L1 Syndrome (L1CAM-related disorder) — Disease Characteristics Research Report
Executive summary
L1 syndrome is an X-linked recessive neurodevelopmental disorder caused by pathogenic variants in L1CAM (Xq28), encompassing a phenotypic spectrum historically described as X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS), MASA syndrome, SPG1 (X-linked spastic paraplegia type 1), and CRASH syndrome. Cardinal features include congenital hydrocephalus/ventriculomegaly (often with aqueductal stenosis), corpus callosum agenesis/hypoplasia, spasticity/spastic paraplegia, intellectual disability, and (in ~45%) adducted thumbs. Disease severity ranges from prenatal-onset lethal hydrocephalus to milder, survivable motor/cognitive phenotypes. (weller2001geneticandclinical pages 1-2)
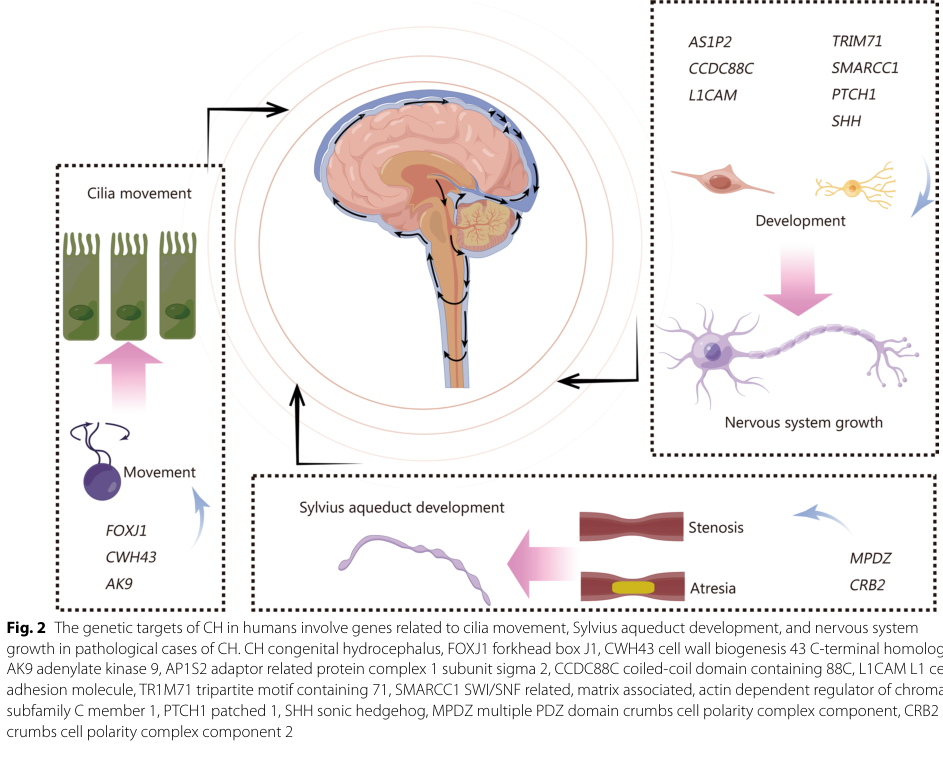
Two 2023–2024 advances particularly relevant to mechanism and translational framing are: (i) a mechanistic connection between L1 processing and autophagy machinery via LC3 binding, required for L1-dependent neurite outgrowth and neuronal survival (loers2023interactionofl1cam pages 14-16, loers2023interactionofl1cam pages 2-4); and (ii) a 2024 synthesis of congenital hydrocephalus genetics that places L1CAM among the small set of confirmed human congenital-hydrocephalus genes and categorizes mechanisms (neurodevelopment, cilia/CSF flow, etc.), including a summary figure (liu2024congenitalhydrocephalusa pages 1-3, liu2024congenitalhydrocephalusa media 5d669376).
Target disease
- Disease name: L1 syndrome
- Category: Mendelian
- MONDO ID: MONDO:0017140 (OpenTargets Search: L1 syndrome-L1CAM)
1. Disease Information
1.1 Definition and overview
L1 syndrome (also termed “L1 disease” in foundational literature) is a group of overlapping X-linked phenotypes caused by L1CAM mutations and classically including HSAS, MASA, SPG1, and X-linked agenesis of the corpus callosum; the broad clinical spectrum is often referred to as CRASH syndrome (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 1-2).
1.2 Key identifiers and synonyms
- MONDO: MONDO:0017140 (OpenTargets Search: L1 syndrome-L1CAM)
- Gene/OMIM note: L1CAM is noted as OMIM 308840 in a prenatal-exome case series (li2020l1cammutationsin pages 1-3).
- Spectrum terms/synonyms used clinically: HSAS, MASA, CRASH, SPG1, X-linked ACC (li2020l1cammutationsin pages 1-3, silan2005anovell1cam pages 1-2, weller2001geneticandclinical pages 1-2).
1.3 Evidence type
The knowledge base–relevant information is drawn from: - Aggregated disease-level resources and curated databases (e.g., Open Targets disease mapping to MONDO) (OpenTargets Search: L1 syndrome-L1CAM) - Human clinical genetics reviews and case reports (prenatal and postnatal) (weller2001geneticandclinical pages 1-2, ochando2016prenataldiagnosisof pages 2-3, li2020l1cammutationsin pages 1-3) - Model organism and mechanistic experimental studies (mouse/in vitro) (loers2023interactionofl1cam pages 14-16, jiang2024singlenucleotidepolymorphism pages 1-2).
URL examples: - Weller & Gärtner 2001 (Human Mutation; 2001-07; DOI URL): https://doi.org/10.1002/humu.1144 (weller2001geneticandclinical pages 1-2) - Liu et al. 2024 (Military Medical Research; 2024-08; DOI URL): https://doi.org/10.1186/s40779-024-00560-5 (liu2024congenitalhydrocephalusa pages 1-3)
2. Etiology
2.1 Disease causal factors
Primary cause: pathogenic germline variation in L1CAM (Xq28), an Ig-superfamily neuronal cell adhesion molecule required for nervous system development and multiple neurodevelopmental processes (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 1-2).
Inheritance: typically X-linked recessive; affected individuals are usually hemizygous males, while heterozygous females are often asymptomatic but can occasionally show manifestations, plausibly due to skewed X-inactivation (silan2005anovell1cam pages 2-3).
