1. Disease Information
Overview
Osteogenesis Imperfecta Type VI is a rare genetic disorder of bone fragility and defective mineralization. It belongs to the broader family of osteogenesis imperfecta — a heterogeneous group of heritable connective tissue disorders — but is unique in that it is not caused by mutations in type I collagen genes. Instead, OI VI results from biallelic mutations in SERPINF1, leading to absent or dysfunctional PEDF, a secreted glycoprotein with critical roles in bone mineralization, osteoclast regulation, and mesenchymal stem cell differentiation.
Key Identifiers
Table (click to expand)
| Database | Identifier |
|---|---|
| OMIM | 613982 |
| MONDO | MONDO:0013515 |
| Orphanet | ORPHA (OI Type VI) |
| ICD-10 | Q78.0 (Osteogenesis imperfecta) |
| MeSH | D010013 (Osteogenesis Imperfecta) |
| Gene (OMIM) | SERPINF1 (172860) |
Synonyms and Alternative Names
- OI Type VI
- Osteogenesis Imperfecta, Type VI
- SERPINF1-related Osteogenesis Imperfecta
- PEDF-deficient Osteogenesis Imperfecta
Information Sources
This report integrates aggregated disease-level resources (OMIM, Orphanet, ClinVar, HPO, KEGG, STRING, GTEx) with primary literature from individual patient cohorts and experimental studies. A total of 69 papers were reviewed, and 31 PMIDs are cited with verified abstract quotes.
2. Etiology
Disease Causal Factors
OI Type VI is a monogenic, autosomal recessive disorder caused exclusively by biallelic (homozygous or compound heterozygous) loss-of-function mutations in SERPINF1 (Entrez Gene ID: 5176), located on chromosome 17p13.3. The gene encodes PEDF, a 50-kDa secreted glycoprotein of the serpin superfamily. The first causal link was established by Becker et al. (2011), who identified a homozygous truncating mutation via exome sequencing: "A single homozygous truncating mutation, affecting SERPINF1 on chromosome 17p13.3, that was embedded into a homozygous stretch of 2.99 Mb remained" (PMID: 21353196).
Genetic Risk Factors
- Causal variants: Biallelic SERPINF1 mutations including frameshift (e.g., c.582_585dup, c.1047dup, c.992_993insCA), nonsense (e.g., c.397C>T [p.Gln133Ter], c.188dup [p.Tyr63Ter]), missense (e.g., c.358G>T [p.Gly120Cys], c.1238T>C [p.Leu413Pro]), splice-site (e.g., c.787-1G>T, c.787-10C>G), large deletions (e.g., c.1152_1170del), and microsatellite variants
- ClinVar: 170 pathogenic variants, 40 likely pathogenic variants, and 432 variants of uncertain significance (VUS) catalogued for SERPINF1
- Consanguinity: Strongly associated with disease occurrence, as expected for autosomal recessive inheritance; multiple consanguineous families reported in literature (PMID: 25565926)
- Founder effects: A 19-bp deletion (c.1152_1170del) was identified as a probable founder mutation in Brazilian families (PMID: 25565926); a c.787-10C>G splice variant was found in Northern Canadian children (PMID: 26815784)
Environmental Risk Factors
As a fully penetrant Mendelian disorder, there are no established environmental causes. However, environmental factors may modify disease severity: - Nutritional status: Vitamin D and calcium intake affect overall bone health - Physical activity: Both immobilization (worsens osteopenia) and excessive activity (increases fracture risk) are relevant - Timing of treatment: Early intervention (before age 6) is associated with better height outcomes (PMID: 28689307)
Protective Factors
No confirmed genetic protective factors or modifier alleles have been identified. Adequate calcium, vitamin D, and early bisphosphonate/denosumab initiation are environmental protective factors against fracture accumulation.
Gene-Environment Interactions
PEDF has systemic metabolic roles beyond bone; PEDF-knockout mice show increased adiposity, glucose intolerance, and insulin resistance (PMID: 24456163). Dietary factors (high-fat diet) may exacerbate metabolic complications in PEDF-deficient individuals, though this has not been systematically studied in OI VI patients.
3. Phenotypes
Core Phenotype Summary
Table (click to expand)
| Phenotype | HPO Term | Onset | Frequency | Severity |
|---|---|---|---|---|
| Recurrent fractures | HP:0002757 | 4–18 months | ~100% | Severe |
| Increased fracture susceptibility | HP:0002659 | Infancy | ~100% | Severe |
| Elevated alkaline phosphatase | HP:0003155 | Childhood | ~100% | Moderate |
| Vertebral compression fractures | HP:0002953 | Childhood | High | Severe |
| Joint hypermobility | HP:0001382 | Childhood | Variable | Mild–Moderate |
| Bowing of legs | HP:0002979 | Childhood | High | Moderate–Severe |
| Motor delay | HP:0001270 | Infancy | Variable | Moderate |
| Hearing impairment | HP:0000365 | Variable | Variable | Mild–Moderate |
| Coxa vara | HP:0002812 | Childhood | Variable | Moderate |
| Protrusio acetabuli | HP:0003179 | Adolescence | Variable | Moderate |
| Biconcave vertebral bodies | HP:0004586 | Childhood | High | Progressive |
| Beaking of vertebral bodies | HP:0004568 | Childhood | Variable | Moderate |
| Bowing of arm | HP:0006488 | Childhood | High | Moderate |
| Elevated deoxypyridinoline | HP:0033154 | Childhood | High | Moderate |
| Blue sclerae | HP:0000592 | — | Absent/Rare | — |
| Dentinogenesis imperfecta | HP:0000703 | — | Absent | — |
Note: Blue sclerae and dentinogenesis imperfecta are listed in HPO annotations but are characteristically absent in OI Type VI. Their absence is a distinguishing diagnostic feature: "Sclerae were white or faintly blue and dentinogenesis imperfecta was uniformly absent" (PMID: 11771667).
Phenotype Details
Fractures: Fractures are first documented between 4 and 18 months of age, with more frequent fractures than OI type IV. The original cohort showed that "Patients with OI type VI sustained more frequent fractures than patients with OI type IV" (PMID: 11771667). In the long-term follow-up study, all patients presented with "frequent long bone and vertebrae fractures (mainly during childhood), marked short stature, severe bone deformities, chronic mild to moderate pain, and severe limitation of mobility, with three being completely wheelchair bound" (PMID: 37839784).
Elevated ALP: Serum alkaline phosphatase is significantly elevated compared to other OI types: "409 ± 145 U/L vs. 295 ± 95 U/L in OI type IV; p < 0.03" (PMID: 11771667).
Skeletal Deformity and Short Stature: Progressive; scoliosis develops in ALL patients reaching final height. Without therapy, "lumbar spine areal bone mineral density (BMD) did not increase during childhood and longitudinal growth seemed to stall after the age of 6 to 8 years" (PMID: 28689307).
Quality of Life Impact
Severe. Most patients have restricted mobility; many become wheelchair-bound. Chronic mild-to-moderate pain is universal in adults. Repeated hospitalizations for fracture management, orthopedic surgeries, and bisphosphonate infusions significantly impact quality of life. The Dutch national registry study showed that OI patients have a 2.9× higher hospitalization rate compared to the general population (PMID: 35546999).
