8) * Subtle cerebellar vermis atrophy on MRI (UBERON: cerebellar vermis) (ganne2022gm2gangliosidosisab pages 5-8)
3.2 Quality of life impact
Formal QoL instruments were not reported in the retrieved evidence. Functional impairment is implied by progressive neurologic decline and loss of mobility/vision in infantile forms, and by progressive ataxia/LMN dysfunction in later-onset disease. (ganne2022gm2gangliosidosisab pages 1-5, ganne2022gm2gangliosidosisab pages 5-8)
4. Genetic / molecular information
4.1 Causal gene
- GM2A (ganglioside GM2 activator). (ganne2022gm2gangliosidosisab pages 1-5)
Role: GM2AP is described as a non-enzymatic lipid-binding cofactor that enables HexA to interact with GM2 ganglioside; a review states GM2-AP “forms a complex with GM2 ganglioside allowing interaction between hexosaminidase A and GM2 ganglioside.” (ganne2022gm2gangliosidosisab pages 1-5)
4.2 Pathogenic variant types and examples
Reported AB-variant disease alleles include: * Nonsense: GM2A c.472G>T (p.E158X) (infantile case report; predicted truncation and loss of function) (sheth2016gm2gangliosidosisab pages 3-4) * Structural/copy-number variant: homozygous exon 2 deletion in GM2A (requires CNV analysis; first whole-exon deletion reported) (hall2017gm2activatordeficiency pages 1-3) * Compound heterozygosity: c.79A>T (p.Lys27*) with c.415C>T (p.Pro139Ser) in a late-onset case; the missense allele was discussed as potentially hypomorphic and extremely rare in population databases. (ganne2022gm2gangliosidosisab pages 5-8, ganne2022gm2gangliosidosisab pages 14-17)
Population frequency notes: for the late-onset case, the p.Pro139Ser allele was reported with no homozygotes in gnomAD/ExAC/TOPMed and only one prior report in a Latin American individual. (ganne2022gm2gangliosidosisab pages 14-17)
4.3 Modifier genes / pathways
NEU3 (neuraminidase 3) acts as a compensatory enzyme in mice, providing an alternative GM2 breakdown route (GM2 → GA2) and attenuating the phenotype of Gm2a−/− mice. This is a key translational consideration and a model-based modifier mechanism. (deschenes2023characterizationofa pages 1-2, vyas2023efficacyofadenoassociated pages 9-10)
4.4 Epigenetic information / chromosomal abnormalities
Not reported in retrieved evidence for AB variant. (sheth2016gm2gangliosidosisab pages 1-3)
5. Environmental information
No non-genetic environmental, lifestyle, or infectious contributors were identified in the retrieved evidence; AB variant is a monogenic lysosomal storage disorder. (sheth2016gm2gangliosidosisab pages 1-3)
6. Mechanism / pathophysiology
6.1 Causal chain (trigger → molecular defect → clinical manifestations)
- GM2A pathogenic variants reduce/abolish functional GM2 activator protein (GM2AP). (sheth2016gm2gangliosidosisab pages 3-4, hall2017gm2activatordeficiency pages 1-3)
- Without GM2AP, HexA cannot efficiently hydrolyze GM2 ganglioside in lysosomes (despite normal HexA/HexB activity on synthetic substrates). (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 1-3)
- GM2 accumulates intralysosomally, particularly in CNS neurons, driving progressive neurodegeneration and neurologic decline (developmental regression, seizures, ataxia, etc.). (sheth2016gm2gangliosidosisab pages 1-3, ganne2022gm2gangliosidosisab pages 1-5)
6.2 Pathways, processes, compartments
- Biological pathway: GM2 ganglioside catabolism (lysosomal glycosphingolipid degradation) (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 1-3)
- Subcellular compartment: lysosome (GO Cellular Component suggestion: GO:0005764 lysosome) (sheth2016gm2gangliosidosisab pages 1-3)
- Cell types: neurons (CL suggestion: CL:0000540 neuron); also glial responses are plausible but not directly evidenced in retrieved AB-variant human data. (sheth2016gm2gangliosidosisab pages 1-3)
Suggested GO Biological Process terms (mechanism-level): * GO:0006687 glycosphingolipid metabolic process * GO:0046467 membrane lipid metabolic process * GO:0006508 proteolysis (relevant for degradation of misfolded/truncated proteins described in some AB-variant mutation mechanisms)
6.3 Model-organism mechanistic insight (species difference)
