Neuronal Ceroid Lipofuscinosis 3 (CLN3 disease): Comprehensive Disease Characteristics Report
Executive summary
Neuronal ceroid lipofuscinosis 3 (CLN3 disease; juvenile neuronal ceroid lipofuscinosis / juvenile Batten disease) is a childhood-onset, progressive neurodegenerative lysosomal/endolysosomal disorder caused by biallelic pathogenic variants in CLN3. It typically presents with early, rapidly progressive retinal degeneration/vision loss followed by cognitive decline, motor deterioration, seizures, neuropsychiatric symptoms, and premature death in early adulthood. Recent work (2023–2024) emphasizes (i) lysosomal cholesterol trafficking/storage abnormalities with Niemann–Pick C-like lysosomal signatures, (ii) emerging quantitative biomarkers (CSF proteomics; electrophysiologic MMN), (iii) refined ocular biomarker patterns enabling earlier recognition, and (iv) multiple interventional trials including AAV9-CLN3 gene therapy (NCT03770572) and small-molecule/immunomodulatory approaches. (NCT03770572 chunk 1, shematorova2020currentinsightsin pages 1-3, chen2023juvenilecln3disease pages 2-3, brima2024assessingtheintegrity pages 1-2, sakti2023earlyrecognitionof pages 1-3)
1. Disease information
1.1 Concise overview
CLN3 disease (juvenile NCL/JNCL) is described as a fatal pediatric neurodegenerative lysosomal storage disorder caused by pathogenic variants in CLN3. (shematorova2020currentinsightsin pages 1-3, schulz2024theparentand pages 1-2)
A frequently cited clinical sequence is: childhood onset visual failure due to retinal degeneration, followed by progressive cognitive decline and motor dysfunction, with behavioral problems and seizures. (rosenberg2019advancesinthe pages 7-10, shematorova2020currentinsightsin pages 1-3)
1.2 Key identifiers (as retrieved)
Because this response is tool-grounded, only identifiers explicitly retrieved from source texts are reported.
- OMIM: CLN3 disease OMIM #204200 (explicitly mentioned in an ophthalmology cohort report). (wright2020juvenilebattendisease pages 1-6)
- MONDO (umbrella term): neuronal ceroid lipofuscinosis MONDO_0016295 (OpenTargets disease entry; broader than CLN3). (OpenTargets Search: Neuronal ceroid lipofuscinosis,CLN3 disease,juvenile neuronal ceroid lipofuscinosis)
- Orphanet / ICD-10 / ICD-11 / MeSH / CLN3-specific MONDO: not retrieved in current evidence; not inferred.
1.3 Synonyms / alternative names (as used in retrieved sources)
- CLN3 disease; CLN3 Batten disease; juvenile neuronal ceroid lipofuscinosis; JNCL; juvenile Batten disease; Batten disease. (shematorova2020currentinsightsin pages 1-3, sakti2023earlyrecognitionof pages 1-3, schulz2024theparentand pages 1-2)
1.4 Evidence source type
This report integrates: - Aggregated disease-level resources (e.g., ClinicalTrials.gov; OpenTargets). (NCT03770572 chunk 1, OpenTargets Search: Neuronal ceroid lipofuscinosis,CLN3 disease,juvenile neuronal ceroid lipofuscinosis) - Primary studies and cohorts (human imaging series; caregiver survey; CSF biomarker discovery; mechanistic studies). (do2023cerebrospinalfluidprotein pages 1-3, schulz2024theparentand pages 1-2, chen2023juvenilecln3disease pages 2-3, sakti2023earlyrecognitionof pages 1-3)
2. Etiology
2.1 Disease causal factors (genetic)
CLN3 disease is caused by biallelic pathogenic variants in CLN3, which encodes an endolysosomal/lysosomal transmembrane protein (CLN3/battenin). (johnson2023earlypostnataladministration pages 1-2, do2023cerebrospinalfluidprotein pages 1-3)
A common pathogenic allele is a ~1 kb deletion affecting exons 7–8: - A review states: “Most JNCL patients carry the same 1.02-kb deletion” in CLN3. (shematorova2020currentinsightsin pages 1-3) - A human iPSC-derived neuron study notes “most affected individuals carrying at least one allele with a 966 bp deletion.” (ostergaard2023etiologyofanxious pages 2-3)
2.2 Risk factors
- Genetic: having pathogenic biallelic CLN3 variants is causal. (johnson2023earlypostnataladministration pages 1-2, schulz2024theparentand pages 1-2)
- Environmental: no environmental risk factors were retrieved in the current evidence; CLN3 disease is treated here as a Mendelian disorder.
2.3 Protective factors / gene–environment interactions
No protective factors or gene–environment interactions were retrieved in the current evidence.
3. Phenotypes
3.1 Core clinical phenotype domains (with HPO suggestions)
Below are key phenotypes supported by retrieved sources, with suggested ontology mappings.