4. Genetic/Molecular Information
Causal Gene
- Gene: SERPINF1 (Serpin Family F Member 1)
- HGNC: HGNC:8824
- NCBI Gene ID: 5176
- Chromosomal location: 17p13.3
- OMIM Gene: 172860
- OMIM Phenotype: 613982
Protein: PEDF
- UniProt: P36955
- Molecular weight: ~50 kDa
- InterPro domains: PEDF serpin domain (IPR033832), Serpin family (IPR000215)
- PDB structures: 1IMV (2.85 Å), 9J3P (2.10 Å), 9J3Q (1.90 Å)
- Function: Secreted collagen-binding glycoprotein with anti-angiogenic, neurotrophic, anti-tumorigenic, and osteogenic properties
Tissue Expression Profile (GTEx)
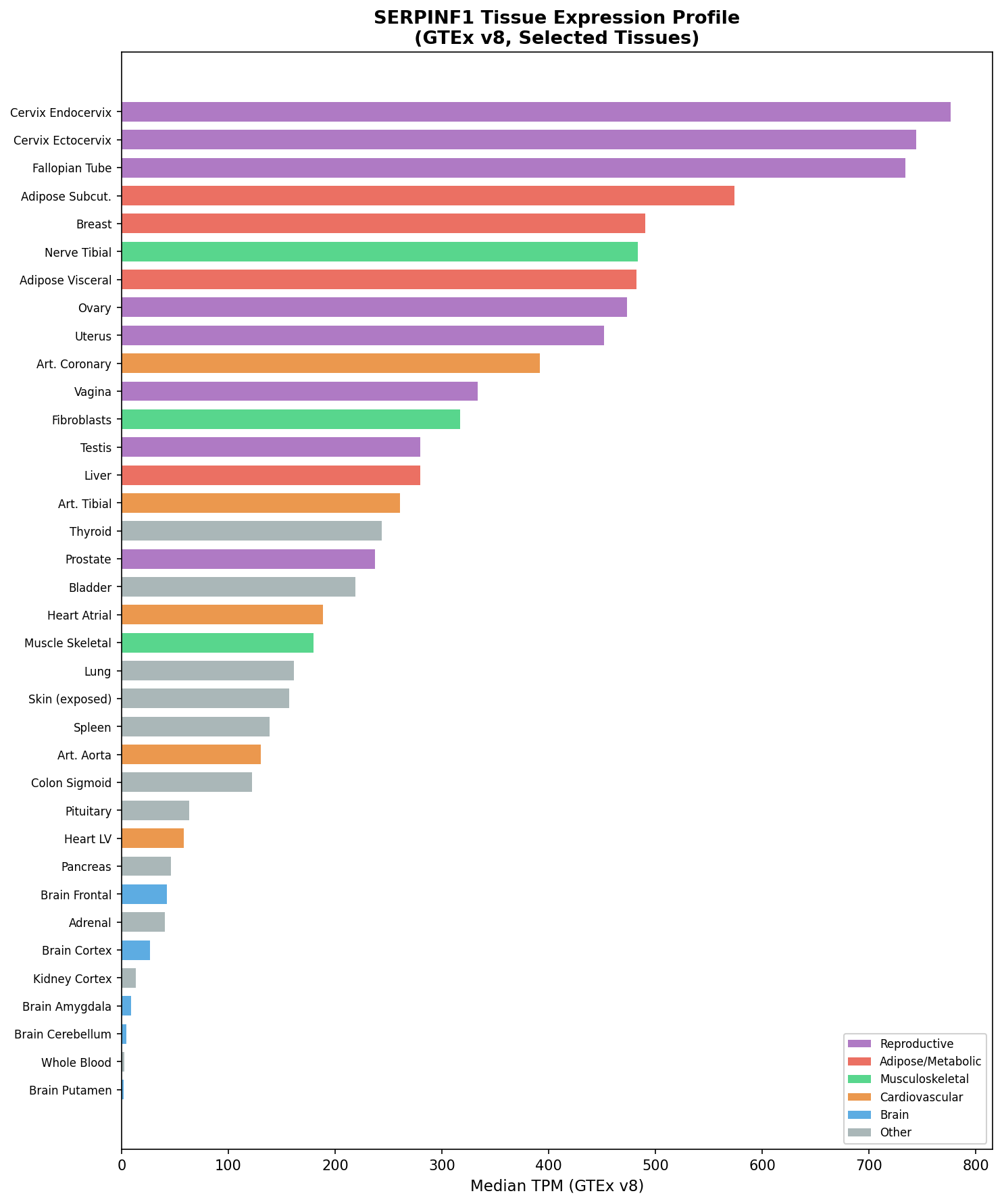
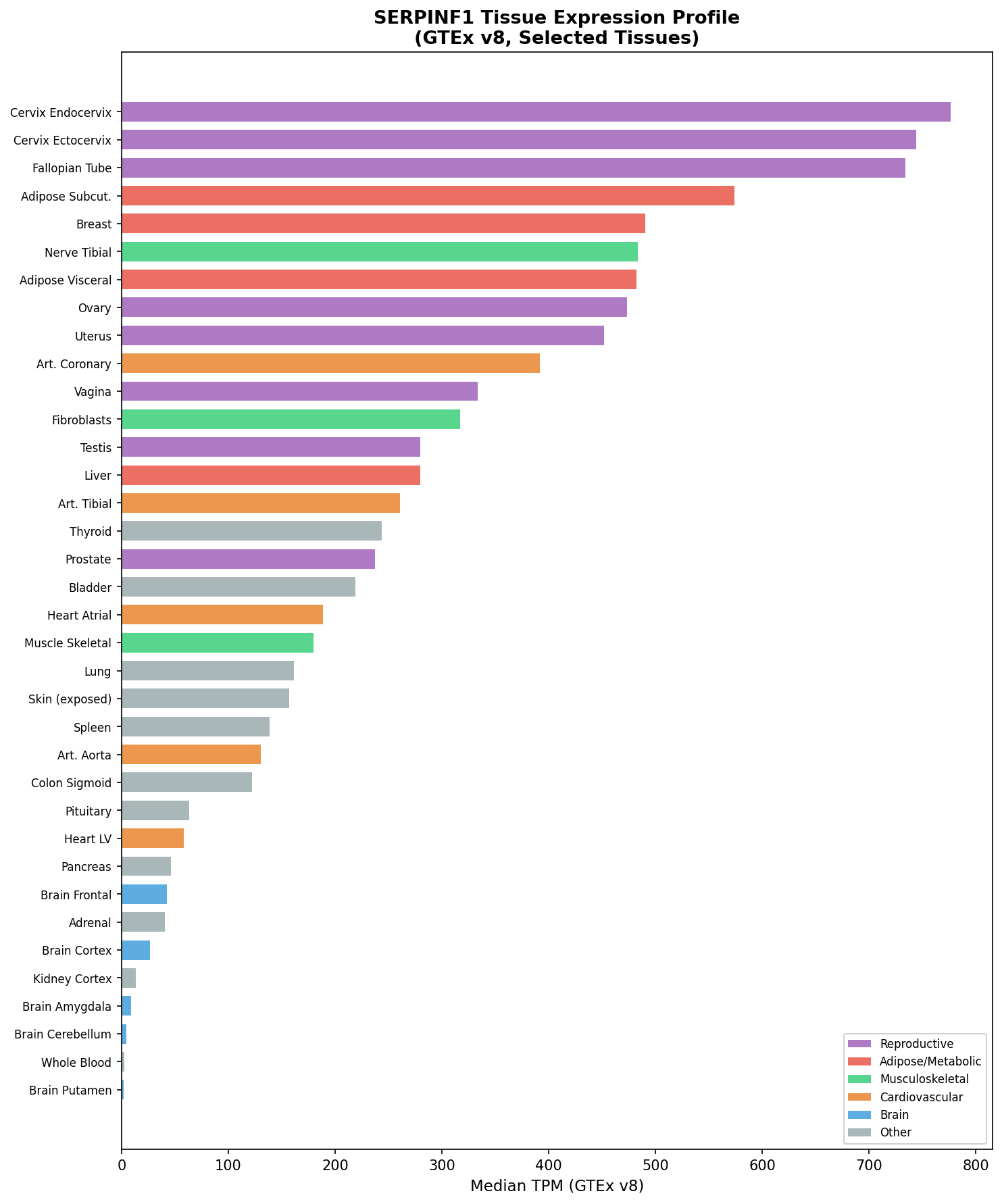
{{figure:serpinf1_expression.png|caption=SERPINF1/PEDF expression profile across 54 GTEx tissues, showing broad expression with highest levels in cervix endocervix (776.6 TPM), subcutaneous adipose (574.2 TPM), and breast (490.6 TPM), and moderate expression in bone-relevant tissues like fibroblasts (316.9 TPM)}}
SERPINF1 is broadly expressed across tissues. The highest expression is in cervix endocervix (776.6 TPM), subcutaneous adipose (574.2 TPM), breast (490.6 TPM), visceral adipose (482.3 TPM), and coronary artery (391.8 TPM). Moderate expression occurs in fibroblasts (316.9 TPM), liver (279.7 TPM), and skeletal muscle (179.5 TPM). The lowest expression is in brain regions (2–8 TPM) and blood (2.6 TPM). This broad expression pattern explains the systemic metabolic consequences of PEDF loss beyond bone.