A major translational insight is that mice have stronger NEU3-mediated alternative GM2 catabolism, which masks severity in Gm2a−/− mice. A 2023 study generated Gm2a−/−Neu3−/− mice and showed (relative to Gm2a−/− alone) markedly increased CNS GM2 accumulation, neurodegenerative pathology, and shortened lifespan, supporting the causal role of GM2 storage and providing a more severe preclinical model. (deschenes2023characterizationofa pages 8-10, deschenes2023characterizationofa pages 1-2)
7. Anatomical structures affected
7.1 Organ/system level
- Central nervous system (primary): progressive neurodegeneration (UBERON suggestion: UBERON:0000955 brain; UBERON:0001017 central nervous system). (sheth2016gm2gangliosidosisab pages 1-3, ganne2022gm2gangliosidosisab pages 1-5)
- Eye/retina (common clinical sign): cherry-red macula (UBERON:0000966 retina; UBERON:0001768 macula). (sheth2016gm2gangliosidosisab pages 1-3)
7.2 Tissue/cell level
- Neural tissue; neuronal storage pathology (CL:0000540 neuron). (sheth2016gm2gangliosidosisab pages 1-3)
7.3 Subcellular level
- Lysosomal storage (GO:0005764 lysosome). (sheth2016gm2gangliosidosisab pages 1-3)
8. Temporal development
8.1 Onset
- Infantile: first year of life (often 3–12 months in summaries/cases). (deschenes2023characterizationofa pages 1-2, ganne2022gm2gangliosidosisab pages 1-5)
- Juvenile: 2–10 years (summary). (ganne2022gm2gangliosidosisab pages 1-5)
- Late-onset: heterogenous; AB-variant late-onset case began with gait disorder at ~10 years. (ganne2022gm2gangliosidosisab pages 5-8)
8.2 Progression
- Infantile AB variant is typically rapidly progressive with early childhood mortality in summaries. (ganne2022gm2gangliosidosisab pages 1-5, noites2025gm2gangliosidosisab pages 2-3)
- Later-onset disease appears more slowly progressive and heterogeneous, but AB-variant-specific natural history datasets remain extremely limited. (ganne2022gm2gangliosidosisab pages 5-8, ganne2022gm2gangliosidosisab pages 1-5)
9. Inheritance and population
9.1 Inheritance
Autosomal recessive (biallelic GM2A). (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 3-4)
9.2 Epidemiology
Robust population prevalence/incidence estimates for AB variant are not available in the retrieved evidence. Instead, rarity is described by case counts, e.g.: * “Only seven mutations in nine cases have been reported…” in a 2016 review/case report, with their report adding a tenth molecularly confirmed case. (sheth2016gm2gangliosidosisab pages 3-4) * A 2022 review states AB variant is the rarest GM2 subtype with “less than thirty cases described in the literature.” (ganne2022gm2gangliosidosisab pages 1-5) * A 2022 Indian clinical series identified 1 AB-variant case among 32 gangliosidosis patients in that center-based cohort (not population incidence). (gowda2022clinicalandlaboratory pages 1-2)
9.3 Populations / founder effects
No clear AB-variant founder mutation pattern was established in the retrieved evidence; reported cases span diverse ancestries (e.g., Indian, Saudi, Spanish, US Black, Laotian/Hmong). (sheth2016gm2gangliosidosisab pages 1-3)
10. Diagnostics
10.1 Clinical and biochemical testing
Key diagnostic clue: Tay–Sachs-like phenotype with normal leukocyte HexA and total hexosaminidase activity should prompt evaluation for GM2AP deficiency / GM2A variants. (sheth2016gm2gangliosidosisab pages 3-4)
Case-report example enzyme pattern (infantile AB variant): normal Hex-A and total-Hex activities were documented despite a classic clinical presentation. (sheth2016gm2gangliosidosisab pages 3-4)
10.2 Genetic testing strategy
Recommended approach (from case-based evidence): 1. If GM2 gangliosidosis is clinically suspected, start with HexA/HexB enzymology. 2. If HexA/HexB results are non-diagnostic/normal but phenotype is strong, perform GM2A sequencing. 3. If sequencing is negative but suspicion remains, perform exon-level copy number analysis because exon deletions can be missed by routine sequencing (e.g., exon 2 deletion). (hall2017gm2activatordeficiency pages 1-3)
Advanced confirmatory methods (used in a late-onset case): plasma GM2 quantification by LC–MS/MS with reported sensitivity/specificity 100% at specific cutoffs; fibroblast studies by EM/IF; and GM2A expression studies (RT-qPCR/cDNA sequencing) to help classify variants. (ganne2022gm2gangliosidosisab pages 5-8, ganne2022gm2gangliosidosisab pages 8-14)
10.3 Differential diagnosis
- Tay–Sachs (HEXA): low HexA activity.