2.2 Risk factors
For a Mendelian disease, the major risk factor is carrier status in the mother and familial history of affected males; prenatal case reports emphasize recurrence in pedigrees and the role of targeted L1CAM analysis (ochando2016prenataldiagnosisof pages 2-3, ochando2016prenataldiagnosisof pages 1-2).
2.3 Protective factors and gene–environment interaction
No protective genetic variants or environmental protective factors were identified in the retrieved evidence. L1 syndrome is not typically conceptualized as gene–environment driven; however, severity may be modulated by variant class/domain and potentially genetic background (model systems show allele/background dependence for hydrocephalus phenotypes) (congiu2023micemutatedin pages 16-17).
3. Phenotypes
3.1 Core phenotype spectrum (clinical)
Commonly described features across the L1 spectrum include: - Hydrocephalus/ventriculomegaly, often congenital and sometimes prenatal-onset (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4) - Agenesis/hypoplasia of the corpus callosum (weller2001geneticandclinical pages 1-2, ochando2016prenataldiagnosisof pages 2-3) - Spastic paraplegia/spasticity with gait disturbance (weller2001geneticandclinical pages 1-2, gasser2010efnsguidelineson pages 8-9) - Intellectual disability/developmental delay (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4) - Adducted thumbs (~45% in a foundational review) (weller2001geneticandclinical pages 1-2) - MASA-associated features including aphasia/speech impairment and shuffling gait (li2020l1cammutationsin pages 1-3, gasser2010efnsguidelineson pages 8-9)
3.2 Prenatal imaging phenotypes
Prenatal ultrasound and fetal MRI findings reported in L1CAM-related disease include severe ventriculomegaly/hydrocephalus, third ventricle dilation, and corpus callosum agenesis; absent cavum septum pellucidum has been described in affected fetuses (li2020l1cammutationsin pages 3-4, ochando2016prenataldiagnosisof pages 1-2).
3.3 HPO mapping
A structured HPO mapping is provided below.
Table (click to expand)
| Phenotype | Suggested HPO term(s) | Onset / frequency / notes | Evidence sources |
|---|---|---|---|
| Hydrocephalus / ventriculomegaly | HP:0000238 Hydrocephalus; HP:0002119 Ventriculomegaly | Often prenatal or congenital; may begin in utero and range from severe fetal hydrocephalus to milder ventricular enlargement; core hallmark of L1 syndrome/HSAS (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4, li2020l1cammutationsin pages 1-3) | (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4, li2020l1cammutationsin pages 1-3) |
| Aqueductal stenosis | HP:0002620 Stenosis of the cerebral aqueduct | Classic feature of HSAS/X-linked hydrocephalus; commonly inferred from prenatal/postnatal neuroimaging and disease definition (weller2001geneticandclinical pages 1-2, varagur2022syndromichydrocephalus. pages 1-3, varagur2022syndromichydrocephalus. pages 19-23) | (weller2001geneticandclinical pages 1-2, varagur2022syndromichydrocephalus. pages 1-3, varagur2022syndromichydrocephalus. pages 19-23) |
| Corpus callosum agenesis / hypoplasia | HP:0001274 Agenesis of the corpus callosum; HP:0002079 Hypoplasia of the corpus callosum | Frequently detected prenatally or congenitally; part of CRASH/L1 spectrum; may be complete or partial (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4, ochando2016prenataldiagnosisof pages 2-3, li2020l1cammutationsin pages 1-3) | (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4, ochando2016prenataldiagnosisof pages 2-3, li2020l1cammutationsin pages 1-3) |
| Adducted thumbs | HP:0001182 Adducted thumb | Characteristic but not universal; reported in ~45% of cases in a foundational review; can sometimes be detected prenatally (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4) | (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4) |
| Spastic paraplegia / spasticity | HP:0001258 Spasticity; HP:0007021 Spastic paraplegia | Usually childhood-onset in SPG1/MASA end of spectrum; chronic motor disability affecting gait and mobility; severity variable (weller2001geneticandclinical pages 1-2, gasser2010efnsguidelineson pages 8-9, kutlubaeva2024hereditaryspasticparaplegias pages 1-2) | (weller2001geneticandclinical pages 1-2, gasser2010efnsguidelineson pages 8-9, kutlubaeva2024hereditaryspasticparaplegias pages 1-2) |
| Intellectual disability / developmental delay | HP:0001249 Intellectual disability; HP:0001263 Global developmental delay | Common across the spectrum; severity ranges from mild learning impairment to severe developmental disability (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4, silan2005anovell1cam pages 1-2) | (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 3-4, silan2005anovell1cam pages 1-2) |
| Aphasia / speech impairment | HP:0002381 Aphasia; HP:0002167 Delayed speech and language development | Included in MASA acronym; speech/language impairment may be part of milder surviving phenotypes (li2020l1cammutationsin pages 1-3, gasser2010efnsguidelineson pages 8-9) | (li2020l1cammutationsin pages 1-3, gasser2010efnsguidelineson pages 8-9) |
| Shuffling gait / gait abnormality | HP:0002362 Shuffling gait; HP:0001288 Gait disturbance | Included in MASA acronym; usually reflects corticospinal tract dysfunction/spastic paraparesis with childhood onset or progressive mobility limitation (li2020l1cammutationsin pages 1-3, gasser2010efnsguidelineson pages 8-9, awuah2024hereditaryspasticparaplegia pages 1-2) | (li2020l1cammutationsin pages 1-3, gasser2010efnsguidelineson pages 8-9, awuah2024hereditaryspasticparaplegia pages 1-2) |
| White matter abnormalities / hypomyelination | HP:0002500 Abnormal cerebral white matter morphology; HP:0003429 Hypomyelination | Reported as reduced white matter / hypomyelination in L1 spectrum and model data; may contribute to motor/cognitive dysfunction (silan2005anovell1cam pages 1-2, varagur2022syndromichydrocephalus. pages 1-3, awuah2024hereditaryspasticparaplegia pages 8-10) | (silan2005anovell1cam pages 1-2, varagur2022syndromichydrocephalus. pages 1-3, awuah2024hereditaryspasticparaplegia pages 8-10) |
| Corticospinal tract hypoplasia | HP:0031887 Corticospinal tract hypoplasia | Described as part of the core neuroanatomic spectrum and likely underlies spastic paraplegia/spastic gait (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 1-2) | (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 1-2) |
| Macrocephaly | HP:0000256 Macrocephaly | May accompany ventriculomegaly/hydrocephalus, especially in affected males; secondary to CSF accumulation rather than a universal primary trait (silan2005anovell1cam pages 1-2) | (silan2005anovell1cam pages 1-2) |
| Dilated third ventricle (prenatal ultrasound/MRI finding) | HP:0003370 Enlarged third ventricle | Prenatal imaging clue; reported with severe fetal hydrocephalus/ventriculomegaly and corpus callosum anomalies (li2020l1cammutationsin pages 3-4, ochando2016prenataldiagnosisof pages 1-2, silan2005anovell1cam pages 2-3) | (li2020l1cammutationsin pages 3-4, ochando2016prenataldiagnosisof pages 1-2, silan2005anovell1cam pages 2-3) |
| Absent cavum septum pellucidum (prenatal imaging finding) | HP:0012113 Absent septum pellucidum | Prenatal imaging clue that may accompany corpus callosum agenesis and ventriculomegaly in affected male fetuses (ochando2016prenataldiagnosisof pages 2-3, ochando2016prenataldiagnosisof pages 1-2) | (ochando2016prenataldiagnosisof pages 2-3, ochando2016prenataldiagnosisof pages 1-2) |
Table: This table maps the principal clinical and prenatal imaging features of L1 syndrome to suggested HPO terms, with brief notes on onset and frequency where available. It is useful for structured phenotype annotation in a disease knowledge base.