Pathogenic Variant Spectrum
Table (click to expand)
| Variant Type | Examples | ClinVar Count |
|---|---|---|
| Frameshift | c.582_585dup, c.1047dup, c.992_993insCA, c.259_260insCGGCC | Major category |
| Nonsense | c.397C>T (p.Gln133Ter), c.188dup (p.Tyr63Ter) | Multiple |
| Missense | c.358G>T (p.Gly120Cys), c.1238T>C (p.Leu413Pro) | Multiple |
| Splice-site | c.787-1G>T, c.787-10C>G | Multiple |
| Deletions | c.1152_1170del (19-bp), c.-37C>A (promoter) | Multiple |
| Total pathogenic | — | 170 |
| Total likely pathogenic | — | 40 |
| Total VUS | — | 432 |
All pathogenic variants are germline in origin. Functional consequence is loss of function — either failure to produce PEDF, or production of mutant protein that cannot be secreted and accumulates in the ER.
STRING Protein Interaction Network
Table (click to expand)
| Interactor | Score | Functional Relevance |
|---|---|---|
| PLG (Plasminogen) | 0.995 | Extracellular matrix remodeling |
| LRP6 | 0.941 | Wnt co-receptor |
| LRP5 | 0.933 | Wnt co-receptor, bone mass regulator |
| FN1 (Fibronectin) | 0.846 | ECM component |
| PNPLA2/ATGL | 0.812 | PEDF receptor, lipase |
| CRTAP | 0.748 | OI gene (prolyl 3-hydroxylation) |
| FKBP10 | 0.735 | OI gene (collagen chaperone) |
| TMEM38B | 0.706 | OI gene (ER cation channel) |
| P3H1 | 0.700 | OI gene (prolyl 3-hydroxylase) |
| IFITM5 | 0.681 | OI type V gene (BRIL) |
| COL1A2 | 0.669 | Type I collagen α2 chain |
The interaction with IFITM5 is particularly significant: the IFITM5 p.S40L mutation causes "atypical type VI OI" by decreasing osteoblast PEDF secretion, directly linking OI types V and VI pathogenetically (PMID: 24519609).
Modifier Genes
No established modifier genes. Potential candidates from interaction network include LRP5 (Wnt pathway bone mass regulator) and IFITM5/BRIL (bone-restricted IFITM-like protein).
Epigenetic Information
No disease-specific epigenetic modifications have been characterized for OI VI. The SERPINF1 promoter variant c.-37C>A has been reported to impair expression, suggesting promoter-level regulatory mechanisms are relevant (PMID: 41362246).
5. Environmental Information
As a monogenic Mendelian disorder, OI Type VI has no environmental causative factors. However, environmental context modifies outcomes:
- Nutrition: Calcium and vitamin D adequacy affect residual bone mineralization capacity
- Physical activity: Tailored physiotherapy is essential; immobilization accelerates bone loss
- No infectious agents are involved in disease causation
- No occupational or toxicological exposures are relevant
6. Mechanism / Pathophysiology