- Sandhoff (HEXB): low total hexosaminidase activity and often more systemic involvement.
- AB variant (GM2A): normal HexA/HexB activity on routine assays with GM2 gangliosidosis phenotype. (sheth2016gm2gangliosidosisab pages 3-4, hall2017gm2activatordeficiency pages 1-3)
10.4 Screening (carrier/newborn)
The retrieved evidence supports that molecular diagnosis (gene-based) is essential for AB-variant detection and family counseling because enzyme testing may not be informative for GM2A deficiency. (hall2017gm2activatordeficiency pages 1-3, sheth2016gm2gangliosidosisab pages 3-4)
11. Outcome / prognosis
- Infantile: severe, progressive neurodegeneration; reviews describe death in early childhood (~4–5 years) (ganne2022gm2gangliosidosisab pages 1-5)
- Juvenile: progressive decline with death before adulthood in summary descriptions (ganne2022gm2gangliosidosisab pages 1-5)
- Late-onset: better prognosis but heterogeneous; AB-variant-specific data remain extremely limited. (ganne2022gm2gangliosidosisab pages 1-5, ganne2022gm2gangliosidosisab pages 5-8)
12. Treatment
12.1 Standard of care
No disease-modifying standard therapy was identified in the retrieved AB-variant evidence; care is generally supportive and multidisciplinary given progressive neurodegeneration. (ganne2022gm2gangliosidosisab pages 1-5)
Suggested MAXO terms (supportive care examples): * MAXO:0000756 palliative care * MAXO:0000917 physical therapy * MAXO:0000918 occupational therapy * MAXO:0000919 speech therapy * MAXO:0001020 seizure management (antiseizure pharmacotherapy)
12.2 Recent developments (prioritize 2023–2024)
12.2.1 AB-variant–focused gene therapy (preclinical; 2023)
Two 2023 studies provide AB-variant-specific proof-of-concept GM2A gene replacement:
-
Systemic IV ssAAV9-GM2A in Gm2a−/− mice at a single dose reported as 1 × 10^14 vg/kg (and also described as 1 × 10^11 vg/mouse in study methods), delivered at postnatal day 1 or 6 weeks, led to detectable transgene persistence in brain and liver, reduced GM2 accumulation at 20 weeks, and improved rotarod performance in the 6-week-treated cohort; longer-term biochemical correction at 60 weeks was less robust. (vyas2023efficacyofadenoassociated pages 1-2, vyas2023efficacyofadenoassociated pages 7-9, vyas2023efficacyofadenoassociated pages 9-10)
-
Intrathecal scAAV9.hGM2A in Gm2a−/− mice prevented GM2 accumulation, with dose-escalation (0.5, 1.0, 2.0 × 10^11 vg/mouse) showing dose-responsive biochemical correction and long-term persistence up to 104 weeks, with no severe adverse events reported. (deschenes2023biochemicalcorrectionof pages 1-2, deschenes2023biochemicalcorrectionof pages 2-4)
Real-world implementation status: These are preclinical mouse studies; no human AB-variant GM2A gene therapy trial was identified in retrieved clinical trial records. (vyas2023efficacyofadenoassociated pages 1-2, deschenes2023biochemicalcorrectionof pages 1-2)
12.2.2 Related (non–AB-variant-specific) gene therapy clinical trials for GM2 gangliosidoses
Although not AB-variant-specific, the current clinical development pipeline for GM2 gangliosidoses includes: * TSHA-101 (AAV9-HEXA/HEXB bicistronic) intrathecal gene therapy trial in infantile-onset GM2 gangliosidosis: NCT04798235 (Active, not recruiting; start 2021-03-12). (NCT04798235 chunk 1) * AXO-AAV-GM2 gene therapy trial for Tay–Sachs or Sandhoff: NCT04669535 (terminated in retrieved trial metadata; pediatric surgical delivery includes bilateral thalamic and intracisternal/intrathecal routes). (NCT04669535 chunk 2) * Long-term follow-up of AXO-AAV-GM2-treated subjects: NCT06614569 (Active, not recruiting; start 2024-09-17). (NCT06614569 chunk 1)
12.2.3 Substrate reduction therapy (SRT) concept (review-level; 2024)
A 2024 review frames AB variant within GM2 disorders and discusses targeting GM2 synthesis pathways as an SRT approach, while noting the AB variant is due to GM2AP deficiency (not HexA deficiency). (abidi2024metabolismofglycosphingolipids pages 19-24)
12.3 Expert opinion and analysis (from authoritative sources in retrieved evidence)
- Diagnostic experts emphasize persistence despite normal enzyme tests: one report states diagnosis “requires a high degree of suspicion and persistence, despite consistently normal or uninformative results,” highlighting a recurring clinical pitfall. (hall2017gm2activatordeficiency pages 1-3)
- Translational experts emphasize model validity: multiple 2023 sources argue that the mild Gm2a−/− phenotype is due to NEU3-mediated compensation and recommend double-knockout models (or other strategies) to better match human disease for therapy testing. (vyas2023efficacyofadenoassociated pages 9-10, deschenes2023characterizationofa pages 1-2)
13. Prevention
Primary prevention is genetic: * Carrier testing and reproductive counseling (autosomal recessive inheritance). (sheth2016gm2gangliosidosisab pages 3-4) * Prenatal and preimplantation genetic testing are conceptually applicable where familial GM2A variants are known; however, specific AB-variant screening guideline statements were not present in retrieved evidence.