3.4 Quality of life impact
Direct QoL instruments specific to L1 syndrome were not found in the retrieved primary sources. However, L1 syndrome includes SPG1/MASA phenotypes within the hereditary spastic paraplegia (HSP) spectrum. A 2024 review of HSP emphasizes that HSP “does not reduce a person’s lifespan” but “significantly impairs their quality of life as they age,” reflecting progressive mobility impairment (awuah2024hereditaryspasticparaplegia pages 1-2). This provides indirect but clinically relevant framing for QoL in L1CAM-related spastic paraplegia phenotypes.
4. Genetic / Molecular Information
4.1 Causal gene
L1CAM encodes a ~1275 aa single-pass transmembrane glycoprotein with six Ig-like domains and five FNIII domains and a conserved cytoplasmic tail that includes the ankyrin-binding FIGQY motif (weller2001geneticandclinical pages 1-2).
4.2 Pathogenic variant spectrum
Key points supported by clinical genetics sources: - Reported >280 distinct L1CAM mutations, ~50% missense; many are “private” to individual families (xie2018twonovelpathogenic pages 1-2). - Variant types include missense, nonsense, frameshift, splice-altering variants, and structural variants/CNVs (xie2018twonovelpathogenic pages 1-2, weller2001geneticandclinical pages 10-11).
4.3 Genotype–phenotype correlations (current understanding)
A consistent trend across the clinical literature is: - Truncating variants (especially extracellular-domain truncations leading to absent/non-detectable protein) are associated with severe phenotypes including severe hydrocephalus and higher infant mortality (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 2-3). - Missense variants and cytoplasmic-domain variants often produce milder phenotypes than extracellular truncations (weller2001geneticandclinical pages 1-2, xie2018twonovelpathogenic pages 1-2). - A practical classification framework (Classes I–IV) dividing extracellular truncating vs extracellular missense vs cytoplasmic vs splicing-related groups is described in fetal hydrocephalus case literature (xie2018twonovelpathogenic pages 1-2).
4.4 Allele frequency and constraint notes
A genetics study in another phenotype context (childhood-onset psychosis) reports L1CAM is highly constrained (e.g., pLI=1) and uses gnomAD rarity thresholds for candidate variants; while not L1-syndrome–specific, it underscores strong selective constraint on L1CAM (sato2020hemizygousmutationsin pages 1-6).
4.5 Modifier genes / epigenetics / chromosomal abnormalities
No definitive modifier genes or disease-specific epigenetic signatures were identified in retrieved sources. Some reports acknowledge phenotypic variability within families and across variants, implying possible modifying factors (silan2005anovell1cam pages 2-3).
5. Environmental Information
No established environmental, lifestyle, or infectious causal contributors were identified in the retrieved evidence. L1 syndrome is primarily genetic (X-linked, L1CAM pathogenic variants) (weller2001geneticandclinical pages 1-2).
6. Mechanism / Pathophysiology
6.1 Causal chain (high-level)
L1CAM pathogenic variants → impaired cell adhesion/signaling/trafficking and impaired neurite outgrowth/axon guidance/migration and CNS tract development → neuroanatomic malformations (aqueductal stenosis, ventriculomegaly/hydrocephalus, corpus callosum/corticospinal tract abnormalities) → developmental delay/intellectual disability and motor syndrome (spastic paraplegia/gait disturbance), plus associated features (e.g., adducted thumbs). (weller2001geneticandclinical pages 1-2)
6.2 2023 mechanistic advance: L1–LC3 (autophagy-related) coupling
A 2023 mechanistic study reports that the L1-70 fragment binds LC3 via an extracellular LIR motif in the fourth FNIII domain, and that this interaction is required for L1-mediated neurite outgrowth and neuronal survival (loers2023interactionofl1cam pages 14-16, loers2023interactionofl1cam pages 2-4). This creates a mechanistic bridge between L1CAM processing and autophagy/mitophagy-related pathways.
Direct abstract-supported quote (from abstract text captured): the study states that “L1-70 interacts with LC3 via the extracellular LIR motif in the fourth fibronectin type III domain” and that “the disruption of the L1-LC3 interaction reduces L1-mediated neurite outgrowth and neuronal survival” (loers2023interactionofl1cam pages 1-2).
6.3 2024 congenital hydrocephalus genetics synthesis
A 2024 congenital hydrocephalus review provides epidemiologic framing and pathway categories (cilia movement, neurogenesis/apoptosis, etc.), and explicitly lists L1CAM among the limited number of genes currently associated with congenital hydrocephalus in humans (liu2024congenitalhydrocephalusa pages 1-3).
Direct abstract-supported quote: “The global prevalence rate for congenital hydrocephalus (CH) is approximately one out of every five hundred births” and genetic influences may be involved in “up to 40%” of cases, but the etiology has been pinpointed in “fewer than 5%” of human instances (liu2024congenitalhydrocephalusa pages 1-3).
A key figure from this review summarizes L1CAM within genetic and mechanistic groupings: - Figure shows L1CAM listed under “nervous system growth/development” among CH genetic causes (liu2024congenitalhydrocephalusa media 5d669376).