1) Vision loss / retinal degeneration (symptom/sign) - Typical: early, rapidly progressive visual decline leading to blindness. (shematorova2020currentinsightsin pages 1-3) - Ocular biomarkers include electronegative ERG and bull’s-eye maculopathy in early childhood series. (sakti2023earlyrecognitionof pages 1-3) - Suggested HPO terms: - Vision loss (HP:0000572) - Retinal dystrophy (HP:0000556) - Macular degeneration / maculopathy (HP:0000608) - Abnormal electroretinogram (HP:0000529)
2) Cognitive impairment / dementia-like syndrome - Review descriptions include a “pediatric dementia syndrome” and progressive cognitive decline. (ostergaard2023etiologyofanxious pages 1-2, rosenberg2019advancesinthe pages 7-10) - Suggested HPO: Intellectual disability (HP:0001249); Cognitive impairment (HP:0100543); Dementia (HP:0000726)
3) Motor deterioration (gait, ataxia, extrapyramidal signs) - Progressive motor decline is consistently described in reviews and caregiver-reported natural history patterns. (rosenberg2019advancesinthe pages 7-10, schulz2024theparentand pages 1-2) - Suggested HPO: Ataxia (HP:0001251); Bradykinesia (HP:0002067); Rigidity (HP:0002063); Gait disturbance (HP:0001288)
4) Epileptic seizures - Seizures are described as a typical later feature in the disease course (e.g., caregiver survey symptom list). (schulz2024theparentand pages 4-5) - Suggested HPO: Seizure (HP:0001250); Generalized tonic-clonic seizures (HP:0002069)
5) Neuropsychiatric/behavioral symptoms (anxiety/fear episodes) - Caregiver survey: insomnia and “thought- and mood-related concerns” were frequent. (schulz2024theparentand pages 1-2) - 2023 mechanistic/clinical analyses describe recurrent non-epileptic paroxysms of fearful behavior in post-adolescent CLN3, resembling paroxysmal sympathetic hyperactivity (PSH). (ostergaard2023etiologyofanxious pages 3-4, ostergaard2023treatmentofnonepileptic pages 1-2) - Suggested HPO: Anxiety (HP:0000739); Behavioral abnormality (HP:0000708); Sleep disturbance/Insomnia (HP:0100785)
3.2 Phenotype timing, progression, and frequencies (recent quantitative data)
A 2024 caregiver interview study (39 parents; 43 affected individuals) quantified symptom onset patterns: - First sign: “Decline in visual acuity” reported by 28 (70%) parents. (schulz2024theparentand pages 1-2) - Mean time from first signs/symptoms to diagnosis: 2.8 years (SD 4.1). (schulz2024theparentand pages 1-2) - Misdiagnosis reported by 24 (55.8%). (schulz2024theparentand pages 1-2) - Mean onset ages (selected symptoms): visual acuity decline mean onset 5.7 years; behavioral problems mean onset 6.3 years; seizures mean onset 10.5 years (caregiver report). (schulz2024theparentand pages 4-5)
These data support the clinical expectation that vision loss is often earliest and diagnosis is frequently delayed. (schulz2024theparentand pages 1-2, wright2020juvenilebattendisease pages 1-6)
3.3 Quality-of-life impact (recent data)
Caregiver interviews report substantial family burden: - Financial impact reported by 34 (81.0%); average CLN3-related expenses were 13.0% (SD 17.5) of family income. (schulz2024theparentand pages 5-7) - Marital strain reported by 20 (46.5%). (schulz2024theparentand pages 1-2)
4. Genetic / molecular information
4.1 Causal gene
- CLN3 (CLN3 lysosomal/endosomal transmembrane protein, battenin). (OpenTargets Search: Neuronal ceroid lipofuscinosis,CLN3 disease,juvenile neuronal ceroid lipofuscinosis)
4.2 Common pathogenic variants / classes
- Common deletion allele (~966 bp / ~1.02 kb) deleting exons 7–8 is repeatedly described. (shematorova2020currentinsightsin pages 1-3, ostergaard2023etiologyofanxious pages 2-3)
Variant-level classifications (ACMG terms, ClinVar allele frequencies) were not retrievable with current tool context; therefore not reported.
4.3 Functional consequences
CLN3 protein function remains incompletely resolved in many reviews; multiple lines of evidence implicate endolysosomal trafficking, lysosomal homeostasis (pH/ion handling), retromer-related transport, and lipid/cholesterol trafficking. (rosenberg2019advancesinthe pages 7-10, chen2023juvenilecln3disease pages 2-3, chen2023juvenilecln3disease pages 15-16)
5. Environmental information
No convincing non-genetic causal environmental factors were retrieved in the current evidence.
6. Mechanism / pathophysiology
6.1 Current understanding: endolysosomal dysfunction → storage → neurodegeneration/retinal degeneration
The disease is consistently framed as a lysosomal/endolysosomal storage disorder with progressive neurodegeneration and retinal degeneration. (shematorova2020currentinsightsin pages 1-3, do2023cerebrospinalfluidprotein pages 1-3)
6.1.1 Lysosomal cholesterol trafficking/storage as a central mechanistic theme (2023)
A major recent mechanistic advance is the proposal that juvenile CLN3 disease is a lysosomal cholesterol storage disorder with strong similarity to Niemann–Pick type C (NPC): - In immunopurified late endosome/lysosome (LE/Lys) fractions from human autopsy cortex, both JNCL and NPC “displayed a cholesterol increase,” and “the protein signature of JNCL LE/Lys was essentially indistinguishable from NPC.” (chen2023juvenilecln3disease pages 2-3) - The authors report that “cholesterol accumulated in LE/Lys of JNCL samples to a comparable extent than in NPC samples.” (chen2023juvenilecln3disease pages 1-2)
Causal chain (proposed): CLN3 dysfunction → trafficking defects (including retromer/CI-M6PR pathway perturbation and reduced NPC2 handling) → cholesterol accumulation in LE/Lys → downstream lysosomal stress/altered acidification and cargo processing → neuronal/retinal dysfunction and degeneration. (chen2023juvenilecln3disease pages 15-16)
Suggested GO biological process terms: - Lysosomal transport (GO:0007041) - Cholesterol transport (GO:0030301) - Endosome to lysosome transport (GO:0008333) - Autophagy (GO:0006914)