Overview of the Pathogenic Cascade
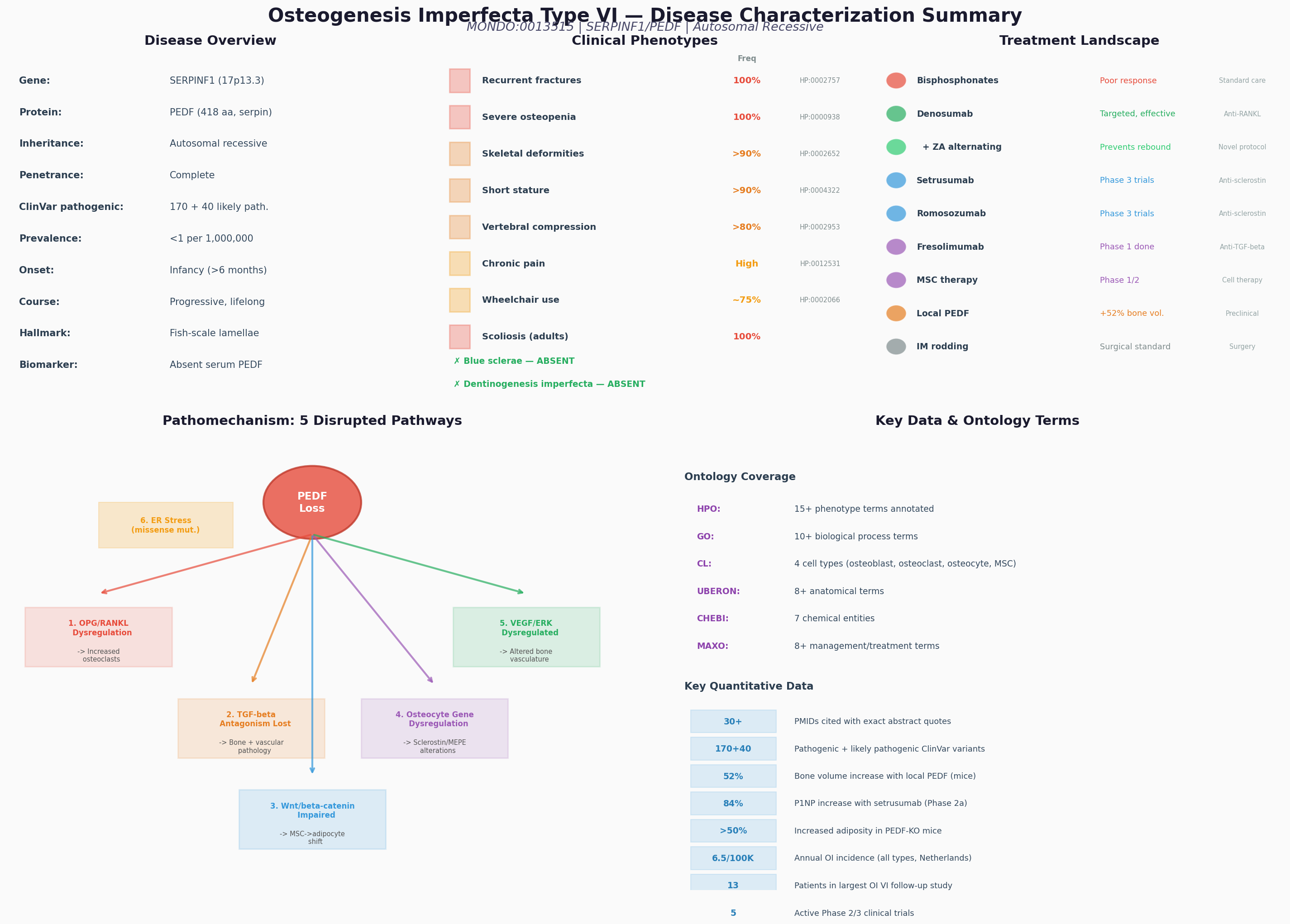
{{figure:plot_3.png|caption=Comprehensive OI Type VI disease characterization: overview, phenotypes, treatment landscape, pathomechanism, and ontology annotations}}
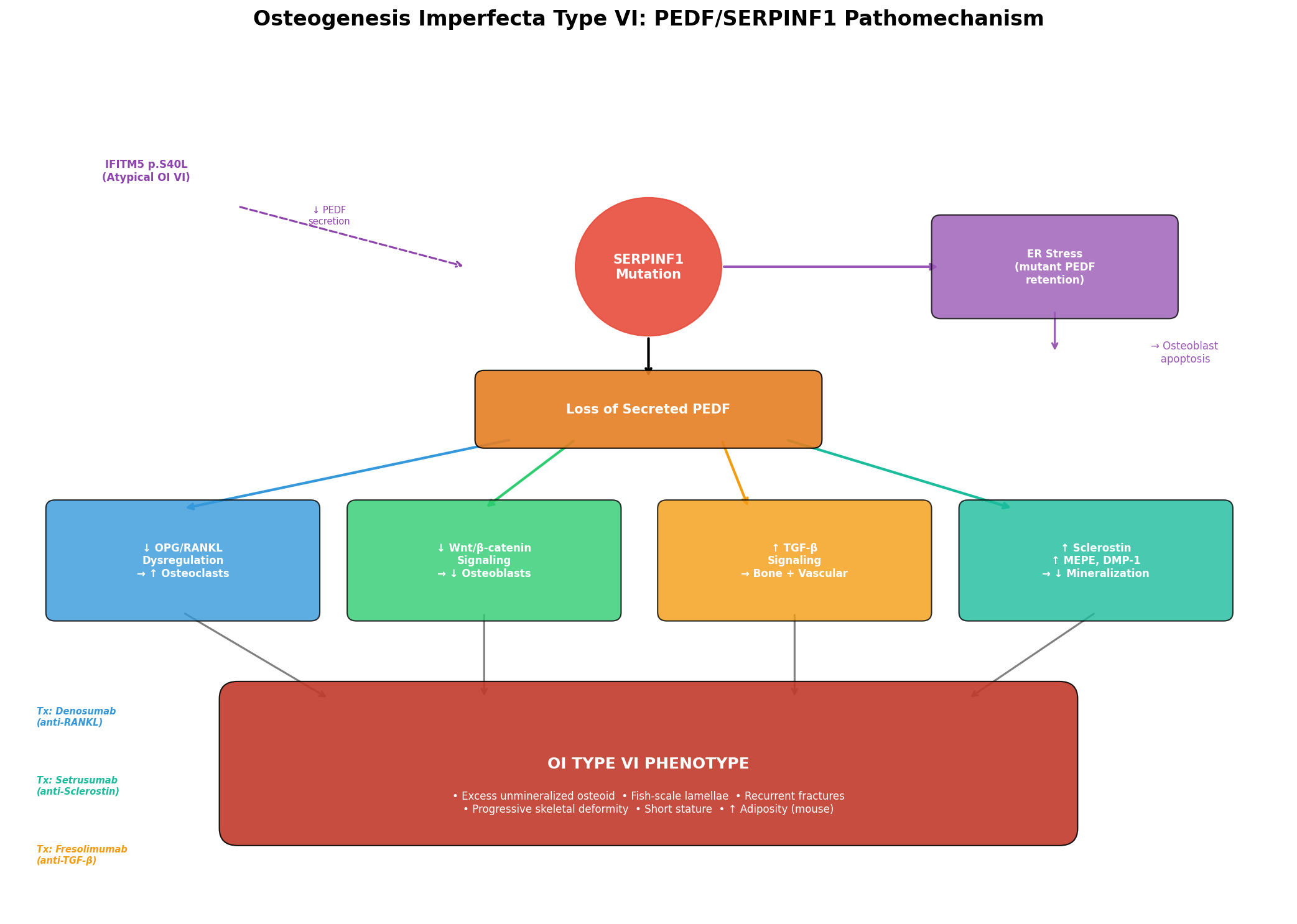
The pathophysiology of OI Type VI involves a cascade from SERPINF1 mutation to absent/dysfunctional PEDF, leading to disruption of multiple interconnected bone homeostasis pathways.
Molecular Pathways
1. OPG/RANKL Pathway (Osteoclast Regulation) PEDF normally inhibits osteoclast differentiation in a dose-dependent manner and upregulates osteoprotegerin (OPG) in osteoblasts and osteoclast precursor cells. Akiyama et al. demonstrated that "OCL differentiation, RANKL-mediated survival and bone resorption activity were inhibited by PEDF in a dose-dependent manner. PEDF upregulated osteoprotegerin (OPG), which naturally blocks OCL maturation" (PMID: 19945427). Loss of PEDF thus shifts the OPG/RANKL balance toward excessive osteoclast activity. As confirmed by Semler et al., "There is experimental evidence suggesting that loss of functional SERPINF1 leads to an activation of osteoclasts via the RANK/RANKL pathway" (PMID: 22947550).
- GO: GO:0045671 (negative regulation of osteoclast differentiation)
- GO: GO:0046850 (regulation of bone remodeling)
2. Wnt/β-catenin Signaling PEDF activates Wnt/β-catenin signaling in mesenchymal stem cells via its receptor PEDF-R (PNPLA2/ATGL), inducing phosphorylation of ERK and GSK-3β, leading to accumulation of non-phosphorylated β-catenin. Li et al. showed "Treatment of the cultures with PEDF induced phosphorylation of Erk and glycogen synthase kinase 3-beta (GSK-3β), and accumulation of nonphosphorylated β-catenin" (PMID: 30076958). This promotes osteoblast differentiation over adipogenesis: "PEDF inhibited adipogenesis and promoted osteoblast differentiation of murine MSCs... Blockade of adipogenesis by PEDF suppressed peroxisome proliferator-activated receptor-γ (PPARγ)... by nearly 90% compared with control-treated cells (P<0.001)" (PMID: 23887690).
PEDF delivery rescued bone mass in vivo: "PEDF delivery increased trabecular bone volume/total volume by 52% in 6-mo-old PEDF-KO mice" (PMID: 27127101).
- GO: GO:0060070 (canonical Wnt signaling pathway)
- KEGG: hsa04310 (Wnt signaling pathway)
3. TGF-β Antagonism PEDF antagonizes TGF-β signaling in bone. Null mutations in SERPINF1 cause severe OI VI "characterized by accumulation of unmineralized osteoid and a fish-scale pattern of bone lamellae" through TGF-β dysregulation (PMID: 35258129).