6.4 Anatomy and cell types implicated
Primary anatomic substrates include: - Brain ventricles and CSF pathways (ventriculomegaly/hydrocephalus; aqueduct) (weller2001geneticandclinical pages 1-2, varagur2022syndromichydrocephalus. pages 1-3) - Corpus callosum and corticospinal tracts (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 1-2)
Suggested ontology mappings: - UBERON: UBERON:0000955 (brain); UBERON:0002285 (corpus callosum); UBERON:0004686 (cerebral aqueduct); UBERON:0002288 (lateral ventricle) (anatomy inferred from phenotypes) (weller2001geneticandclinical pages 1-2, varagur2022syndromichydrocephalus. pages 1-3). - CL (cell types): CL:0000540 (neuron); CL:0000129 (glial cell); CL:0000127 (astrocyte); Schwann cell involvement noted for L1 expression (CL:0000218) (weller2001geneticandclinical pages 1-2). - GO biological processes (examples): neuron migration, axon guidance, neurite outgrowth, cell–cell adhesion, myelination, regulation of autophagy/mitophagy (supported conceptually by L1 functions and L1–LC3 evidence) (weller2001geneticandclinical pages 1-2, loers2023interactionofl1cam pages 14-16).
6.5 Molecular profiling / omics
No disease-specific multi-omics signatures (transcriptomic/proteomic/metabolomic) in patients were found in the retrieved sources.
7. Anatomical Structures Affected
- Primary systems: CNS (ventricular system/CSF flow, midline structures, long tracts) (weller2001geneticandclinical pages 1-2)
- Structures: aqueduct of Sylvius, ventricles (hydrocephalus), corpus callosum, corticospinal tracts (weller2001geneticandclinical pages 1-2, varagur2022syndromichydrocephalus. pages 19-23)
- Peripheral/other: adducted thumbs reflect limb/hand posture abnormalities (weller2001geneticandclinical pages 1-2).
8. Temporal Development
- Onset: commonly prenatal/congenital for severe HSAS; prenatal imaging may detect ventriculomegaly and ACC in the second trimester (ochando2016prenataldiagnosisof pages 1-2, li2020l1cammutationsin pages 3-4).
- Course: severe prenatal hydrocephalus can lead to stillbirth/early infant death; milder variants may survive with chronic neurodevelopmental disability (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 1-2).
9. Inheritance and Population
9.1 Inheritance pattern
- X-linked recessive (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 2-3).
9.2 Epidemiology (statistics)
- HSAS frequency: ~1 in 30,000 live male births (silan2005anovell1cam pages 1-2, xie2018twonovelpathogenic pages 1-2).
- Contribution to congenital hydrocephalus: L1CAM mutations estimated up to ~25% of isolated male congenital hydrocephalus in classic review literature (weller2001geneticandclinical pages 1-2).
- Proportion of congenital hydrocephalus that is X-linked hydrocephalus: ~5% (varagur2022syndromichydrocephalus. pages 1-3).
- Congenital hydrocephalus overall: ~1/500 births globally; genetics may contribute to up to 40% but <5% of cases are genetically solved (liu2024congenitalhydrocephalusa pages 1-3).
10. Diagnostics
10.1 Clinical/imaging
- Prenatal ultrasound: ventriculomegaly/hydrocephalus, third ventricle dilation, corpus callosum agenesis; adducted thumbs can sometimes be detected (li2020l1cammutationsin pages 3-4, silan2005anovell1cam pages 2-3).
- Fetal MRI can complement ultrasound for ventriculomegaly and callosal anomalies (ochando2016prenataldiagnosisof pages 2-3).
- Postnatal MRI is used to assess hydrocephalus/aqueductal stenosis and associated malformations (varagur2022syndromichydrocephalus. pages 19-23).
10.2 Genetic testing approaches (real-world)
- If L1 syndrome is suspected (e.g., male fetus with hydrocephalus + other signs) and karyotype is normal, targeted L1CAM sequencing is used in practice (ochando2016prenataldiagnosisof pages 1-2).
- Medical exome sequencing has been used for fetuses with ultrasound abnormalities to identify L1CAM variants, using ACMG/AMP interpretation; reported detection rates vary strongly by family history (15.7% sporadic vs 74.2% with ≥2 affected relatives) (li2020l1cammutationsin pages 3-4).
10.3 Differential diagnosis (prenatal hydrocephalus)
Differential diagnoses noted in prenatal L1CAM hydrocephalus work include Chiari II malformation, other aqueductal stenosis/gliosis, intrauterine infection, hemorrhage, fetal alcohol syndrome, and chromosomal abnormalities (trisomy 13/18, triploidy) (ochando2016prenataldiagnosisof pages 2-3).
11. Outcome / Prognosis
- Spectrum: ranges from severe prenatal-onset hydrocephalus with stillbirth/early infant death to milder ventricular enlargement compatible with longer survival (weller2001geneticandclinical pages 1-2).
- Severe cases: can have poor neurodevelopmental outcomes even after neurosurgical CSF diversion (e.g., ventriculostomy) because of underlying brain malformations (silan2005anovell1cam pages 1-2).
- Motor disability/QoL: in L1CAM-related spastic paraplegia phenotypes, progressive spasticity and gait impairment can substantially impair QoL; HSP literature suggests lifespan may be preserved in many forms but disability can be significant (awuah2024hereditaryspasticparaplegia pages 1-2, gasser2010efnsguidelineson pages 8-9).
12. Treatment
12.1 Surgical/interventional management (hydrocephalus)
- Syndromic hydrocephalus management commonly includes ventriculoperitoneal shunting; ETV ± CPC is used in some settings but efficacy is variable in young infants (varagur2022syndromichydrocephalus. pages 1-3, varagur2022syndromichydrocephalus. pages 19-23).
- Case literature includes endoscopic ventriculostomy in a severe L1CAM family case (silan2005anovell1cam pages 1-2).
Suggested MAXO terms (examples): - MAXO:0000058 (cerebrospinal fluid shunting procedure; ventriculoperitoneal shunt) — concept mapped to VP shunt use (varagur2022syndromichydrocephalus. pages 19-23) - MAXO:0000756 (endoscopic third ventriculostomy) — concept mapped to ETV (varagur2022syndromichydrocephalus. pages 19-23)
12.2 Pharmacotherapy / disease-modifying therapies
No disease-modifying pharmacotherapy is established for L1 syndrome in the retrieved sources; foundational and prenatal reports emphasize lack of curative therapy and the role of genetic counseling (silan2005anovell1cam pages 1-2, weller2001geneticandclinical pages 1-2).