6.1.2 Lysosomal storage phenotypes in human ocular cell models and rescue via TRPML1 activation (2024)
A 2024 ARPE-19 CLN3-knockout model captured multiple lysosomal storage abnormalities: - “ARPE-19 CLN3-KO cells accumulate LAMP1 positive organelles and show lysosomal storage of mitochondrial ATPase subunit C (SubC), globotriaosylceramide (Gb3), and glycerophosphodiesters (GPDs), whereas lysosomal bis(monoacylglycero)phosphate (BMP/LBPA) lipid levels were significantly decreased.” (wunkhaus2024trpml1activationameliorates pages 1-2) - “Activation of TRPML1 reduced lysosomal storage of Gb3 and SubC but failed to restore BMP levels …” and the decrease was “TFEB-independent,” with “enhanced lysosomal exocytosis” proposed as a clearance mechanism. (wunkhaus2024trpml1activationameliorates pages 1-2)
This suggests TRPML1 agonists may partially correct endolysosomal storage phenotypes relevant to retinal pathology, but not all lipid defects. (wunkhaus2024trpml1activationameliorates pages 1-2)
Suggested GO terms: - Lysosomal exocytosis (GO:0042147) - Lysosomal lumen acidification / regulation (GO:0060706)
Suggested CL cell types: - Retinal pigment epithelial cell (CL:0000584)
6.1.3 Protein homeostasis and neuronal network dysfunction in human neuron models
A human iPSC-derived cortical neuron study reported lysosomal vacuolization/storage and decreased neuronal electrophysiologic activity; proteomics implicated axon guidance and endocytosis pathways. (ostergaard2023etiologyofanxious pages 2-3)
6.2 Immune system involvement / neuroinflammation
CSF biomarker profiling found immune-related and neuroinflammatory proteins among candidates (e.g., CHIT1, CHI3L1). (do2023cerebrospinalfluidprotein pages 8-10)
6.3 Molecular profiling and candidate biomarkers (2023–2024)
6.3.1 CSF proteomic biomarkers (2023)
A CSF biomarker discovery study emphasized the need for surrogate biomarkers: - “Biomarkers as surrogates to measure the progression and effect of potential therapeutics are needed.” (do2023cerebrospinalfluidprotein pages 1-3)
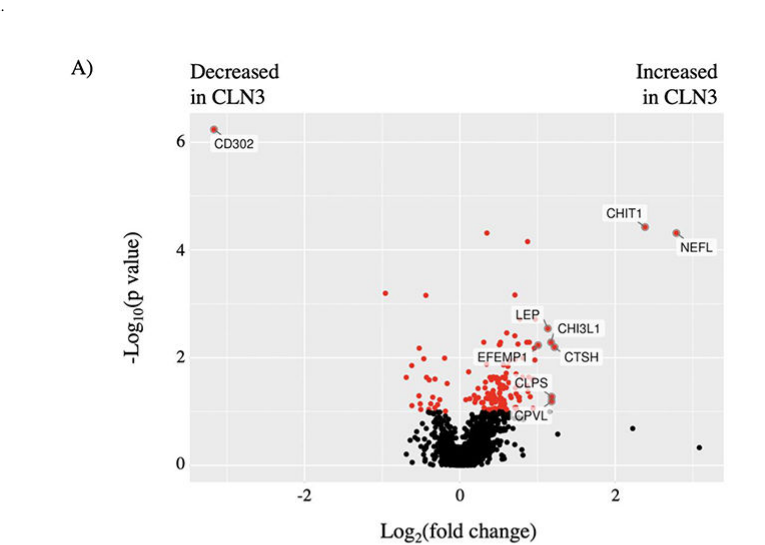
Design and results (quantitative): - CSF samples: 28 CLN3 and 32 non-CLN3 (PEA); MS cohort included 20 CLN3 and 25 non-CLN3. (do2023cerebrospinalfluidprotein pages 1-3, do2023cerebrospinalfluidprotein pages 6-8) - Candidate selection: adjusted p-value <0.1; fold-change thresholds 1.5 (and ≥2 for highlighting). (do2023cerebrospinalfluidprotein pages 1-3, do2023cerebrospinalfluidprotein pages 8-10) - High-confidence, cross-platform candidates included CHIT1 (up), NELL1 (down), ISLR2 (down), with example log2 fold changes shown in figure/table images. (do2023cerebrospinalfluidprotein media a51ca0f6)
6.3.2 Brain-based electrophysiology biomarker: duration-evoked MMN (2024)
A 2024 EEG study proposed MMN as an objective biomarker: - Cohorts: CLN3 n=21 (ages 6–28); controls n=41 (ages 6–26). (brima2024assessingtheintegrity pages 2-4) - Findings: MMN was robust at 900 ms stimulus rate, significantly reduced at the fastest rate, and absent at the slowest rate in CLN3 vs controls. (brima2024assessingtheintegrity pages 1-2)
7. Anatomical structures affected
7.1 Organ/system level
- Central nervous system (progressive neurodegeneration). (shematorova2020currentinsightsin pages 1-3)
- Eye/retina (progressive retinal degeneration; early hallmark). (sakti2023earlyrecognitionof pages 1-3, wright2020juvenilebattendisease pages 1-6)
Suggested UBERON terms: - Brain (UBERON:0000955) - Retina (UBERON:0000966)
7.2 Tissue/cell level (supported examples)
- Retinal dysfunction and likely inner retinal involvement (electronegative ERG; bull’s-eye maculopathy). (sakti2023earlyrecognitionof pages 1-3)
- Retinal pigment epithelium lysosomal storage phenotypes (in vitro). (wunkhaus2024trpml1activationameliorates pages 1-2)
7.3 Subcellular level
- Late endosome/lysosome dysfunction and storage (cholesterol, SubC, lipids). (chen2023juvenilecln3disease pages 2-3, wunkhaus2024trpml1activationameliorates pages 1-2)
Suggested GO cellular component terms: - Lysosome (GO:0005764) - Late endosome (GO:0005770)
8. Temporal development
8.1 Onset
A review describes early vision problems in the majority of cases within childhood: “in more than 80% of patients vision problems begin at age 5–10 years.” (shematorova2020currentinsightsin pages 1-3)
Caregiver-reported symptom timing supports early childhood onset with diagnosis at ~8 years and substantial diagnostic delay. (schulz2024theparentand pages 1-2)
8.2 Progression
Progression is described as relentless and fatal, with progressive visual loss followed by broader neurologic decline and seizures, culminating in severe disability and premature death in young adulthood. (rosenberg2019advancesinthe pages 7-10, shematorova2020currentinsightsin pages 1-3)
9. Inheritance and population
9.1 Inheritance
CLN3 disease is treated as a Mendelian autosomal recessive condition caused by biallelic CLN3 mutations. (johnson2023earlypostnataladministration pages 1-2, schulz2024theparentand pages 1-2)
9.2 Epidemiology
One source summarized NCL epidemiology at a broad level: “all NCL forms are predicted to affect ~1 in 100,000 worldwide” (with CLN3 described as the most common subtype). (schwartz2022improvingaavretinal pages 18-22)
Because this statement is not CLN3-specific and comes from a nonstandard venue (“Unknown journal” in the retrieved record), it should be treated as approximate. (schwartz2022improvingaavretinal pages 18-22)
9.3 Prognosis / life expectancy
A treatment review states typical onset is between 4 and 10 years, with life expectancy into the early 20s. (rosenberg2019advancesinthe pages 7-10)
Another review states death commonly occurs ~20–30 years of age. (shematorova2020currentinsightsin pages 1-3)
10. Diagnostics
10.1 Clinical presentation triggering diagnostic workup
Early ophthalmic presentation is common and can be diagnostically challenging.