Secondary prevention: early diagnosis (including molecular diagnosis when enzyme assays are normal) may support earlier supportive interventions and trial enrollment. (hall2017gm2activatordeficiency pages 1-3)
14. Other species / natural disease
No naturally occurring AB-variant GM2A deficiency in non-human species was identified in the retrieved evidence; the key comparative biology discussion in retrieved sources relates to species differences in GM2 catabolism (NEU3 compensation in mice). (deschenes2023characterizationofa pages 1-2)
15. Model organisms
15.1 Mouse models (highly relevant)
- Gm2a−/−: moderate GM2 accumulation and mild neurological defects; normal lifespan; considered more representative of predicted adult-onset human AB variant because of murine compensatory GM2 catabolism. (vyas2023efficacyofadenoassociated pages 2-4, deschenes2023characterizationofa pages 1-2)
- Gm2a−/−Neu3−/− (double knockout): severe CNS GM2 accumulation, ataxia, reduced mobility/coordination, weight loss, and early lethality (~6–7 months); proposed as a “highly relevant model for pre-clinical drug studies” for severe AB variant. (deschenes2023characterizationofa pages 1-2)
15.2 Model applications and limitations
- Applications: testing gene therapy delivery and biochemical correction; interrogating modifier pathways (NEU3). (vyas2023efficacyofadenoassociated pages 7-9, deschenes2023characterizationofa pages 8-10)
- Limitations: single Gm2a−/− mice may underpredict severity of human infantile AB variant due to mouse-specific alternative catabolism; thus, therapeutic efficacy in Gm2a−/− should be interpreted cautiously. (vyas2023efficacyofadenoassociated pages 9-10, deschenes2023characterizationofa pages 1-2)
Visual evidence (figures)
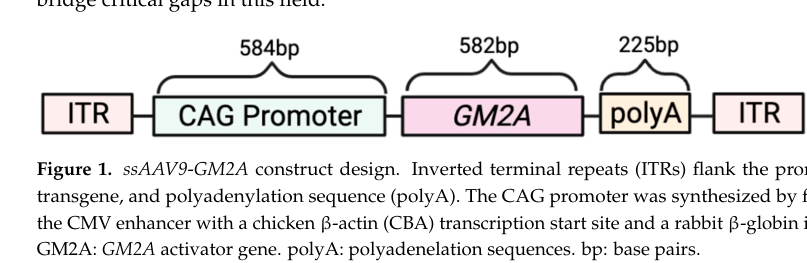
Vyas et al. (2023) provide key preclinical gene-therapy visuals including ssAAV9-GM2A construct, biodistribution, GM2 histology/quantification, and rotarod improvement, supporting translational claims about vector design, tissue distribution, biochemical correction, and functional outcomes. (vyas2023efficacyofadenoassociated media 2eb6c65b, vyas2023efficacyofadenoassociated media 580ab361, vyas2023efficacyofadenoassociated media 8f62a7a9, vyas2023efficacyofadenoassociated media bb08d317)
URLs and publication dates (retrieved sources)
- Sheth et al. 2016-07. BMC Pediatrics. https://doi.org/10.1186/s12887-016-0626-6 (sheth2016gm2gangliosidosisab pages 3-4)
- Hall et al. 2017-05. JIMD Reports. https://doi.org/10.1007/8904_2017_31 (hall2017gm2activatordeficiency pages 1-3)
- Ganne et al. 2022-08. Neurological Sciences. https://doi.org/10.1007/s10072-022-06270-x (ganne2022gm2gangliosidosisab pages 1-5)
- Deschenes et al. 2023-05. Int J Mol Sci. https://doi.org/10.3390/ijms24119217 (deschenes2023biochemicalcorrectionof pages 1-2)
- Vyas et al. 2023-09. Int J Mol Sci. https://doi.org/10.3390/ijms241914611 (vyas2023efficacyofadenoassociated pages 1-2)
- Deschenes et al. 2023-11. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2023.1242814 (deschenes2023characterizationofa pages 1-2)
- ClinicalTrials.gov: NCT04798235 (start 2021-03-12). https://clinicaltrials.gov/study/NCT04798235 (NCT04798235 chunk 1)