12.3 Experimental/advanced therapeutics
Mouse/in vitro work describes L1 mimetics and multiple L1-based strategies in CNS injury contexts (e.g., L1 peptides, function-triggering antibodies, recombinant domains), but these are not clinical L1 syndrome therapies in the retrieved evidence (jiang2024singlenucleotidepolymorphism pages 1-2).
13. Prevention
Primary prevention is genetic: - Genetic counseling and carrier testing in families with known L1CAM variants (varagur2022syndromichydrocephalus. pages 1-3). - Prenatal diagnosis using DNA from chorionic villus sampling or amniocentesis with targeted mutation analysis is emphasized; preimplantation genetic diagnosis/testing is also described as possible (silan2005anovell1cam pages 2-3, ochando2016prenataldiagnosisof pages 2-3).
Suggested MAXO terms (examples): - MAXO:0000079 (genetic counseling) - MAXO:0001002 (prenatal genetic testing) - MAXO:0001184 (preimplantation genetic testing)
14. Other Species / Natural Disease
No naturally occurring veterinary analogs were identified in the retrieved evidence.
15. Model Organisms
Mouse and in vitro models provide substantial mechanistic support: - L1/858–863 knock-in mouse: mutation of a dibasic motif in FNIII domain disrupts cleavage sites and is associated with hippocampal neuronal death, astrogliosis and behavioral alterations (congiu2023micemutatedin pages 1-2, congiu2023micemutatedin pages 2-4). - L1-201 (D201) point-mutant mouse: used as an L1 syndrome model; males show worse learning/memory after experimental TBI; in vitro L1 mimetics normalized neuritogenesis and survival deficits and Schwann-cell process formation (jiang2024singlenucleotidepolymorphism pages 1-2). - L1 deficiency/other alleles: multiple mouse perturbations show axon guidance and tract defects and variable ventriculomegaly/hydrocephalus depending on allele and genetic background (congiu2023micemutatedin pages 16-17). - Mechanistic in vitro assays: ELISA/immunoprecipitation/proximity ligation and neurite outgrowth assays demonstrate L1–LC3 coupling required for neurite outgrowth and neuronal survival (loers2023interactionofl1cam pages 2-4, loers2023interactionofl1cam pages 1-2).
Key knowledge-base summary table
Table (click to expand)
| Category | Key points (concise) | Evidence/notes |
|---|---|---|
| Disease identifier | L1 syndrome; MONDO:0017140 | Open Targets disease mapping links L1 syndrome to MONDO_0017140 and L1CAM as the principal associated target (OpenTargets Search: L1 syndrome-L1CAM) |
| Core synonyms / spectrum terms | Overlapping L1CAM-related phenotypes include X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS), MASA syndrome, CRASH syndrome (corpus callosum hypoplasia, retardation/intellectual disability, adducted thumbs, spastic paraplegia, hydrocephalus), SPG1 (spastic paraplegia type 1), and X-linked agenesis/partial agenesis of the corpus callosum (ACC) | Synonym set and spectrum terminology are consistently described across foundational reviews and case reports (weller2001geneticandclinical pages 1-2, xie2018twonovelpathogenic pages 1-2, li2020l1cammutationsin pages 1-3, silan2005anovell1cam pages 1-2) |
| Causal gene | L1CAM (L1 cell adhesion molecule), Xq28; neural cell-adhesion glycoprotein important for CNS development | L1CAM is the established causal gene for L1 syndrome and related allelic disorders (OpenTargets Search: L1 syndrome-L1CAM, weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 1-3) |
| Inheritance | X-linked recessive; affected individuals are usually hemizygous males; female carriers are often asymptomatic but can occasionally manifest disease with skewed/non-random X-inactivation | Human clinical literature describes typical X-linked transmission and occasional manifesting females (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 2-3, li2020l1cammutationsin pages 3-4) |
| Hallmark phenotypes | Major recurrent features: congenital hydrocephalus/ventriculomegaly (often aqueductal stenosis), adducted thumbs, spasticity/spastic paraplegia, agenesis/hypoplasia of corpus callosum, intellectual disability/developmental delay, gait and speech problems; severity ranges from fetal-lethal hydrocephalus to milder neurodevelopmental disease | Core phenotype spectrum summarized in reviews and prenatal/fetal case series (weller2001geneticandclinical pages 1-2, xie2018twonovelpathogenic pages 1-2, li2020l1cammutationsin pages 3-4, li2020l1cammutationsin pages 1-3) |
| Onset / course | Often prenatal or congenital for severe HSAS; hydrocephalus may start in utero; milder presentations may survive into childhood/adulthood with chronic motor/cognitive disability | Natural-history pattern described in foundational review and fetal studies (weller2001geneticandclinical pages 1-2, li2020l1cammutationsin pages 1-3, xie2018twonovelpathogenic pages 1-2) |
| Key epidemiology: L1 syndrome / HSAS | ~1 in 30,000 live male births; described as the most common inherited form of hydrocephalus | Reported in multiple L1CAM-focused papers (silan2005anovell1cam pages 1-2, xie2018twonovelpathogenic pages 1-2, li2020l1cammutationsin pages 3-4) |
| Key epidemiology: contribution to congenital hydrocephalus | Up to ~25% of isolated male congenital hydrocephalus may be attributable to an X-chromosomal gene mutation in classic literature on L1 disease | Foundational review gives this estimate in the context of L1 disease/X-linked hydrocephalus (weller2001geneticandclinical pages 1-2) |
| Broader congenital hydrocephalus epidemiology | Congenital hydrocephalus (CH) affects ~1/500 births globally; genetic factors may contribute up to 40% of cases, but a precise genetic etiology has been pinpointed in <5% of human cases | Recent CH review provides current epidemiologic framing and highlights limited solved fraction despite substantial genetic contribution (liu2024congenitalhydrocephalusa pages 1-3) |
| Variant spectrum | Broad spectrum with >280 reported L1CAM variants; about ~50% missense; many are private/family-specific; variant types include missense, nonsense, frameshift, splice-site, CNVs, and whole-gene deletions | Mutation spectrum summarized in case literature/reviews; CNVs are also recognized in HSP-related genes including L1CAM (xie2018twonovelpathogenic pages 1-2, weller2001geneticandclinical pages 9-10) |