A 2023 ocular biomarker series concluded that early maculopathy and electrophysiologic signatures should prompt directed evaluation and genetic assessment for CLN3. (sakti2023earlyrecognitionof pages 1-3)
10.2 Ophthalmic testing / imaging (real-world implementation)
In 5 genetically confirmed CLN3 children (median age 6.2 years), ocular workup found: - electronegative ERG in all patients, - bull’s-eye maculopathy in all, - characteristic FAF ring patterns, - OCT ellipsoid-zone disruption in all. (sakti2023earlyrecognitionof pages 1-3)
These modalities are widely available in tertiary ophthalmology centers and are being used to support earlier recognition and monitoring. (sakti2023earlyrecognitionof pages 1-3)
10.3 Laboratory/pathology
A CLN3 ocular cohort emphasized blood film microscopy for vacuolated lymphocytes as an accessible screening step, followed by confirmatory genetic testing. (wright2020juvenilebattendisease pages 1-6)
10.4 Genetic testing
Molecular confirmation (two pathogenic variants, or one variant plus supportive clinical/pathology in some research contexts) is used for trial enrollment and NIH natural history protocols. (NCT03307304 chunk 1)
11. Outcomes / prognosis
11.1 Functional decline
Progressive loss of vision, cognition, and motor function is consistently described; caregiver data highlight insomnia and mood/thought concerns as frequent burdens. (schulz2024theparentand pages 1-2)
11.2 Key complications highlighted in recent clinical analysis
In late disease, recurrent non-epileptic fearful episodes with autonomic signs may occur and can be difficult to manage. (ostergaard2023treatmentofnonepileptic pages 1-2, ostergaard2023etiologyofanxious pages 1-2)
12. Treatment
12.1 Current standard of care
No disease-modifying, approved therapy for CLN3 disease was identified in the retrieved sources; care is largely supportive (symptom management; neuro/vision support; seizure management). (do2023cerebrospinalfluidprotein pages 1-3)
12.2 Interventional and experimental therapies (clinical trials)
12.2.1 AAV9-CLN3 gene replacement (intrathecal) — NCT03770572
ClinicalTrials.gov record describes a Phase 1/2 open-label dose-escalation gene transfer trial delivering CLN3 using self-complementary AAV9 (CLN-301) via intrathecal lumbar injection in children with genetically confirmed CLN3. Primary outcomes include safety and co-primary efficacy on UBDRS physical subscale; follow-up planned up to 5 years with longer-term monitoring. (NCT03770572 chunk 1)
- URL: https://clinicaltrials.gov/study/NCT03770572 (from trial record context) (NCT03770572 chunk 1)
- Trial start date in record: 2018-11-13. (NCT03770572 chunk 1)
Preclinical rationale (mouse): A 2023 study reports that early postnatal AAV9 delivery produced robust CNS expression and “consistently and persistently” rescued multiple hallmarks while being “safe and well-tolerated,” prompting the launch of NCT03770572. (johnson2023earlypostnataladministration pages 1-2)
Suggested MAXO terms: - Gene therapy (MAXO:0001001) - Intrathecal drug administration (MAXO:0000570)
12.2.2 Mycophenolate mofetil (CellCept) immunomodulation — NCT01399047
ClinicalTrials.gov record NCT01399047 captures an interventional study of mycophenolate mofetil with pediatric/young adult eligibility (6–25 years) and extensive safety-related exclusion criteria for immunosuppression. (NCT01399047 chunk 2)
A Lancet Neurology review notes that in a trial of 19 children with CLN3 disease, mycophenolate was well tolerated but there was no clinical benefit. (ostergaard2023etiologyofanxious pages 2-3)
Suggested MAXO: - Immunosuppressive therapy (MAXO:0000648)
12.2.3 Miglustat — NCT05174039
The miglustat trial record (NCT05174039) was retrieved as a CLN3 interventional study (completed status in trial list), but detailed outcomes were not available in the retrieved chunk text; therefore efficacy conclusions are not reported here. (NCT03307304 chunk 1)
12.2.4 PLX-200 (gemfibrozil formulation) — NCT04637282
ClinicalTrials.gov record NCT04637282 describes a Phase 3 randomized placebo-controlled study of PLX-200 with primary outcome change in Hamburg Rating Scale motor score at Week 60; the record version date is 2026-06-12 and the study is not yet recruiting. (NCT04637282 chunk 1, NCT04637282 chunk 2)
Because the record is dated beyond 2024 in the retrieved data, it is included as pipeline context rather than “2023–2024 evidence of efficacy.” (NCT04637282 chunk 2)
12.3 Symptom-focused management: fearful/anxious non-epileptic episodes (2023 expert analysis)
A 2023 clinical analysis reports that recurrent non-epileptic frightened episodes occur in “more than half” of post-adolescent CLN3 patients and resemble PSH after TBI. (ostergaard2023treatmentofnonepileptic pages 1-2)
Management strategies emphasize trigger minimization, analgesia/sedation approaches, and exploration of transcutaneous vagal nerve stimulation to rebalance autonomic disproportion (research recommendation). (ostergaard2023etiologyofanxious pages 1-2, ostergaard2023treatmentofnonepileptic pages 5-7)
Suggested MAXO: - Symptomatic treatment (MAXO:0000011) - Vagus nerve stimulation (MAXO:0000934)
13. Prevention
Primary prevention is not currently feasible outside genetic risk reduction.
Secondary/tertiary prevention approaches supported by evidence include: - earlier recognition of ocular biomarkers to accelerate diagnosis and access to trials/supportive care (sakti2023earlyrecognitionof pages 1-3) - family planning/genetic counseling needs highlighted by caregiver impact studies (schulz2024theparentand pages 1-2)
14. Other species / natural disease
No CLN3-specific naturally occurring nonhuman disease evidence was retrieved in the current context.