- ClinicalTrials.gov: NCT04669535 (start 2021). https://clinicaltrials.gov/study/NCT04669535 (NCT04669535 chunk 2)
- ClinicalTrials.gov: NCT06614569 (start 2024-09-17). https://clinicaltrials.gov/study/NCT06614569 (NCT06614569 chunk 1)
- ClinicalTrials.gov: NCT00668187 (start 2010-12). https://clinicaltrials.gov/study/NCT00668187 (NCT00668187 chunk 1)
Data gaps and limitations
- AB-variant-specific incidence/prevalence, carrier frequency, and founder variants are not established in the retrieved evidence; rarity is primarily captured via reported case counts and center-based cohorts. (ganne2022gm2gangliosidosisab pages 1-5, sheth2016gm2gangliosidosisab pages 1-3, gowda2022clinicalandlaboratory pages 1-2)
- AB-variant-specific prospective natural history and validated clinical endpoints remain sparse; broader GM2 natural history studies exist but may include few/no AB-variant participants. (NCT00668187 chunk 1)
- Orphanet/ICD/MeSH identifiers for AB variant were not recoverable from the retrieved full-text evidence set.
References
-
(ganne2022gm2gangliosidosisab pages 1-5): Benjamin Ganne, Benjamin Dauriat, Laurence Richard, Foudil Lamari, Karima Ghorab, Laurent Magy, Mehdi Benkirane, Alexandre Perani, Valentine Marquet, Patrick Calvas, Catherine Yardin, and Sylvie Bourthoumieu. Gm2 gangliosidosis ab variant: first case of late onset and review of the literature. Neurological Sciences, 43:6517-6527, Aug 2022. URL: https://doi.org/10.1007/s10072-022-06270-x, doi:10.1007/s10072-022-06270-x. This article has 15 citations and is from a peer-reviewed journal.
-
(sheth2016gm2gangliosidosisab pages 1-3): Jayesh Sheth, Chaitanya Datar, Mehul Mistri, Riddhi Bhavsar, Frenny Sheth, and Krati Shah. Gm2 gangliosidosis ab variant: novel mutation from india – a case report with a review. BMC Pediatrics, Jul 2016. URL: https://doi.org/10.1186/s12887-016-0626-6, doi:10.1186/s12887-016-0626-6. This article has 39 citations and is from a peer-reviewed journal.
-
(hall2017gm2activatordeficiency pages 1-3): Patricia L. Hall, Regina Laine, John J. Alexander, Arunkanth Ankala, Lisa A. Teot, Hart G. W. Lidov, and Irina Anselm. Gm2 activator deficiency caused by a homozygous exon 2 deletion in gm2a. JIMD reports, 38:61-65, May 2017. URL: https://doi.org/10.1007/8904_2017_31, doi:10.1007/8904_2017_31. This article has 10 citations and is from a peer-reviewed journal.
-
(deschenes2023characterizationofa pages 8-10): Natalie M. Deschenes, Camilyn Cheng, Prem Khanal, Brianna M. Quinville, Alex E. Ryckman, Melissa Mitchell, Alexey V. Pshezhetsky, and Jagdeep S. Walia. Characterization of a phenotypically severe animal model for human ab-variant gm2 gangliosidosis. Frontiers in Molecular Neuroscience, Nov 2023. URL: https://doi.org/10.3389/fnmol.2023.1242814, doi:10.3389/fnmol.2023.1242814. This article has 5 citations.
-
(vyas2023efficacyofadenoassociated pages 1-2): Meera Vyas, Natalie M. Deschenes, Karlaina J. L. Osmon, Zhilin Chen, Imtiaz Ahmad, Shalini Kot, Patrick Thompson, Chris Richmond, Steven J. Gray, and Jagdeep S. Walia. Efficacy of adeno-associated virus serotype 9-mediated gene therapy for ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:14611, Sep 2023. URL: https://doi.org/10.3390/ijms241914611, doi:10.3390/ijms241914611. This article has 6 citations.