| Genotype–phenotype correlation | General trend: missense variants in extracellular or cytoplasmic regions often produce milder phenotypes, whereas truncating / loss-of-function variants, especially in extracellular domains or with absent protein, are associated with more severe disease including severe hydrocephalus and higher infant mortality | Recurrent genotype–phenotype trend across classic and later reports (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 2-3, li2020l1cammutationsin pages 3-4, xie2018twonovelpathogenic pages 1-2) |
| Mutation classes | Proposed functional classes: Class I extracellular frameshift/nonsense → loss of function, severe; Class II extracellular missense → partial function, variable severity; Class III cytoplasmic variants → signaling defects, usually milder; Class IV extracellular variants associated with aberrant splicing, phenotype less clearly defined | Practical classification summarized in fetal hydrocephalus report/review (xie2018twonovelpathogenic pages 1-2) |
| 2023 mechanistic advance | L1–LC3 interaction: the L1-70 fragment binds LC3 via an extracellular LIR motif in the 4th FNIII domain; disrupting this interaction impairs L1-dependent neurite outgrowth and neuronal survival, linking L1CAM to autophagy/mitophagy-related machinery | Direct mechanistic evidence from 2023 study; important for understanding downstream neuronal vulnerability in L1CAM dysfunction (loers2023interactionofl1cam pages 14-16, loers2023interactionofl1cam pages 1-2, loers2023interactionofl1cam pages 16-17, loers2023interactionofl1cam pages 2-4) |
| 2023-2024 broader mechanism context | L1CAM biology supports cell migration, neurite outgrowth, neuronal survival, myelination, synaptic plasticity; congenital hydrocephalus pathways highlighted in 2024 review include nervous system growth/development, cilia synthesis/movement, ion channels/transport, Reissner’s fiber synthesis, cell apoptosis, and neurogenesis | Recent CH review places L1CAM among the limited confirmed CH genes and Figure 2 summarizes pathway groupings; recent mouse work also supports consequences of extracellular-domain mutations for neuronal death/behavior (liu2024congenitalhydrocephalusa pages 1-3, liu2024congenitalhydrocephalusa media 5d669376, congiu2023micemutatedin pages 20-21, jiang2024singlenucleotidepolymorphism pages 1-2) |
| Diagnostic/KB note | Evidence here is drawn from aggregated disease-level resources and published human case series/reviews, with mechanistic support from mouse/in vitro studies; useful for phenotype, mechanism, and inheritance fields in a knowledge base | Human clinical, review, and model-organism evidence are all represented in the cited contexts (OpenTargets Search: L1 syndrome-L1CAM, weller2001geneticandclinical pages 1-2, liu2024congenitalhydrocephalusa pages 1-3, loers2023interactionofl1cam pages 14-16) |
Table: This table summarizes high-yield knowledge base facts for L1 syndrome, including identifiers, synonyms, inheritance, hallmark phenotypes, epidemiology, genotype-phenotype patterns, and recent mechanistic advances. It is useful as a concise evidence-backed overview of L1CAM-related disease.
Visual evidence (recent review)
Figure evidence supporting L1CAM as a congenital hydrocephalus gene and mechanism category: - A 2024 review figure summarizes genetic causes/pathways of congenital hydrocephalus and includes L1CAM under nervous system growth/development (liu2024congenitalhydrocephalusa media 5d669376).
Notes on limitations of this report
- Disease identifiers beyond MONDO (e.g., Orphanet IDs, ICD-10/ICD-11, MeSH IDs) were not directly retrievable in the current tool context; key synonym/phenotype mapping is nonetheless supported by primary and review literature (weller2001geneticandclinical pages 1-2, silan2005anovell1cam pages 1-2).
- Many 2023–2024 L1 syndrome clinical reports and some systematic reviews were not obtainable in full text via the current retrieval; consequently, some sections (e.g., formal diagnostic criteria, detailed prevalence by region, standardized QoL metrics) remain qualitative. (liu2024congenitalhydrocephalusa pages 1-3, weller2001geneticandclinical pages 1-2)
References
-
(weller2001geneticandclinical pages 1-2): Sabine Weller and Jutta Gärtner. Genetic and clinical aspects of x‐linked hydrocephalus (l1 disease): mutations in the l1cam gene. Human Mutation, 18:1-12, Jul 2001. URL: https://doi.org/10.1002/humu.1144, doi:10.1002/humu.1144. This article has 241 citations and is from a domain leading peer-reviewed journal.
-
(loers2023interactionofl1cam pages 14-16): Gabriele Loers, Ralf Kleene, Viviana Granato, Ute Bork, and Melitta Schachner. Interaction of l1cam with lc3 is required for l1-dependent neurite outgrowth and neuronal survival. International Journal of Molecular Sciences, 24:12531, Aug 2023. URL: https://doi.org/10.3390/ijms241512531, doi:10.3390/ijms241512531. This article has 9 citations.
-
(loers2023interactionofl1cam pages 2-4): Gabriele Loers, Ralf Kleene, Viviana Granato, Ute Bork, and Melitta Schachner. Interaction of l1cam with lc3 is required for l1-dependent neurite outgrowth and neuronal survival. International Journal of Molecular Sciences, 24:12531, Aug 2023. URL: https://doi.org/10.3390/ijms241512531, doi:10.3390/ijms241512531. This article has 9 citations.
-
(liu2024congenitalhydrocephalusa pages 1-3): Xiu-Yun Liu, Xin Song, Marek Czosnyka, Chiara Robba, Zofia Czosnyka, Jennifer Lee Summers, Hui-Jie Yu, Guo-Yi Gao, Peter Smielewski, Fang Guo, Mei-Jun Pang, and Dong Ming. Congenital hydrocephalus: a review of recent advances in genetic etiology and molecular mechanisms. Military Medical Research, Aug 2024. URL: https://doi.org/10.1186/s40779-024-00560-5, doi:10.1186/s40779-024-00560-5. This article has 16 citations and is from a peer-reviewed journal.
-
(liu2024congenitalhydrocephalusa media 5d669376): Xiu-Yun Liu, Xin Song, Marek Czosnyka, Chiara Robba, Zofia Czosnyka, Jennifer Lee Summers, Hui-Jie Yu, Guo-Yi Gao, Peter Smielewski, Fang Guo, Mei-Jun Pang, and Dong Ming. Congenital hydrocephalus: a review of recent advances in genetic etiology and molecular mechanisms. Military Medical Research, Aug 2024. URL: https://doi.org/10.1186/s40779-024-00560-5, doi:10.1186/s40779-024-00560-5. This article has 16 citations and is from a peer-reviewed journal.