15. Model organisms
15.1 Models in current evidence
- Mouse models used for AAV9 gene therapy efficacy/safety studies (preclinical). (johnson2023earlypostnataladministration pages 1-2)
- Human cellular models (ARPE-19 CLN3 knockout RPE; iPSC-derived neurons) capturing lysosomal storage and functional phenotypes. (wunkhaus2024trpml1activationameliorates pages 1-2, ostergaard2023etiologyofanxious pages 2-3)
Limitations noted in broader discussions include difficulty translating endpoints without robust survival phenotypes and the need for biomarkers to support therapeutic trials. (do2023cerebrospinalfluidprotein pages 1-3)
Recent developments (2023–2024 focus)
Table (click to expand)
| Year | Study type | Key finding | Quantitative data | DOI/URL |
|---|---|---|---|---|
| 2023 | Human autopsy brain lipidomics/proteomics | Juvenile CLN3 disease behaves as a lysosomal cholesterol storage disorder with late endosome/lysosome profiles resembling Niemann-Pick type C disease (chen2023juvenilecln3disease pages 2-3, chen2023juvenilecln3disease pages 1-2, chen2023juvenilecln3disease pages 15-16) | Human samples: controls n=6, JNCL n=5, NPC n=4; cholesterol accumulated in LE/Lys of JNCL to a comparable extent to NPC; JNCL and NPC LE/Lys protein signatures were described as essentially indistinguishable (chen2023juvenilecln3disease pages 2-3, chen2023juvenilecln3disease pages 1-2) | 10.1016/j.ebiom.2023.104628 / https://doi.org/10.1016/j.ebiom.2023.104628 |
| 2023 | Human CSF biomarker discovery | CSF proteomics identified candidate surrogate biomarkers for disease progression and therapeutic response in CLN3 (do2023cerebrospinalfluidprotein pages 1-3, do2023cerebrospinalfluidprotein pages 8-10, do2023cerebrospinalfluidprotein media a51ca0f6) | Discovery cohorts: 28 CLN3 and 32 non-CLN3 for PEA; 20 CLN3 and 25 non-CLN3 for MS; PEA profiled 1467 proteins and found 54 candidates at adjusted p<0.1 and fold-change threshold 1.5; MS found 233 candidates; 25 overlapped across methods; key log2 FCs: CHIT1 +2.69 (PEA), +1.50 (MS); NELL1 -1.29 (PEA), -1.10 (MS); ISLR2 -1.28 (PEA), -1.25 (MS); NEFL +2.79 by PEA (do2023cerebrospinalfluidprotein pages 1-3, do2023cerebrospinalfluidprotein pages 8-10, do2023cerebrospinalfluidprotein media a51ca0f6) | 10.1021/acs.jproteome.3c00199 / https://doi.org/10.1021/acs.jproteome.3c00199 |
| 2024 | Human electrophysiology biomarker study | Duration-evoked MMN ERP shows impaired auditory sensory-memory processing and potential as a brain-based biomarker in CLN3 (brima2024assessingtheintegrity pages 1-2, brima2024assessingtheintegrity pages 2-4) | Final analyzed cohort: CLN3 n=21 (age 6-28 y), controls n=41 (age 6-26 y); MMN robust at 900 ms SOA, significantly reduced at 450 ms, and not detectable at 1800 ms in CLN3 relative to controls (brima2024assessingtheintegrity pages 1-2, brima2024assessingtheintegrity pages 2-4) | 10.1186/s11689-023-09515-8 / https://doi.org/10.1186/s11689-023-09515-8 |
| 2023 | Human ocular phenotype/diagnostic biomarker series | Early ocular biomarkers can facilitate recognition of CLN3, especially electronegative ERG plus characteristic multimodal retinal imaging abnormalities (sakti2023earlyrecognitionof pages 1-3, sakti2023earlyrecognitionof pages 9-11, sakti2023earlyrecognitionof pages 13-14) | 5 unrelated children; 4 females/1 male; median age 6.2 y (range 4.6-11.7); BCVA 0.18-0.88 logMAR at first presentation; electronegative ERG in all; bull's-eye maculopathy in all; FAF hyper-autofluorescent ring around hypo-autofluorescent fovea; OCT foveal ellipsoid-zone disruption in all (sakti2023earlyrecognitionof pages 1-3, sakti2023earlyrecognitionof pages 9-11) | 10.1007/s10633-023-09930-1 / https://doi.org/10.1007/s10633-023-09930-1 |
| 2024 | In vitro human RPE CLN3-KO model | TRPML1 activation partially rescues lysosomal storage phenotypes in CLN3-deficient retinal pigment epithelial cells (wunkhaus2024trpml1activationameliorates pages 1-2, wunkhaus2024trpml1activationameliorates pages 11-12, wunkhaus2024trpml1activationameliorates pages 7-8, wunkhaus2024trpml1activationameliorates pages 4-5) | CLN3-KO ARPE-19 cells accumulated LAMP1+ organelles, SubC, Gb3, and GPDs, with decreased BMP/LBPA; TRPML1 agonist ML-SA5 reduced Gb3 and SubC and rapidly lowered GPDs (many significantly reduced by 90 min, further by 72 h), but did not normalize BMP/LBPA; rescue was TFEB-independent and linked to enhanced lysosomal exocytosis (wunkhaus2024trpml1activationameliorates pages 1-2, wunkhaus2024trpml1activationameliorates pages 11-12, wunkhaus2024trpml1activationameliorates pages 7-8) | 10.1038/s41598-024-67479-8 / https://doi.org/10.1038/s41598-024-67479-8 |
Table: This table compiles recent 2023-2024 CLN3 disease studies spanning human biomarker, imaging, electrophysiology, and mechanistic cell-model research. It is useful for quickly comparing the strongest recent quantitative findings and their translational relevance.
Key identifiers / nomenclature summary
Table (click to expand)
| Disease / scope | Common synonyms in current evidence | Causal gene | Inheritance | OMIM / disease number in evidence | MONDO ID in current evidence | Key clinical trial IDs in current evidence | Notes |
|---|---|---|---|---|---|---|---|
| CLN3 disease | CLN3 disease; CLN3 Batten disease; juvenile neuronal ceroid lipofuscinosis; JNCL; juvenile Batten disease; Batten disease (shematorova2020currentinsightsin pages 1-3, brima2024assessingtheintegrity pages 1-2, sakti2023earlyrecognitionof pages 1-3, schulz2024theparentand pages 1-2) | CLN3 (shematorova2020currentinsightsin pages 1-3, johnson2023earlypostnataladministration pages 1-2, schulz2024theparentand pages 1-2) | Autosomal recessive / caused by biallelic CLN3 variants (shematorova2020currentinsightsin pages 1-3, johnson2023earlypostnataladministration pages 1-2, schulz2024theparentand pages 1-2) | OMIM #204200 for CLN3 disease was explicitly mentioned in current evidence (wright2020juvenilebattendisease pages 1-6) | CLN3-specific MONDO not extracted in current evidence; use broader NCL MONDO below if needed (OpenTargets Search: Neuronal ceroid lipofuscinosis,CLN3 disease,juvenile neuronal ceroid lipofuscinosis) | NCT03770572; NCT03307304; NCT01399047; NCT05174039; NCT04637282; NCT01873924 (NCT03770572 chunk 1, NCT03307304 chunk 1, NCT04637282 chunk 1, NCT01399047 chunk 2) | Most affected individuals carry a common ~1 kb / 966 bp / 1.02 kb CLN3 deletion affecting exons 7-8 in the cited literature (shematorova2020currentinsightsin pages 1-3, johnson2023earlypostnataladministration pages 1-2) |
| Neuronal ceroid lipofuscinosis (broader disease family entry relevant to CLN3) | neuronal ceroid lipofuscinosis; NCL; Batten disease (shematorova2020currentinsightsin pages 1-3, brima2024assessingtheintegrity pages 1-2) | Multiple genes in broader family; CLN3 is one supported associated target for this family-level term (OpenTargets Search: Neuronal ceroid lipofuscinosis,CLN3 disease,juvenile neuronal ceroid lipofuscinosis) | not retrieved in current evidence for the family-level umbrella term | not retrieved in current evidence | MONDO_0016295 (neuronal ceroid lipofuscinosis) (OpenTargets Search: Neuronal ceroid lipofuscinosis,CLN3 disease,juvenile neuronal ceroid lipofuscinosis) | not retrieved in current evidence for the umbrella term | This MONDO is broader than CLN3 disease and should not be assumed CLN3-specific (OpenTargets Search: Neuronal ceroid lipofuscinosis,CLN3 disease,juvenile neuronal ceroid lipofuscinosis) |
Table: This table summarizes the disease names, synonyms, gene, inheritance, identifiers, and clinical trial IDs for CLN3 disease using only retrieved evidence. It is useful for harmonizing nomenclature and database mapping while clearly distinguishing supported identifiers from those not retrieved.