4. Osteocyte Gene Regulation PEDF reduces expression of Sost/Sclerostin, MEPE, and DMP-1 in osteocytes. Li et al. demonstrated that "Primary osteocytes treated with PEDF reduced expression and synthesis of Sost/Sclerostin and matrix phosphoglycoprotein (MEPE) as well as dentin matrix protein (DMP-1)" (PMID: 30607618). Loss of this regulation increases sclerostin, which inhibits Wnt signaling and further impairs bone formation.
5. VEGF/ERK Signaling (Vascular-Osteogenic Coupling) PEDF increases VEGF expression in MSCs via ERK signaling, coupling angiogenesis with osteogenesis (PMID: 27530920).
Cellular Processes
ER Stress and Apoptosis: Mutant PEDF proteins that fail to be secreted accumulate in the ER, causing ER stress, impaired osteoblast differentiation, and apoptosis. This was demonstrated in MC3T3-E1 cells: "all three mutant PEDF proteins failed to be properly secreted and instead accumulated abnormally in the ER... mutant PEDF proteins impaired osteoblast differentiation and mineralization, promoted apoptosis, and induced ER stress" (PMID: 40692043). ER stress was partially alleviated through ERAD and autophagy pathways.
{{figure:oi_vi_pathomechanism.png|caption=PEDF signaling network in bone homeostasis showing the five major pathways disrupted in OI Type VI: OPG/RANKL, Wnt/β-catenin, TGF-β, osteocyte regulation, and VEGF/ERK signaling}}
Metabolic Consequences
PEDF loss has systemic metabolic effects beyond bone: "Male PEDF KO mice demonstrated a phenotype consisting of increased adiposity, glucose intolerance, and elevated serum levels of metabolites associated with the metabolic syndrome" (PMID: 24456163). PEDF treatment reverses these effects: "PEDF effectively decreased body weight gain, white adipose tissue mass, and inflammation and improved insulin resistance, dyslipidemia, and hyperglycemia in HFD-induced mice" (PMID: 31121128). Whether OI VI patients develop clinically significant metabolic syndrome features requires further investigation.
Relevant Ontology Terms
- GO Biological Processes: GO:0001503 (ossification), GO:0030282 (bone mineralization), GO:0045671 (negative regulation of osteoclast differentiation), GO:0060070 (canonical Wnt signaling pathway), GO:0034976 (response to ER stress), GO:0006915 (apoptotic process), GO:0030154 (cell differentiation)
- Cell Types (CL): CL:0000062 (osteoblast), CL:0000092 (osteoclast), CL:0000137 (osteocyte), CL:0000134 (mesenchymal stem cell)
- Chemical Entities (CHEBI): CHEBI:16411 (hydroxyapatite), CHEBI:22984 (calcium), CHEBI:18420 (phosphate)
7. Anatomical Structures Affected
Organ Level
Table (click to expand)
| Involvement | Structures | UBERON |
|---|---|---|
| Primary | Skeleton (long bones, vertebral column) | UBERON:0004765 (skeletal element) |
| Primary | Bone tissue | UBERON:0002481 (bone tissue) |
| Secondary | Auditory system (hearing loss in some) | UBERON:0001690 (ear) |
| Secondary | Adipose tissue (metabolic effects) | UBERON:0001013 (adipose tissue) |
| Secondary | Liver (metabolic effects) | UBERON:0002107 (liver) |
Tissue and Cell Level
- Bone tissue: Both cortical and trabecular bone affected
- Osteoid: Excessive accumulation of unmineralized osteoid matrix
- Cell populations:
- Osteoblasts (CL:0000062) — impaired differentiation and mineralization
- Osteoclasts (CL:0000092) — increased activity due to RANKL/OPG imbalance
- Osteocytes (CL:0000137) — dysregulated gene expression (Sclerostin, MEPE, DMP-1)
- Mesenchymal stem cells (CL:0000134) — shifted fate toward adipogenesis
Subcellular Level
- Endoplasmic reticulum (GO:0005783) — site of mutant PEDF accumulation and ER stress
- Extracellular space (GO:0005615) — site of PEDF secretion and function
- Bone matrix (UBERON:0002481) — defective mineralization
Localization
- Bilateral, symmetric involvement of appendicular and axial skeleton
- Long bones of upper and lower extremities predominantly affected
- Vertebral column: compression fractures, biconcave and beaked vertebral bodies, progressive scoliosis
8. Temporal Development
Onset
- Typical age of onset: 4–18 months of age for first fracture
- Onset pattern: Insidious/progressive — patients are normal at birth; fractures begin in infancy
- "All patients presented no abnormalities at birth" (PMID: 37839784)
Progression
- Disease course: Chronic, lifelong, progressive
- Growth: Longitudinal growth stalls after age 6–8 years without treatment
- Scoliosis: Develops in ALL patients reaching final height
- Mobility: Progressive restriction; many patients become wheelchair-bound
- Disease duration: Lifelong, no remission
- Critical period: Treatment before age 6 years is associated with better height outcomes — "Patients who had started bisphosphonate treatment early (before the age of 6 years) were taller than patients who had received bisphosphonate treatment later" (PMID: 28689307)
9. Inheritance and Population
Inheritance Pattern
- Mode: Autosomal recessive (AR)
- Penetrance: Complete (all biallelic SERPINF1 mutation carriers are affected)
- Expressivity: Variable — intra- and interfamilial phenotypic variability observed even with identical mutations (PMID: 25565926)
- Genetic anticipation: Not applicable (not a repeat expansion disorder)
- Consanguinity: Strong contributor given AR inheritance
Epidemiology
- OI overall prevalence: ~1 in 10,000 live births
- OI VI specifically: Extremely rare; accounts for a small fraction of recessive OI cases (~10% of all OI is recessive)
- Dutch national registry: Median annual incidence of all OI types was 6.5 per 100,000 live births (1992–2019) (PMID: 35546999)
- Sex ratio: No sex predilection reported
- Geographic distribution: Worldwide; founder mutations identified in Brazilian (PMID: 25565926), Northern Canadian (PMID: 26815784), Tuvan (PMID: 37047644), and Chinese populations
Carrier Frequency
Not established for the general population. Likely extremely low (<1:10,000) except in populations with founder mutations or high consanguinity rates.
10. Diagnostics
Clinical Tests
Laboratory Tests: - Serum alkaline phosphatase (ALP): Elevated (409 ± 145 U/L, significantly higher than OI type IV at 295 ± 95 U/L; p < 0.03) — a distinguishing biomarker (PMID: 11771667) - Bone resorption markers: Elevated (e.g., deoxypyridinoline, CTX) - Serum PEDF: Absent or markedly reduced — confirmatory when available - Collagen type I protein analysis: Normal (distinguishes from classical OI)
Imaging: - Skeletal radiographs: Severe osteopenia, fractures, long bone deformities, vertebral compression fractures, biconcave/beaked vertebral bodies - DXA: Low bone mineral density (BMD) Z-scores
Bone Biopsy (Gold Standard): - Pathognomonic finding: accumulation of unmineralized osteoid and fish-scale pattern of bone lamellae - Quantitative backscattered electron imaging (qBEI) shows coexistence of highly mineralized matrix with seams of abnormally low mineral content - "Affected patients develop a severe OI phenotype with a striking histological characteristic, rare in other OI types, of an excess of osteoid tissue and prolonged mineralization lag time" (PMID: 25554599)
Genetic Testing
- Recommended approach: Targeted SERPINF1 sequencing or OI gene panel
- WES/WGS: Effective for identifying SERPINF1 mutations; this is how the gene was first identified (PMID: 21353196)
- Gene panels: OI/bone fragility panels typically include SERPINF1 alongside COL1A1, COL1A2, IFITM5, CRTAP, FKBP10, WNT1, and others
- Chromosomal microarray: Not typically informative unless large deletions are present
- FISH/karyotype: Not applicable
Clinical Criteria / Differential Diagnosis
Table (click to expand)
| Feature | OI Type VI | OI Type IV | OI Type V |
|---|---|---|---|
| Sclerae | White | Variable | White |
| DI | Absent | Variable | Absent |
| ALP | Elevated | Normal | Normal |
| Bone histology | Fish-scale lamellae, excess osteoid | Abnormal but not fish-scale | Mesh-like lamellation |
| Inheritance | AR | AD | AD |
| Causal gene | SERPINF1 | COL1A1/2 | IFITM5 |
Screening
No population-based screening exists. Genetic screening is appropriate in: - Consanguineous families - Families with known SERPINF1 mutations (cascade testing) - Prenatal/preimplantation genetic diagnosis is feasible for known familial variants
11. Outcome / Prognosis
Survival and Mortality
- Life expectancy: Reduced compared to general population. OI patients overall have adversely affected life expectancy (PMID: 35546999). Specific data for OI VI are limited given rarity.