-
(OpenTargets Search: L1 syndrome-L1CAM): Open Targets Query (L1 syndrome-L1CAM, 7 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(silan2005anovell1cam pages 1-2): Fatma Silan, Ismail Ozdemir, and Willy Lissens. A novel l1cam mutation with l1 spectrum disorders. Prenatal Diagnosis, 25:57-59, Jan 2005. URL: https://doi.org/10.1002/pd.978, doi:10.1002/pd.978. This article has 16 citations and is from a peer-reviewed journal.
-
(li2020l1cammutationsin pages 1-3): Ying-Ting Li, Jing-Si Chen, Wei Jian, Yi-Duo He, Nan Li, Yi-Nong Xie, Jing Wang, Victor Wei Zhang, Wei-Ran Huang, Fu-man Jiang, Xiao-Qing Ye, Dun-Jin Chen, and Min Chen. L1cam mutations in three fetuses diagnosed by medical exome sequencing. Taiwanese journal of obstetrics & gynecology, 59 3:451-455, May 2020. URL: https://doi.org/10.1016/j.tjog.2020.03.022, doi:10.1016/j.tjog.2020.03.022. This article has 12 citations and is from a peer-reviewed journal.
-
(ochando2016prenataldiagnosisof pages 2-3): I. Ochando, V. Vidal, J. Gascon, M. Acién, Antonio Urbano, and J. Rueda. Prenatal diagnosis of x-linked hydrocephalus in a family with a novel mutation in l1cam gene. Journal of Obstetrics and Gynaecology, 36:403-405, Apr 2016. URL: https://doi.org/10.3109/01443615.2015.1086982, doi:10.3109/01443615.2015.1086982. This article has 14 citations and is from a peer-reviewed journal.
-
(jiang2024singlenucleotidepolymorphism pages 1-2): Haoyu Jiang, Anna O. Giarratana, Thomas Theis, Vini Nagaraj, Xiaofeng Zhou, Smita Thakker-Varia, Melitta Schachner, and Janet Alder. Single nucleotide polymorphism in cell adhesion molecule l1 affects learning and memory in a mouse model of traumatic brain injury. International Journal of Molecular Sciences, 25:3043, Mar 2024. URL: https://doi.org/10.3390/ijms25053043, doi:10.3390/ijms25053043. This article has 3 citations.
-
(silan2005anovell1cam pages 2-3): Fatma Silan, Ismail Ozdemir, and Willy Lissens. A novel l1cam mutation with l1 spectrum disorders. Prenatal Diagnosis, 25:57-59, Jan 2005. URL: https://doi.org/10.1002/pd.978, doi:10.1002/pd.978. This article has 16 citations and is from a peer-reviewed journal.
-
(ochando2016prenataldiagnosisof pages 1-2): I. Ochando, V. Vidal, J. Gascon, M. Acién, Antonio Urbano, and J. Rueda. Prenatal diagnosis of x-linked hydrocephalus in a family with a novel mutation in l1cam gene. Journal of Obstetrics and Gynaecology, 36:403-405, Apr 2016. URL: https://doi.org/10.3109/01443615.2015.1086982, doi:10.3109/01443615.2015.1086982. This article has 14 citations and is from a peer-reviewed journal.
-
(congiu2023micemutatedin pages 16-17): Ludovica Congiu, Viviana Granato, Igor Jakovcevski, Ralf Kleene, Luciana Fernandes, Sandra Freitag, Matthias Kneussel, Melitta Schachner, and Gabriele Loers. Mice mutated in the third fibronectin domain of l1 show enhanced hippocampal neuronal cell death, astrogliosis and alterations in behavior. Biomolecules, 13:776, Apr 2023. URL: https://doi.org/10.3390/biom13050776, doi:10.3390/biom13050776. This article has 2 citations.
-
(li2020l1cammutationsin pages 3-4): Ying-Ting Li, Jing-Si Chen, Wei Jian, Yi-Duo He, Nan Li, Yi-Nong Xie, Jing Wang, Victor Wei Zhang, Wei-Ran Huang, Fu-man Jiang, Xiao-Qing Ye, Dun-Jin Chen, and Min Chen. L1cam mutations in three fetuses diagnosed by medical exome sequencing. Taiwanese journal of obstetrics & gynecology, 59 3:451-455, May 2020. URL: https://doi.org/10.1016/j.tjog.2020.03.022, doi:10.1016/j.tjog.2020.03.022. This article has 12 citations and is from a peer-reviewed journal.
-
(gasser2010efnsguidelineson pages 8-9): T. Gasser, J. Finsterer, J. Baets, C. Van Broeckhoven, S. Di Donato, B. Fontaine, P. De Jonghe, A. Lossos, T. Lynch, C. Mariotti, L. Schöls, A. Spinazzola, Z. Szolnoki, S. J. Tabrizi, C.M.E. Tallaksen, M. Zeviani, J‐M. Burgunder, and H. F. Harbo. Efns guidelines on the molecular diagnosis of ataxias and spastic paraplegias. European Journal of Neurology, 17:179-188, Feb 2010. URL: https://doi.org/10.1111/j.1468-1331.2009.02873.x, doi:10.1111/j.1468-1331.2009.02873.x. This article has 74 citations and is from a domain leading peer-reviewed journal.
-
(varagur2022syndromichydrocephalus. pages 1-3): Kaamya Varagur, Sai Anusha Sanka, and Jennifer M. Strahle. Syndromic hydrocephalus. Neurosurgery clinics of North America, 33 1:67-79, Jan 2022. URL: https://doi.org/10.1016/j.nec.2021.09.006, doi:10.1016/j.nec.2021.09.006. This article has 38 citations and is from a peer-reviewed journal.
-
(varagur2022syndromichydrocephalus. pages 19-23): Kaamya Varagur, Sai Anusha Sanka, and Jennifer M. Strahle. Syndromic hydrocephalus. Neurosurgery clinics of North America, 33 1:67-79, Jan 2022. URL: https://doi.org/10.1016/j.nec.2021.09.006, doi:10.1016/j.nec.2021.09.006. This article has 38 citations and is from a peer-reviewed journal.
-
(kutlubaeva2024hereditaryspasticparaplegias pages 1-2): R. F. Kutlubaeva, M. Kutlubaev, R. V. Magzhanov, E. V. Sayfullina, and I. Khidiyatova. Hereditary spastic paraplegias. Neuromuscular Diseases, Jan 2024. URL: https://doi.org/10.17650/2222-8721-2023-13-4-74-82, doi:10.17650/2222-8721-2023-13-4-74-82. This article has 5 citations.