Visual evidence (biomarker figure/table)
Do et al. (2023) provide volcano plots and a table of cross-platform CSF biomarker candidates, including CHIT1 (increased) and ISLR2/NELL1 (decreased), with reported log2 fold changes in both PEA and MS platforms. (do2023cerebrospinalfluidprotein media a51ca0f6)
Notes on evidence gaps and tool-grounding
- Several requested identifiers (Orphanet, ICD-10/ICD-11, MeSH, CLN3-specific MONDO) were not retrievable in the current tool context and therefore are not reported.
- Variant allele frequencies (gnomAD) and ClinVar/ClinGen assertions were not available in retrieved evidence.
- Some ClinicalTrials.gov records were retrieved only partially (e.g., miglustat), preventing outcome reporting.
References
-
(NCT03770572 chunk 1): Gene Therapy for Children With CLN3 Batten Disease. Neela Therapeutics. 2018. ClinicalTrials.gov Identifier: NCT03770572
-
(shematorova2020currentinsightsin pages 1-3): Elena K. Shematorova and George V. Shpakovski. Current insights in elucidation of possible molecular mechanisms of the juvenile form of batten disease. International Journal of Molecular Sciences, 21:8055, Oct 2020. URL: https://doi.org/10.3390/ijms21218055, doi:10.3390/ijms21218055. This article has 12 citations.
-
(chen2023juvenilecln3disease pages 2-3): Jacinda Chen, Rajesh Kumar Soni, Yimeng Xu, Sabrina Simoes, Feng-Xia Liang, Laura DeFreitas, Robert Hwang, Jorge Montesinos, Joseph H. Lee, Estela Area-Gomez, Renu Nandakumar, Badri Vardarajan, and Catherine Marquer. Juvenile cln3 disease is a lysosomal cholesterol storage disorder: similarities with niemann-pick type c disease. eBioMedicine, 92:104628, Jun 2023. URL: https://doi.org/10.1016/j.ebiom.2023.104628, doi:10.1016/j.ebiom.2023.104628. This article has 13 citations and is from a peer-reviewed journal.
-
(brima2024assessingtheintegrity pages 1-2): Tufikameni Brima, Edward G. Freedman, Kevin D. Prinsloo, Erika F. Augustine, Heather R. Adams, Kuan Hong Wang, Jonathan W. Mink, Luke H. Shaw, Emma P. Mantel, and John J. Foxe. Assessing the integrity of auditory sensory memory processing in cln3 disease (juvenile neuronal ceroid lipofuscinosis (batten disease)): an auditory evoked potential study of the duration-evoked mismatch negativity (mmn). Journal of Neurodevelopmental Disorders, Jan 2024. URL: https://doi.org/10.1186/s11689-023-09515-8, doi:10.1186/s11689-023-09515-8. This article has 11 citations and is from a peer-reviewed journal.
-
(sakti2023earlyrecognitionof pages 1-3): Dhimas H. Sakti, Elisa E. Cornish, Clare L. Fraser, Benjamin M. Nash, Trent M. Sandercoe, Michael M. Jones, Neil A. Rowe, Robyn V. Jamieson, Alexandra M. Johnson, and John R. Grigg. Early recognition of cln3 disease facilitated by visual electrophysiology and multimodal imaging. Documenta Ophthalmologica. Advances in Ophthalmology, 146:241-256, Mar 2023. URL: https://doi.org/10.1007/s10633-023-09930-1, doi:10.1007/s10633-023-09930-1. This article has 14 citations.
-
(schulz2024theparentand pages 1-2): Angela Schulz, Nita Patel, Jon J. Brudvig, Frank Stehr, Jill M. Weimer, and Erika F. Augustine. The parent and family impact of cln3 disease: an observational survey-based study. Orphanet Journal of Rare Diseases, Mar 2024. URL: https://doi.org/10.1186/s13023-024-03119-8, doi:10.1186/s13023-024-03119-8. This article has 16 citations and is from a peer-reviewed journal.
-
(rosenberg2019advancesinthe pages 7-10): Jonathan B. Rosenberg, Alvin Chen, Stephen M. Kaminsky, Ronald G. Crystal, and Dolan Sondhi. Advances in the treatment of neuronal ceroid lipofuscinosis. Nov 2019. URL: https://doi.org/10.1080/21678707.2019.1684258, doi:10.1080/21678707.2019.1684258. This article has 43 citations.
-
(wright2020juvenilebattendisease pages 1-6): Genevieve A. Wright, Michalis Georgiou, Anthony G. Robson, Naser Ali, Ambreen Kalhoro, SM Kleine Holthaus, Nikolas Pontikos, Ngozi Oluonye, Emanuel R. de Carvalho, Magella M. Neveu, Richard G. Weleber, and Michel Michaelides. Juvenile batten disease (cln3): detailed ocular phenotype, novel observations, delayed diagnosis, masquerades, and prospects for therapy. Ophthalmology Retina, 4(4):433-445, Apr 2020. URL: https://doi.org/10.1016/j.oret.2019.11.005, doi:10.1016/j.oret.2019.11.005. This article has 68 citations and is from a peer-reviewed journal.
-
(OpenTargets Search: Neuronal ceroid lipofuscinosis,CLN3 disease,juvenile neuronal ceroid lipofuscinosis): Open Targets Query (Neuronal ceroid lipofuscinosis,CLN3 disease,juvenile neuronal ceroid lipofuscinosis, 21 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(do2023cerebrospinalfluidprotein pages 1-3): An N. Dang Do, David E. Sleat, Kiersten Campbell, Nicholas L. Johnson, Haiyan Zheng, Christopher A. Wassif, Ryan K. Dale, and Forbes D. Porter. Cerebrospinal fluid protein biomarker discovery in cln3. Journal of proteome research, 22:2493-2508, Jun 2023. URL: https://doi.org/10.1021/acs.jproteome.3c00199, doi:10.1021/acs.jproteome.3c00199. This article has 7 citations and is from a peer-reviewed journal.