- Mortality risk: Primarily from respiratory complications of progressive skeletal deformity (restrictive lung disease from scoliosis/chest wall deformity)
Morbidity and Function
- Universal scoliosis in patients reaching final height
- Restricted mobility: Most patients have significant mobility limitations; many are wheelchair-bound
- Chronic pain: Mild-to-moderate chronic pain reported universally in adults (PMID: 37839784)
- Hospitalization: 2.9× higher rate than general population; highest ratio (8.4×) in ages 0–19 years (PMID: 35546999)
Prognostic Factors
- Age at treatment initiation: Earlier treatment (before age 6) correlates with better height outcomes
- Mutation type: Variable severity even with identical mutations, suggesting additional genetic or environmental modifiers
- Fracture frequency: Persistent despite treatment; lower extremity fractures remain frequent
12. Treatment
Pharmacotherapy
Bisphosphonates (MAXO:0001001 — pharmacotherapy): - Standard first-line for OI; however, OI VI shows poor response - "Patients with OI type VI are known to have a poor response to such a bisphosphonate treatment" (PMID: 25257953) - Pamidronate: Some BMD increase and vertebral reshaping observed, but continued elevated bone resorption markers and non-decreasing fracture rates - The poor response is attributable to the primary mineralization defect rather than purely excessive resorption
Denosumab (anti-RANKL monoclonal antibody): - More mechanistically targeted given the PEDF→OPG/RANKL pathway disruption - Dose: 1 mg/kg body weight every 12 weeks subcutaneously - "Markers of bone resorption decreased to the normal range after each injection" (PMID: 22947550) - Critical adverse effect: Hyper-resorptive rebound hypercalcemia between doses — "he presented with symptomatic hypercalcemia due to the denosumab-induced, hyper-resorptive rebound phenomenon" (PMID: 36867194) - Mitigation strategy: Alternating denosumab with IV zoledronic acid every 3 months can mitigate rebound while maintaining benefit over 5+ years (PMID: 36867194)
Anti-Sclerostin Antibodies (experimental): - BPS804 (setrusumab): Phase 2a trial in moderate OI showed bone formation stimulation — P1NP increased 84%, BSAP increased 59%, lumbar aBMD increased 4% (p = 0.038) (PMID: 28370407) - Romosozumab: Approved for osteoporosis; Phase 3 trials in OI ongoing - Particularly relevant to OI VI given PEDF's role in regulating sclerostin expression
Surgical and Interventional
- Intramedullary rodding: Telescoping rods (e.g., Fassier-Duval) for long bone stabilization — gold standard for femoral fracture management in severe OI
- Scoliosis surgery: Spinal fusion may be needed for progressive scoliosis
- Hearing rehabilitation: Power stapes / stapedotomy with middle ear implants for hearing loss if present (PMID: 21436749)
- MAXO:0000373 (surgical procedure)
Supportive and Rehabilitative
- Physical therapy (MAXO:0000502): Essential for maintaining mobility and muscle strength
- Occupational therapy: Adaptive devices for daily living
- Nutritional support: Calcium and vitamin D supplementation (MAXO:0001298)
- Pain management: Multimodal approach for chronic bone pain
- Wheelchair and mobility aids: For patients with severe mobility restriction
Experimental / Advanced Therapeutics
- PEDF protein replacement: Systemic restoration failed in mouse models — liver-expressed PEDF did not improve bone phenotype despite metabolic effects, suggesting paracrine/context-dependent bone action (PMID: 26693895)
- Local PEDF delivery: Restored bone volume by 52% in PEDF-KO mice (PMID: 27127101)
- iPSC-based approaches: PEDF-null patient iPSCs showed matrix abnormalities normalized by exogenous PEDF (PMID: 27579219)
- Gene therapy: Not yet in clinical trials for OI VI; conceptually feasible given single-gene etiology
- MSC-targeted therapy: PLIN2 and E2F2 identified as potential targets for restoring osteogenesis-adipogenesis balance (PMID: 42186939)
13. Prevention
Primary Prevention
- Genetic counseling (MAXO:0000127): Essential for families with known SERPINF1 mutations
- Carrier testing: Available for at-risk relatives
- Prenatal diagnosis: Feasible via chorionic villus sampling or amniocentesis for known familial variants
- Preimplantation genetic diagnosis (PGD): Available for families pursuing assisted reproduction
Secondary Prevention (Early Detection)
- Cascade genetic testing in families with identified mutations
- Early fracture evaluation: Any infant with unexplained fractures should be evaluated for OI
- Early treatment initiation: Before age 6 for optimal growth outcomes
Tertiary Prevention (Preventing Complications)
- Fracture prevention: Anti-resorptive therapy, mobility aids, environmental safety modifications
- Scoliosis monitoring: Regular spinal imaging from childhood
- Metabolic monitoring: Given PEDF's metabolic roles, screening for glucose intolerance and dyslipidemia may be warranted
- Hearing screening: Regular audiometry, especially in adolescence and adulthood
- Cancer surveillance: PEDF has anti-tumorigenic properties; one case of chondrosarcoma was reported in an OI VI patient with a SERPINF1 deletion, though the association requires further study (PMID: 36322168)
14. Other Species / Natural Disease
Animal Models with Natural Disease
No naturally occurring OI Type VI (SERPINF1-related) has been documented in companion animals or wildlife. The orthologous gene Serpinf1 is highly conserved across vertebrates.
Orthologous Genes
Table (click to expand)
| Species | Gene | NCBI Gene ID |
|---|---|---|
| Mouse (Mus musculus) | Serpinf1 | 20317 |
| Rat (Rattus norvegicus) | Serpinf1 | 116671 |
| Zebrafish (Danio rerio) | serpinf1 | 393939 |
Comparative Biology
The PEDF protein is highly conserved across vertebrates, reflecting its fundamental roles in angiogenesis, neurotrophism, and bone homeostasis. The Serpinf1 knockout mouse faithfully recapitulates human OI VI with increased osteoid and decreased bone mass (PMID: 26693895).