-
(awuah2024hereditaryspasticparaplegia pages 1-2): Wireko Andrew Awuah, Joecelyn Kirani Tan, Anastasiia D Shkodina, Tomas Ferreira, Favour Tope Adebusoye, Adele Mazzoleni, Jack Wellington, Lian David, Ellie Chilcott, Helen Huang, Toufik Abdul-Rahman, Vallabh Shet, Oday Atallah, Jacob Kalmanovich, Riaz Jiffry, Divine Elizabeth Madhu, Kateryna Sikora, Oleksii Kmyta, and Mykhailo Yu Delva. Hereditary spastic paraplegia: novel insights into the pathogenesis and management. SAGE Open Medicine, Dec 2024. URL: https://doi.org/10.1177/20503121231221941, doi:10.1177/20503121231221941. This article has 31 citations.
-
(awuah2024hereditaryspasticparaplegia pages 8-10): Wireko Andrew Awuah, Joecelyn Kirani Tan, Anastasiia D Shkodina, Tomas Ferreira, Favour Tope Adebusoye, Adele Mazzoleni, Jack Wellington, Lian David, Ellie Chilcott, Helen Huang, Toufik Abdul-Rahman, Vallabh Shet, Oday Atallah, Jacob Kalmanovich, Riaz Jiffry, Divine Elizabeth Madhu, Kateryna Sikora, Oleksii Kmyta, and Mykhailo Yu Delva. Hereditary spastic paraplegia: novel insights into the pathogenesis and management. SAGE Open Medicine, Dec 2024. URL: https://doi.org/10.1177/20503121231221941, doi:10.1177/20503121231221941. This article has 31 citations.
-
(xie2018twonovelpathogenic pages 1-2): Bobo Xie, Jingsi Luo, Yaqin Lei, Qi Yang, Mengting Li, Shang Yi, Shiyu Luo, Jin Wang, Zailong Qin, Zuojian Yang, Hongwei Wei, and Xin Fan. Two novel pathogenic variants of l1cam gene in two fetuses with isolated x‑linked hydrocephaly: a case report. Molecular Medicine Reports, 18 6:5760-5764, Oct 2018. URL: https://doi.org/10.3892/mmr.2018.9583, doi:10.3892/mmr.2018.9583. This article has 6 citations and is from a peer-reviewed journal.
-
(weller2001geneticandclinical pages 10-11): Sabine Weller and Jutta Gärtner. Genetic and clinical aspects of x‐linked hydrocephalus (l1 disease): mutations in the l1cam gene. Human Mutation, 18:1-12, Jul 2001. URL: https://doi.org/10.1002/humu.1144, doi:10.1002/humu.1144. This article has 241 citations and is from a domain leading peer-reviewed journal.
-
(sato2020hemizygousmutationsin pages 1-6): Mitra S. Sato, Marinos Kyriakopoulos, Anthony James, Susanne Marwedel, Clare Borsay, Armandina Almanza Gutierrez, Alexandra I. Blakemore, and Anna C. Need. Hemizygous mutations in l1cam in two unrelated male probands with childhood onset psychosis. Psychiatric Genetics, 30:73-82, Jun 2020. URL: https://doi.org/10.1097/ypg.0000000000000253, doi:10.1097/ypg.0000000000000253. This article has 7 citations and is from a peer-reviewed journal.
-
(loers2023interactionofl1cam pages 1-2): Gabriele Loers, Ralf Kleene, Viviana Granato, Ute Bork, and Melitta Schachner. Interaction of l1cam with lc3 is required for l1-dependent neurite outgrowth and neuronal survival. International Journal of Molecular Sciences, 24:12531, Aug 2023. URL: https://doi.org/10.3390/ijms241512531, doi:10.3390/ijms241512531. This article has 9 citations.
-
(congiu2023micemutatedin pages 1-2): Ludovica Congiu, Viviana Granato, Igor Jakovcevski, Ralf Kleene, Luciana Fernandes, Sandra Freitag, Matthias Kneussel, Melitta Schachner, and Gabriele Loers. Mice mutated in the third fibronectin domain of l1 show enhanced hippocampal neuronal cell death, astrogliosis and alterations in behavior. Biomolecules, 13:776, Apr 2023. URL: https://doi.org/10.3390/biom13050776, doi:10.3390/biom13050776. This article has 2 citations.
-
(congiu2023micemutatedin pages 2-4): Ludovica Congiu, Viviana Granato, Igor Jakovcevski, Ralf Kleene, Luciana Fernandes, Sandra Freitag, Matthias Kneussel, Melitta Schachner, and Gabriele Loers. Mice mutated in the third fibronectin domain of l1 show enhanced hippocampal neuronal cell death, astrogliosis and alterations in behavior. Biomolecules, 13:776, Apr 2023. URL: https://doi.org/10.3390/biom13050776, doi:10.3390/biom13050776. This article has 2 citations.
-
(weller2001geneticandclinical pages 9-10): Sabine Weller and Jutta Gärtner. Genetic and clinical aspects of x‐linked hydrocephalus (l1 disease): mutations in the l1cam gene. Human Mutation, 18:1-12, Jul 2001. URL: https://doi.org/10.1002/humu.1144, doi:10.1002/humu.1144. This article has 241 citations and is from a domain leading peer-reviewed journal.
-
(loers2023interactionofl1cam pages 16-17): Gabriele Loers, Ralf Kleene, Viviana Granato, Ute Bork, and Melitta Schachner. Interaction of l1cam with lc3 is required for l1-dependent neurite outgrowth and neuronal survival. International Journal of Molecular Sciences, 24:12531, Aug 2023. URL: https://doi.org/10.3390/ijms241512531, doi:10.3390/ijms241512531. This article has 9 citations.
-
(congiu2023micemutatedin pages 20-21): Ludovica Congiu, Viviana Granato, Igor Jakovcevski, Ralf Kleene, Luciana Fernandes, Sandra Freitag, Matthias Kneussel, Melitta Schachner, and Gabriele Loers. Mice mutated in the third fibronectin domain of l1 show enhanced hippocampal neuronal cell death, astrogliosis and alterations in behavior. Biomolecules, 13:776, Apr 2023. URL: https://doi.org/10.3390/biom13050776, doi:10.3390/biom13050776. This article has 2 citations.