-
(johnson2023earlypostnataladministration pages 1-2): Tyler B. Johnson, Jon J. Brudvig, Shibi Likhite, Melissa A. Pratt, Katherine A. White, Jacob T. Cain, Clarissa D. Booth, Derek J. Timm, Samantha S. Davis, Brandon Meyerink, Ricardo Pineda, Cassandra Dennys-Rivers, Brian K. Kaspar, Kathrin Meyer, and Jill M. Weimer. Early postnatal administration of an aav9 gene therapy is safe and efficacious in cln3 disease. Frontiers in Genetics, Mar 2023. URL: https://doi.org/10.3389/fgene.2023.1118649, doi:10.3389/fgene.2023.1118649. This article has 25 citations and is from a peer-reviewed journal.
-
(ostergaard2023etiologyofanxious pages 2-3): John R. Ostergaard. Etiology of anxious and fearful behavior in juvenile neuronal ceroid lipofuscinosis (cln3 disease). Frontiers in Psychiatry, Apr 2023. URL: https://doi.org/10.3389/fpsyt.2023.1059082, doi:10.3389/fpsyt.2023.1059082. This article has 11 citations.
-
(ostergaard2023etiologyofanxious pages 1-2): John R. Ostergaard. Etiology of anxious and fearful behavior in juvenile neuronal ceroid lipofuscinosis (cln3 disease). Frontiers in Psychiatry, Apr 2023. URL: https://doi.org/10.3389/fpsyt.2023.1059082, doi:10.3389/fpsyt.2023.1059082. This article has 11 citations.
-
(schulz2024theparentand pages 4-5): Angela Schulz, Nita Patel, Jon J. Brudvig, Frank Stehr, Jill M. Weimer, and Erika F. Augustine. The parent and family impact of cln3 disease: an observational survey-based study. Orphanet Journal of Rare Diseases, Mar 2024. URL: https://doi.org/10.1186/s13023-024-03119-8, doi:10.1186/s13023-024-03119-8. This article has 16 citations and is from a peer-reviewed journal.
-
(ostergaard2023etiologyofanxious pages 3-4): John R. Ostergaard. Etiology of anxious and fearful behavior in juvenile neuronal ceroid lipofuscinosis (cln3 disease). Frontiers in Psychiatry, Apr 2023. URL: https://doi.org/10.3389/fpsyt.2023.1059082, doi:10.3389/fpsyt.2023.1059082. This article has 11 citations.
-
(ostergaard2023treatmentofnonepileptic pages 1-2): John R. Ostergaard. Treatment of non-epileptic episodes of anxious, fearful behavior in adolescent juvenile neuronal ceroid lipofuscinosis (cln3 disease). Frontiers in Neurology, Sep 2023. URL: https://doi.org/10.3389/fneur.2023.1216861, doi:10.3389/fneur.2023.1216861. This article has 8 citations and is from a peer-reviewed journal.
-
(schulz2024theparentand pages 5-7): Angela Schulz, Nita Patel, Jon J. Brudvig, Frank Stehr, Jill M. Weimer, and Erika F. Augustine. The parent and family impact of cln3 disease: an observational survey-based study. Orphanet Journal of Rare Diseases, Mar 2024. URL: https://doi.org/10.1186/s13023-024-03119-8, doi:10.1186/s13023-024-03119-8. This article has 16 citations and is from a peer-reviewed journal.
-
(chen2023juvenilecln3disease pages 15-16): Jacinda Chen, Rajesh Kumar Soni, Yimeng Xu, Sabrina Simoes, Feng-Xia Liang, Laura DeFreitas, Robert Hwang, Jorge Montesinos, Joseph H. Lee, Estela Area-Gomez, Renu Nandakumar, Badri Vardarajan, and Catherine Marquer. Juvenile cln3 disease is a lysosomal cholesterol storage disorder: similarities with niemann-pick type c disease. eBioMedicine, 92:104628, Jun 2023. URL: https://doi.org/10.1016/j.ebiom.2023.104628, doi:10.1016/j.ebiom.2023.104628. This article has 13 citations and is from a peer-reviewed journal.
-
(chen2023juvenilecln3disease pages 1-2): Jacinda Chen, Rajesh Kumar Soni, Yimeng Xu, Sabrina Simoes, Feng-Xia Liang, Laura DeFreitas, Robert Hwang, Jorge Montesinos, Joseph H. Lee, Estela Area-Gomez, Renu Nandakumar, Badri Vardarajan, and Catherine Marquer. Juvenile cln3 disease is a lysosomal cholesterol storage disorder: similarities with niemann-pick type c disease. eBioMedicine, 92:104628, Jun 2023. URL: https://doi.org/10.1016/j.ebiom.2023.104628, doi:10.1016/j.ebiom.2023.104628. This article has 13 citations and is from a peer-reviewed journal.
-
(wunkhaus2024trpml1activationameliorates pages 1-2): D. Wünkhaus, R. Tang, K. Nyame, N. N. Laqtom, M. Schweizer, A. Scotto Rosato, E. K. Krogsæter, C. Wollnik, M. Abu-Remaileh, C. Grimm, G. Hermey, R. Kuhn, D. Gruber-Schoffnegger, and S. Markmann. Trpml1 activation ameliorates lysosomal phenotypes in cln3 deficient retinal pigment epithelial cells. Scientific Reports, Jul 2024. URL: https://doi.org/10.1038/s41598-024-67479-8, doi:10.1038/s41598-024-67479-8. This article has 18 citations and is from a peer-reviewed journal.
-
(do2023cerebrospinalfluidprotein pages 8-10): An N. Dang Do, David E. Sleat, Kiersten Campbell, Nicholas L. Johnson, Haiyan Zheng, Christopher A. Wassif, Ryan K. Dale, and Forbes D. Porter. Cerebrospinal fluid protein biomarker discovery in cln3. Journal of proteome research, 22:2493-2508, Jun 2023. URL: https://doi.org/10.1021/acs.jproteome.3c00199, doi:10.1021/acs.jproteome.3c00199. This article has 7 citations and is from a peer-reviewed journal.