-
(OpenTargets Search: GM2 gangliosidosis,Tay-Sachs disease-GM2A,HEXA,HEXB): Open Targets Query (GM2 gangliosidosis,Tay-Sachs disease-GM2A,HEXA,HEXB, 15 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(sheth2016gm2gangliosidosisab pages 3-4): Jayesh Sheth, Chaitanya Datar, Mehul Mistri, Riddhi Bhavsar, Frenny Sheth, and Krati Shah. Gm2 gangliosidosis ab variant: novel mutation from india – a case report with a review. BMC Pediatrics, Jul 2016. URL: https://doi.org/10.1186/s12887-016-0626-6, doi:10.1186/s12887-016-0626-6. This article has 39 citations and is from a peer-reviewed journal.
-
(gualdronfrias2019taysachsdisease pages 3-4): Carlos Andrés Gualdrón-Frías and Laura Tatiana Calderón-Nossa. Tay-sachs disease. Revista de la Facultad de Medicina, 67:323-329, Jul 2019. URL: https://doi.org/10.15446/revfacmed.v67n3.69742, doi:10.15446/revfacmed.v67n3.69742. This article has 3 citations.
-
(ganne2022gm2gangliosidosisab pages 5-8): Benjamin Ganne, Benjamin Dauriat, Laurence Richard, Foudil Lamari, Karima Ghorab, Laurent Magy, Mehdi Benkirane, Alexandre Perani, Valentine Marquet, Patrick Calvas, Catherine Yardin, and Sylvie Bourthoumieu. Gm2 gangliosidosis ab variant: first case of late onset and review of the literature. Neurological Sciences, 43:6517-6527, Aug 2022. URL: https://doi.org/10.1007/s10072-022-06270-x, doi:10.1007/s10072-022-06270-x. This article has 15 citations and is from a peer-reviewed journal.
-
(ganne2022gm2gangliosidosisab pages 8-14): Benjamin Ganne, Benjamin Dauriat, Laurence Richard, Foudil Lamari, Karima Ghorab, Laurent Magy, Mehdi Benkirane, Alexandre Perani, Valentine Marquet, Patrick Calvas, Catherine Yardin, and Sylvie Bourthoumieu. Gm2 gangliosidosis ab variant: first case of late onset and review of the literature. Neurological Sciences, 43:6517-6527, Aug 2022. URL: https://doi.org/10.1007/s10072-022-06270-x, doi:10.1007/s10072-022-06270-x. This article has 15 citations and is from a peer-reviewed journal.
-
(deschenes2023characterizationofa pages 1-2): Natalie M. Deschenes, Camilyn Cheng, Prem Khanal, Brianna M. Quinville, Alex E. Ryckman, Melissa Mitchell, Alexey V. Pshezhetsky, and Jagdeep S. Walia. Characterization of a phenotypically severe animal model for human ab-variant gm2 gangliosidosis. Frontiers in Molecular Neuroscience, Nov 2023. URL: https://doi.org/10.3389/fnmol.2023.1242814, doi:10.3389/fnmol.2023.1242814. This article has 5 citations.
-
(noites2025gm2gangliosidosisab pages 2-3): Inês Noites, Ana Sofia Coelho, Catarina Magalhães, Sandra Ramos, Francisco Laranjeira, Lúcia Lacerda, Ricardo Taipa, Cristina Garrido, Teresa Temudo, and Sónia Figueiroa. Gm2 gangliosidosis ab variant: a hidden truth. Cureus, Sep 2025. URL: https://doi.org/10.7759/cureus.92445, doi:10.7759/cureus.92445. This article has 0 citations.
-
(ganne2022gm2gangliosidosisab pages 14-17): Benjamin Ganne, Benjamin Dauriat, Laurence Richard, Foudil Lamari, Karima Ghorab, Laurent Magy, Mehdi Benkirane, Alexandre Perani, Valentine Marquet, Patrick Calvas, Catherine Yardin, and Sylvie Bourthoumieu. Gm2 gangliosidosis ab variant: first case of late onset and review of the literature. Neurological Sciences, 43:6517-6527, Aug 2022. URL: https://doi.org/10.1007/s10072-022-06270-x, doi:10.1007/s10072-022-06270-x. This article has 15 citations and is from a peer-reviewed journal.
-
(gowda2022clinicalandlaboratory pages 1-2): Vykuntaraju K. Gowda, Priya Gupta, Narmadham K. Bharathi, Maya Bhat, Sanjay K. Shivappa, and Naveen Benakappa. Clinical and laboratory profile of gangliosidosis from southern part of india. Journal of Pediatric Genetics, 11:034-041, Oct 2022. URL: https://doi.org/10.1055/s-0040-1718726, doi:10.1055/s-0040-1718726. This article has 10 citations and is from a peer-reviewed journal.