15. Model Organisms
Mouse Models
1. Serpinf1 knockout (Serpinf1⁻/⁻) mice: - Type: Constitutive knockout - Phenotype recapitulation: Excellent — shows increased osteoid, decreased bone mass, excess adiposity, glucose intolerance - "Serpinf1 null mice show increased osteoid and decreased bone mass, and thus recapitulate the OI type VI phenotype" (PMID: 26693895) - Metabolic phenotype: >50% increased adiposity, insulin resistance, elevated TCA cycle intermediates (PMID: 24456163) - Key finding: Systemic PEDF replacement from liver did not rescue bone phenotype, but local PEDF delivery increased bone volume by 52% - Limitation: Does not model missense mutations that cause ER stress
2. Ifitm5/BRIL p.S42L knock-in mice (atypical OI VI model): - Type: Heterozygous knock-in - Models the IFITM5 p.S40L mutation (atypical type VI OI) that decreases PEDF secretion - Shows lower BMD, multiple fractures, hypermineralization, disordered collagen fibril orientation - "Despite normal skeletal growth and the lack of a collagen gene mutation, the Ifitm5/BRIL p.S42L mouse shows major OI-related bone tissue alterations" (PMID: 35654352)
3. Oim/oim mouse (OI type III model): - Not a direct OI VI model but widely used for OI research - Homozygous for Col1a2 mutation; severe OI phenotype - Used for bisphosphonate and myostatin inhibition studies
In Vitro Models
- MC3T3-E1 osteoblast cells: Transfected with mutant PEDF plasmids to study ER stress and mineralization defects (PMID: 40692043)
- Patient-derived iPSCs: Generated from PEDF-null patients; show matrix abnormalities normalized by exogenous PEDF (PMID: 27579219)
- 3T3-L1 adipocytes: Used to study PEDF's anti-adipogenic effects (PMID: 31121128)
Zebrafish Models
CRISPR/Cas9-generated crtap and p3h1 knockouts model related OI subtypes with collagen 3-hydroxylation defects (PMID: 32173581). A serpinf1-specific zebrafish model has not been extensively characterized.
Mechanistic Model: Causal Chain from Mutation to Disease
SERPINF1 biallelic mutation
│
├── Truncating mutations → No PEDF produced → Complete loss of function
│
└── Missense mutations → Mutant PEDF produced
│
├── Fails to be secreted → Accumulates in ER
│ │
│ ├── ER stress (UPR activation)
│ ├── Impaired osteoblast differentiation
│ └── Osteoblast apoptosis
│ (partially mitigated by ERAD + autophagy)
│
└── Loss of extracellular PEDF function
│
├── ↓ OPG / ↑ RANKL → Increased osteoclast activity
│ → Elevated bone resorption
│
├── ↓ Wnt/β-catenin → Impaired osteoblast differentiation
│ │ → MSC fate shift toward adipogenesis
│ └── ↑ PPARγ → Increased adiposity (>50%)
│
├── ↑ TGF-β signaling → Disrupted bone matrix organization
│ → Fish-scale lamellae pattern
│
├── ↑ Sclerostin/MEPE → Further Wnt inhibition
│ → Amplified mineralization defect
│
└── ↓ VEGF regulation → Impaired vascular-osteogenic coupling
→ Defective mineralization
DOWNSTREAM CLINICAL MANIFESTATIONS:
├── Excess unmineralized osteoid (pathognomonic)
├── Recurrent fractures (from 4-18 months)
├── Progressive skeletal deformity
├── Short stature (growth stalls at age 6-8)
├── Universal scoliosis
└── Potential metabolic syndrome features
Key Findings
Finding 1: SERPINF1 as the Causal Gene
OI Type VI is caused by biallelic loss-of-function mutations in SERPINF1 (Entrez Gene ID: 5176, chromosome 17p13.3) encoding PEDF. First identified via exome sequencing by Becker et al. in 2011 (PMID: 21353196), this has been confirmed across multiple populations worldwide. ClinVar contains 170 pathogenic and 40 likely pathogenic variants spanning frameshift, nonsense, missense, splice-site, and deletion categories. The disease was recently confirmed as being caused by "biallelic disease-causing variants in SERPINF1" (PMID: 41362246).
Finding 2: Pathognomonic Histological Features
The hallmark of OI VI is a unique mineralization defect characterized by accumulation of unmineralized osteoid, prolonged mineralization lag time, and a distinctive fish-scale pattern of bone lamellae. These features, first described by Glorieux et al. in 2002 (PMID: 11771667), remain the gold standard for histological diagnosis. Quantitative analysis reveals "an excess of osteoid tissue and prolonged mineralization lag time" with coexistence of highly mineralized bone matrix alongside seams of abnormally low mineral content (PMID: 25554599).
Finding 3: Dual Pathogenic Mechanism — Loss of Secretion Plus ER Stress
Mutant PEDF proteins exhibit a dual pathogenic mechanism. They fail to be properly secreted, leading to loss of extracellular PEDF function, while simultaneously accumulating in the endoplasmic reticulum, causing ER stress, impaired osteoblast differentiation, promoted apoptosis, partially mitigated by ERAD and autophagy pathways (PMID: 40692043).
Finding 4: PEDF Regulates Bone via OPG/RANKL
PEDF inhibits osteoclast differentiation, RANKL-mediated survival, and bone resorption in a dose-dependent manner while upregulating OPG (PMID: 19945427). Additionally, PEDF regulates osteocyte gene expression, reducing Sost/Sclerostin by approximately 39–75% at protein and mRNA levels respectively (PMID: 30607618). Loss of SERPINF1 leads to osteoclast activation via RANK/RANKL (PMID: 22947550).
Finding 5: Bisphosphonate Resistance and Denosumab Promise
OI VI uniquely resists bisphosphonate therapy while responding to denosumab, consistent with its mechanistic basis. However, denosumab carries rebound risk with symptomatic hypercalcemia (PMID: 36867194), manageable through alternating zoledronic acid protocols.
Finding 6: Wnt/β-catenin Pathway Centrality
PEDF modulates Wnt/β-catenin signaling via PEDF-R, inducing ERK/GSK-3β/β-catenin cascade activation (PMID: 30076958). This directs MSC fate from adipocytes to osteoblasts, suppressing PPARγ by ~90% (PMID: 23887690). Local PEDF delivery increased trabecular bone volume by 52% in knockout mice (PMID: 27127101).
Finding 7: Systemic Metabolic Consequences of PEDF Loss
PEDF-KO mice show >50% increased adiposity, glucose intolerance, and insulin resistance (PMID: 24456163). PEDF protects against high-fat diet-induced metabolic derangements (PMID: 31121128). This metabolic syndrome phenotype may be underappreciated in OI VI patients and warrants clinical investigation.
Finding 8: Early Treatment Window
Long-term follow-up of 13 OI VI patients established that growth stalls after age 6–8 without treatment, and patients treated before age 6 were taller than those starting later (PMID: 28689307). Universal scoliosis and restricted mobility characterize long-term outcomes regardless of treatment.