-
(do2023cerebrospinalfluidprotein pages 6-8): An N. Dang Do, David E. Sleat, Kiersten Campbell, Nicholas L. Johnson, Haiyan Zheng, Christopher A. Wassif, Ryan K. Dale, and Forbes D. Porter. Cerebrospinal fluid protein biomarker discovery in cln3. Journal of proteome research, 22:2493-2508, Jun 2023. URL: https://doi.org/10.1021/acs.jproteome.3c00199, doi:10.1021/acs.jproteome.3c00199. This article has 7 citations and is from a peer-reviewed journal.
-
(do2023cerebrospinalfluidprotein media a51ca0f6): An N. Dang Do, David E. Sleat, Kiersten Campbell, Nicholas L. Johnson, Haiyan Zheng, Christopher A. Wassif, Ryan K. Dale, and Forbes D. Porter. Cerebrospinal fluid protein biomarker discovery in cln3. Journal of proteome research, 22:2493-2508, Jun 2023. URL: https://doi.org/10.1021/acs.jproteome.3c00199, doi:10.1021/acs.jproteome.3c00199. This article has 7 citations and is from a peer-reviewed journal.
-
(brima2024assessingtheintegrity pages 2-4): Tufikameni Brima, Edward G. Freedman, Kevin D. Prinsloo, Erika F. Augustine, Heather R. Adams, Kuan Hong Wang, Jonathan W. Mink, Luke H. Shaw, Emma P. Mantel, and John J. Foxe. Assessing the integrity of auditory sensory memory processing in cln3 disease (juvenile neuronal ceroid lipofuscinosis (batten disease)): an auditory evoked potential study of the duration-evoked mismatch negativity (mmn). Journal of Neurodevelopmental Disorders, Jan 2024. URL: https://doi.org/10.1186/s11689-023-09515-8, doi:10.1186/s11689-023-09515-8. This article has 11 citations and is from a peer-reviewed journal.
-
(schwartz2022improvingaavretinal pages 18-22): MK Schwartz. Improving aav retinal gene therapy for batten disease. Unknown journal, 2022.
-
(NCT03307304 chunk 1): Investigations of Juvenile Neuronal Ceroid Lipofuscinosis. Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). 2017. ClinicalTrials.gov Identifier: NCT03307304
-
(NCT01399047 chunk 2): Erika Augustine. Cellcept for Treatment of Juvenile Neuronal Ceroid Lipofuscinosis. University of Rochester. 2011. ClinicalTrials.gov Identifier: NCT01399047
-
(NCT04637282 chunk 1): Safety, Tolerability, and Efficacy of PLX-200 in Patients With CLN3. Polaryx Therapeutics, Inc.. 2026. ClinicalTrials.gov Identifier: NCT04637282
-
(NCT04637282 chunk 2): Safety, Tolerability, and Efficacy of PLX-200 in Patients With CLN3. Polaryx Therapeutics, Inc.. 2026. ClinicalTrials.gov Identifier: NCT04637282
-
(ostergaard2023treatmentofnonepileptic pages 5-7): John R. Ostergaard. Treatment of non-epileptic episodes of anxious, fearful behavior in adolescent juvenile neuronal ceroid lipofuscinosis (cln3 disease). Frontiers in Neurology, Sep 2023. URL: https://doi.org/10.3389/fneur.2023.1216861, doi:10.3389/fneur.2023.1216861. This article has 8 citations and is from a peer-reviewed journal.
-
(sakti2023earlyrecognitionof pages 9-11): Dhimas H. Sakti, Elisa E. Cornish, Clare L. Fraser, Benjamin M. Nash, Trent M. Sandercoe, Michael M. Jones, Neil A. Rowe, Robyn V. Jamieson, Alexandra M. Johnson, and John R. Grigg. Early recognition of cln3 disease facilitated by visual electrophysiology and multimodal imaging. Documenta Ophthalmologica. Advances in Ophthalmology, 146:241-256, Mar 2023. URL: https://doi.org/10.1007/s10633-023-09930-1, doi:10.1007/s10633-023-09930-1. This article has 14 citations.
-
(sakti2023earlyrecognitionof pages 13-14): Dhimas H. Sakti, Elisa E. Cornish, Clare L. Fraser, Benjamin M. Nash, Trent M. Sandercoe, Michael M. Jones, Neil A. Rowe, Robyn V. Jamieson, Alexandra M. Johnson, and John R. Grigg. Early recognition of cln3 disease facilitated by visual electrophysiology and multimodal imaging. Documenta Ophthalmologica. Advances in Ophthalmology, 146:241-256, Mar 2023. URL: https://doi.org/10.1007/s10633-023-09930-1, doi:10.1007/s10633-023-09930-1. This article has 14 citations.
-
(wunkhaus2024trpml1activationameliorates pages 11-12): D. Wünkhaus, R. Tang, K. Nyame, N. N. Laqtom, M. Schweizer, A. Scotto Rosato, E. K. Krogsæter, C. Wollnik, M. Abu-Remaileh, C. Grimm, G. Hermey, R. Kuhn, D. Gruber-Schoffnegger, and S. Markmann. Trpml1 activation ameliorates lysosomal phenotypes in cln3 deficient retinal pigment epithelial cells. Scientific Reports, Jul 2024. URL: https://doi.org/10.1038/s41598-024-67479-8, doi:10.1038/s41598-024-67479-8. This article has 18 citations and is from a peer-reviewed journal.
-
(wunkhaus2024trpml1activationameliorates pages 7-8): D. Wünkhaus, R. Tang, K. Nyame, N. N. Laqtom, M. Schweizer, A. Scotto Rosato, E. K. Krogsæter, C. Wollnik, M. Abu-Remaileh, C. Grimm, G. Hermey, R. Kuhn, D. Gruber-Schoffnegger, and S. Markmann. Trpml1 activation ameliorates lysosomal phenotypes in cln3 deficient retinal pigment epithelial cells. Scientific Reports, Jul 2024. URL: https://doi.org/10.1038/s41598-024-67479-8, doi:10.1038/s41598-024-67479-8. This article has 18 citations and is from a peer-reviewed journal.
-
(wunkhaus2024trpml1activationameliorates pages 4-5): D. Wünkhaus, R. Tang, K. Nyame, N. N. Laqtom, M. Schweizer, A. Scotto Rosato, E. K. Krogsæter, C. Wollnik, M. Abu-Remaileh, C. Grimm, G. Hermey, R. Kuhn, D. Gruber-Schoffnegger, and S. Markmann. Trpml1 activation ameliorates lysosomal phenotypes in cln3 deficient retinal pigment epithelial cells. Scientific Reports, Jul 2024. URL: https://doi.org/10.1038/s41598-024-67479-8, doi:10.1038/s41598-024-67479-8. This article has 18 citations and is from a peer-reviewed journal.