-
(noites2025gm2gangliosidosisab pages 1-2): Inês Noites, Ana Sofia Coelho, Catarina Magalhães, Sandra Ramos, Francisco Laranjeira, Lúcia Lacerda, Ricardo Taipa, Cristina Garrido, Teresa Temudo, and Sónia Figueiroa. Gm2 gangliosidosis ab variant: a hidden truth. Cureus, Sep 2025. URL: https://doi.org/10.7759/cureus.92445, doi:10.7759/cureus.92445. This article has 0 citations.
-
(deschenes2023biochemicalcorrectionof pages 1-2): Natalie M. Deschenes, Camilyn Cheng, Alex E. Ryckman, Brianna M. Quinville, Prem Khanal, Melissa Mitchell, Zhilin Chen, Waheed Sangrar, Steven J. Gray, and Jagdeep S. Walia. Biochemical correction of gm2 ganglioside accumulation in ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:9217, May 2023. URL: https://doi.org/10.3390/ijms24119217, doi:10.3390/ijms24119217. This article has 8 citations.
-
(deschenes2023biochemicalcorrectionof pages 2-4): Natalie M. Deschenes, Camilyn Cheng, Alex E. Ryckman, Brianna M. Quinville, Prem Khanal, Melissa Mitchell, Zhilin Chen, Waheed Sangrar, Steven J. Gray, and Jagdeep S. Walia. Biochemical correction of gm2 ganglioside accumulation in ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:9217, May 2023. URL: https://doi.org/10.3390/ijms24119217, doi:10.3390/ijms24119217. This article has 8 citations.
-
(vyas2023efficacyofadenoassociated pages 7-9): Meera Vyas, Natalie M. Deschenes, Karlaina J. L. Osmon, Zhilin Chen, Imtiaz Ahmad, Shalini Kot, Patrick Thompson, Chris Richmond, Steven J. Gray, and Jagdeep S. Walia. Efficacy of adeno-associated virus serotype 9-mediated gene therapy for ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:14611, Sep 2023. URL: https://doi.org/10.3390/ijms241914611, doi:10.3390/ijms241914611. This article has 6 citations.
-
(deschenes2023biochemicalcorrectionof pages 14-15): Natalie M. Deschenes, Camilyn Cheng, Alex E. Ryckman, Brianna M. Quinville, Prem Khanal, Melissa Mitchell, Zhilin Chen, Waheed Sangrar, Steven J. Gray, and Jagdeep S. Walia. Biochemical correction of gm2 ganglioside accumulation in ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:9217, May 2023. URL: https://doi.org/10.3390/ijms24119217, doi:10.3390/ijms24119217. This article has 8 citations.
-
(vyas2023efficacyofadenoassociated pages 11-13): Meera Vyas, Natalie M. Deschenes, Karlaina J. L. Osmon, Zhilin Chen, Imtiaz Ahmad, Shalini Kot, Patrick Thompson, Chris Richmond, Steven J. Gray, and Jagdeep S. Walia. Efficacy of adeno-associated virus serotype 9-mediated gene therapy for ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:14611, Sep 2023. URL: https://doi.org/10.3390/ijms241914611, doi:10.3390/ijms241914611. This article has 6 citations.
-
(vyas2023efficacyofadenoassociated pages 2-4): Meera Vyas, Natalie M. Deschenes, Karlaina J. L. Osmon, Zhilin Chen, Imtiaz Ahmad, Shalini Kot, Patrick Thompson, Chris Richmond, Steven J. Gray, and Jagdeep S. Walia. Efficacy of adeno-associated virus serotype 9-mediated gene therapy for ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:14611, Sep 2023. URL: https://doi.org/10.3390/ijms241914611, doi:10.3390/ijms241914611. This article has 6 citations.
-
(vyas2023efficacyofadenoassociated pages 9-10): Meera Vyas, Natalie M. Deschenes, Karlaina J. L. Osmon, Zhilin Chen, Imtiaz Ahmad, Shalini Kot, Patrick Thompson, Chris Richmond, Steven J. Gray, and Jagdeep S. Walia. Efficacy of adeno-associated virus serotype 9-mediated gene therapy for ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:14611, Sep 2023. URL: https://doi.org/10.3390/ijms241914611, doi:10.3390/ijms241914611. This article has 6 citations.