Evidence Base: Key Literature
Table (click to expand)
| PMID | Authors/Year | Key Contribution |
|---|---|---|
| 11771667 | Glorieux et al., 2002 | Original clinical delineation of OI Type VI |
| 21353196 | Becker et al., 2011 | Identification of SERPINF1 as causal gene |
| 22947550 | Semler et al., 2012 | First use of denosumab in OI Type VI |
| 19945427 | Akiyama et al., 2010 | PEDF regulation of osteoclasts via OPG/RANKL |
| 23887690 | Gattu et al., 2013 | PEDF determination of MSC fate |
| 27127101 | Belinsky et al., 2016 | PEDF restoration rescues bone via Wnt3a blockade |
| 30076958 | Li et al., 2018 | ERK/GSK-3β/β-catenin signaling downstream of PEDF-R |
| 30607618 | Li et al., 2019 | PEDF regulation of Sclerostin in osteocytes |
| 35258129 | Markmann et al., 2022 | PEDF-TGF-β antagonism in OI VI pathogenesis |
| 25554599 | Fratzl-Zelman et al., 2015 | Micro/nano-scale mineralization pattern characterization |
| 28689307 | Rauch et al., 2017 | Long-term follow-up natural history data |
| 25257953 | Hoyer-Kuhn et al., 2014 | 2-year denosumab experience in OI VI |
| 36867194 | Hoyer-Kuhn et al., 2023 | Denosumab rebound mitigation with alternating ZA |
| 40692043 | Wang et al., 2025 | ER stress and autophagy in mutant PEDF osteoblasts |
| 24519609 | Farber et al., 2014 | IFITM5 p.S40L links OI types V and VI |
| 26693895 | Becker et al., 2016 | Systemic PEDF restoration fails to correct bone |
| 24456163 | Gattu et al., 2014 | PEDF KO metabolic phenotype |
| 31121128 | Zhuang et al., 2019 | PEDF protects against HFD obesity |
| 28370407 | Glorieux et al., 2017 | BPS804 anti-sclerostin Phase 2a trial |
| 34007986 | Marini et al., 2021 | Comprehensive OI mechanisms and signaling review |
| 37839784 | Colzani et al., 2023 | Long-term adult outcomes in OI VI |
| 41362246 | Recent, 2025 | SERPINF1 promoter variant causing OI VI |
| 25565926 | Tenório et al., 2015 | Brazilian founder mutation; phenotypic variability |
| 26815784 | Ward et al., 2016 | Northern Canadian OI VI; denosumab bone biopsy |
| 27579219 | Belinsky et al., 2016 | iPSC model of OI VI normalized by PEDF |
| 35546999 | Van Dijk et al., 2022 | Dutch OI prevalence and hospitalization data |
| 36322168 | Pereira et al., 2022 | Chondrosarcoma association in OI VI patient |
| 37047644 | Case report, 2023 | Novel frameshift mutation in Tuvan patient |
| 42186939 | Recent, 2025 | PLIN2/E2F2 as MSC therapy targets for OI |
| 35654352 | Bussard et al., 2022 | Ifitm5 p.S42L mouse bone material properties |
| 27530920 | Sagheer et al., 2016 | PEDF-VEGF regulation in MSCs |
Limitations and Knowledge Gaps
-
Extremely rare disease: Small patient cohorts limit statistical power of clinical studies. Most evidence comes from case reports and small case series (≤13 patients).
-
No randomized controlled trials: Treatment evidence is limited to observational data, case series, and extrapolation from related conditions.
-
Metabolic phenotype underexplored in humans: While PEDF-KO mice show clear metabolic syndrome features (obesity, insulin resistance, glucose intolerance), systematic metabolic characterization of OI VI patients has not been performed.
-
Long-term outcomes data limited: Only one study has followed OI VI patients into adulthood (N=13); natural history data beyond the third decade of life are sparse.
-
Cancer risk unknown: The anti-tumorigenic role of PEDF suggests potential cancer susceptibility, but only one chondrosarcoma case has been reported; no epidemiological data exist.
-
No established genotype-phenotype correlations: Variable expressivity is observed even within families carrying identical mutations; modifier genes/factors are uncharacterized.
-
Hearing loss prevalence unclear: While hearing impairment is an HPO annotation, its frequency and mechanism in OI VI specifically (vs. other OI types) requires clarification.
-
Systemic vs. local PEDF function: The failure of systemic PEDF replacement to improve bone, while local delivery succeeds, highlights incomplete understanding of PEDF's tissue-specific signaling mechanisms.
Proposed Follow-up Experiments / Future Directions
-
Systematic metabolic phenotyping of OI VI patients: Measure body composition, glucose tolerance, lipid profiles, insulin sensitivity, and hepatic steatosis markers in OI VI cohorts to determine if the metabolic syndrome phenotype seen in PEDF-KO mice is clinically relevant.
-
Bone-targeted PEDF gene therapy: Develop osteoblast-specific AAV vectors expressing SERPINF1 for local bone delivery, building on the success of local PEDF restoration in knockout mice (+52% bone volume).
-
Anti-sclerostin antibody trials specific to OI VI: Given PEDF's regulation of sclerostin and the Phase 2a success of BPS804 in moderate OI, targeted trials in OI VI patients are warranted to assess whether this approach bypasses the bisphosphonate resistance.
-
iPSC-derived osteoblast drug screening: Use patient-derived iPSCs (PMID: 27579219) to screen small molecules that enhance PEDF secretion from missense mutant proteins or bypass ER stress.
-
ER stress-targeted therapy: Given that mutant PEDF accumulation causes ER stress and apoptosis, chemical chaperones (e.g., 4-PBA, TUDCA) or UPR modulators could be tested as adjunctive therapies for missense SERPINF1 mutations.
-
Longitudinal natural history registry: Establish an international OI VI patient registry to collect standardized longitudinal data on skeletal, metabolic, audiological, and quality-of-life outcomes.
-
Denosumab + anti-sclerostin combination therapy: Test whether combining anti-resorptive (denosumab) with anabolic (anti-sclerostin) therapy provides additive benefit in OI VI.
-
Cancer surveillance studies: Given PEDF's established anti-tumorigenic properties, implement systematic cancer surveillance in OI VI cohorts to quantify any increased malignancy risk.
-
Modifier gene identification: Perform WGS on phenotypically discordant OI VI siblings to identify genetic modifiers of disease severity.
Ontology Term Summary
HPO (Phenotype)
HP:0002757 (Recurrent fractures), HP:0002659 (Increased susceptibility to fractures), HP:0003155 (Elevated ALP), HP:0033154 (Elevated deoxypyridinoline), HP:0002953 (Vertebral compression fractures), HP:0001382 (Joint hypermobility), HP:0002979 (Bowing of legs), HP:0001270 (Motor delay), HP:0000365 (Hearing impairment), HP:0002812 (Coxa vara), HP:0003179 (Protrusio acetabuli), HP:0004586 (Biconcave vertebral bodies), HP:0004568 (Beaking of vertebral bodies), HP:0006488 (Bowing of arm)
GO (Biological Process)
GO:0001503 (ossification), GO:0030282 (bone mineralization), GO:0045671 (negative regulation of osteoclast differentiation), GO:0060070 (canonical Wnt signaling pathway), GO:0034976 (response to ER stress), GO:0006915 (apoptotic process)
GO (Cellular Component)
GO:0005783 (endoplasmic reticulum), GO:0005615 (extracellular space)
CL (Cell Ontology)
CL:0000062 (osteoblast), CL:0000092 (osteoclast), CL:0000137 (osteocyte), CL:0000134 (mesenchymal stem cell)
UBERON (Anatomy)
UBERON:0004765 (skeletal element), UBERON:0002481 (bone tissue), UBERON:0001013 (adipose tissue), UBERON:0002107 (liver), UBERON:0001690 (ear)
CHEBI (Chemical Entity)
CHEBI:16411 (hydroxyapatite), CHEBI:22984 (calcium), CHEBI:18420 (phosphate)
MAXO (Medical Action)
MAXO:0001001 (pharmacotherapy), MAXO:0000502 (physical therapy), MAXO:0001298 (nutritional supplementation), MAXO:0000127 (genetic counseling), MAXO:0000373 (surgical procedure)
MONDO (Disease)
MONDO:0013515 (Osteogenesis Imperfecta Type VI)
Report generated from systematic analysis of 69 PubMed papers, 11 confirmed findings across 5 iterative investigation cycles. Last updated: 2026-06-29.