-
(vyas2023efficacyofadenoassociated media 2eb6c65b): Meera Vyas, Natalie M. Deschenes, Karlaina J. L. Osmon, Zhilin Chen, Imtiaz Ahmad, Shalini Kot, Patrick Thompson, Chris Richmond, Steven J. Gray, and Jagdeep S. Walia. Efficacy of adeno-associated virus serotype 9-mediated gene therapy for ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:14611, Sep 2023. URL: https://doi.org/10.3390/ijms241914611, doi:10.3390/ijms241914611. This article has 6 citations.
-
(deschenes2023characterizationofa pages 2-3): Natalie M. Deschenes, Camilyn Cheng, Prem Khanal, Brianna M. Quinville, Alex E. Ryckman, Melissa Mitchell, Alexey V. Pshezhetsky, and Jagdeep S. Walia. Characterization of a phenotypically severe animal model for human ab-variant gm2 gangliosidosis. Frontiers in Molecular Neuroscience, Nov 2023. URL: https://doi.org/10.3389/fnmol.2023.1242814, doi:10.3389/fnmol.2023.1242814. This article has 5 citations.
-
(deschenes2023characterizationofa pages 3-4): Natalie M. Deschenes, Camilyn Cheng, Prem Khanal, Brianna M. Quinville, Alex E. Ryckman, Melissa Mitchell, Alexey V. Pshezhetsky, and Jagdeep S. Walia. Characterization of a phenotypically severe animal model for human ab-variant gm2 gangliosidosis. Frontiers in Molecular Neuroscience, Nov 2023. URL: https://doi.org/10.3389/fnmol.2023.1242814, doi:10.3389/fnmol.2023.1242814. This article has 5 citations.
-
(NCT04798235 chunk 1): Dr. Anupam Sehgal. First-in-Human Study of TSHA-101 Gene Therapy for Treatment of Infantile Onset GM2 Gangliosidosis. Dr. Anupam Sehgal. 2021. ClinicalTrials.gov Identifier: NCT04798235
-
(NCT04669535 chunk 2): Terence Flotte. A Dose-escalation and Safety & Efficacy Study of AXO-AAV-GM2 in Tay-Sachs or Sandhoff Disease. Terence Flotte. 2021. ClinicalTrials.gov Identifier: NCT04669535
-
(NCT06614569 chunk 1): Terence Flotte. Long-Term Follow-Up of Subjects Treated With AXO-AAV-GM2 for Tay-Sachs or Sandhoff Disease. Terence Flotte. 2024. ClinicalTrials.gov Identifier: NCT06614569
-
(NCT00668187 chunk 1): A Natural History Study of the Gangliosidoses. University of Minnesota. 2010. ClinicalTrials.gov Identifier: NCT00668187
-
(abidi2024metabolismofglycosphingolipids pages 19-24): I Abidi. Metabolism of glycosphingolipids and targeting gm2 synthesis pathway to develop substrate reduction approach in tay-sachs and sandhoff disorders. Unknown journal, 2024.
-
(vyas2023efficacyofadenoassociated media 580ab361): Meera Vyas, Natalie M. Deschenes, Karlaina J. L. Osmon, Zhilin Chen, Imtiaz Ahmad, Shalini Kot, Patrick Thompson, Chris Richmond, Steven J. Gray, and Jagdeep S. Walia. Efficacy of adeno-associated virus serotype 9-mediated gene therapy for ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:14611, Sep 2023. URL: https://doi.org/10.3390/ijms241914611, doi:10.3390/ijms241914611. This article has 6 citations.
-
(vyas2023efficacyofadenoassociated media 8f62a7a9): Meera Vyas, Natalie M. Deschenes, Karlaina J. L. Osmon, Zhilin Chen, Imtiaz Ahmad, Shalini Kot, Patrick Thompson, Chris Richmond, Steven J. Gray, and Jagdeep S. Walia. Efficacy of adeno-associated virus serotype 9-mediated gene therapy for ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:14611, Sep 2023. URL: https://doi.org/10.3390/ijms241914611, doi:10.3390/ijms241914611. This article has 6 citations.
-
(vyas2023efficacyofadenoassociated media bb08d317): Meera Vyas, Natalie M. Deschenes, Karlaina J. L. Osmon, Zhilin Chen, Imtiaz Ahmad, Shalini Kot, Patrick Thompson, Chris Richmond, Steven J. Gray, and Jagdeep S. Walia. Efficacy of adeno-associated virus serotype 9-mediated gene therapy for ab-variant gm2 gangliosidosis. International Journal of Molecular Sciences, 24:14611, Sep 2023. URL: https://doi.org/10.3390/ijms241914611, doi:10.3390/ijms241914611. This article has 6 citations.