Kennedy Disease (Spinal and Bulbar Muscular Atrophy; SBMA): Comprehensive Disease Characteristics Report
Summary (knowledge-base ready)
Kennedy disease, also called spinal and bulbar muscular atrophy (SBMA), is an adult-onset, slowly progressive, X-linked neuromuscular disorder primarily affecting males and caused by a CAG trinucleotide repeat expansion in the androgen receptor (AR) gene leading to an expanded polyglutamine tract in AR protein, with androgen-dependent toxicity involving both lower motor neurons (spinal cord/brainstem) and skeletal muscle. (chang2024theroleof pages 11-12, cantara2024antisenseoligonucleotides(asos) pages 14-16, prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5)
Table (click to expand)
| Category | Key values |
|---|---|
| Disease name / definition | Kennedy disease; spinal and bulbar muscular atrophy (SBMA); an adult-onset, slowly progressive, X-linked neuromuscular / lower motor neuron disease caused by polyglutamine-expanded androgen receptor, with both motor neuron and skeletal muscle involvement (chang2024theroleof pages 11-12, cantara2024antisenseoligonucleotides(asos) pages 14-16, prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5) |
| Identifiers | MONDO: MONDO_0010735 (OpenTargets disease association for Kennedy disease) (OpenTargets Search: Spinal and bulbar muscular atrophy,Kennedy disease-AR); OMIM: 313200 (reported as “X-linked spinal and bulbar muscular atrophy (SMAX1, Kennedy disease, OMIM 313200)”) (wang2020apathogenicmissense pages 1-2); MeSH: Bulbo-Spinal Atrophy, X-Linked D055534 (ClinicalTrials.gov MeSH mapping) (NCT06411912 chunk 2, NCT06169046 chunk 1, NCT00303446 chunk 3) |
| Synonyms / alternative names | Spinal and bulbar muscular atrophy; SBMA; Kennedy disease / Kennedy’s disease; X-linked spinal and bulbar muscular atrophy; X-linked recessive bulbospinal neuronopathy; progressive proximal spinal and bulbar muscular atrophy of late onset (lee2024morethanautophony pages 1-2, debartolo2024differentiallydisruptedspinal pages 14-15, wang2020apathogenicmissense pages 1-2, cantara2024antisenseoligonucleotides(asos) pages 14-16) |
| Causal gene / locus | Gene: AR (androgen receptor); location: Xq11-12 / Xq12; disease caused by CAG trinucleotide expansion in exon 1 producing an expanded polyglutamine tract in AR protein (cantara2024antisenseoligonucleotides(asos) pages 14-16, prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, lee2024morethanautophony pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5) |
| Pathogenic repeat ranges / thresholds | Common pathogenic thresholds in evidence: ≥38 CAG in trial eligibility and mechanistic literature (NCT06169046 chunk 1, prakasam2023lsd1prmt6targetinggenetherapy pages 1-2); review evidence gives normal 9–36 CAG vs SBMA 39–72 CAG (cantara2024antisenseoligonucleotides(asos) pages 14-16); AJ201 trial used ≥36 repeats for enrollment (NCT05517603 chunk 1) |
| Inheritance / sex bias | X-linked recessive / sex-linked disorder; primarily affects adult males; females are usually carriers and may be mildly affected; androgen dependence explains marked male predominance (cantara2024antisenseoligonucleotides(asos) pages 14-16, prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, NCT00303446 chunk 1, hashizume2020diseasemechanismbiomarker pages 1-5) |
| Epidemiology | Prevalence estimates in cited sources: 1–2 per 100,000 (review) (cantara2024antisenseoligonucleotides(asos) pages 14-16, hashizume2020diseasemechanismbiomarker pages 1-5); 2–5 per 100,000 worldwide (mechanistic study intro) (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2); Italy trial record: about 1,000 affected individuals, prevalence 1.5/100,000, annual incidence 0.19/100,000 males (NCT06169046 chunk 1) |
| Typical onset / course | Adult onset, often 30–40 years (range 18–64 in one review); slowly progressive; tremor may precede weakness by >10 years; wheelchair often needed 10–15 years after onset in some reports (cantara2024antisenseoligonucleotides(asos) pages 14-16, hashizume2020diseasemechanismbiomarker pages 1-5, iijima2023longtermeffectsof pages 1-2) |
| Hallmark phenotypes | Progressive limb and bulbar weakness/atrophy, fasciculations, cramps, postural hand tremor, dysarthria, dysphagia, laryngospasm, reduced/absent reflexes, distal vibration sensory loss; androgen-insensitivity features including gynecomastia, testicular atrophy, erectile dysfunction, infertility/decreased fertility; aspiration pneumonia is a major cause of death (lee2024morethanautophony pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5, rhodes2009clinicalfeaturesof pages 6-8) |
| Quality-of-life / functional impact | ADL domains notably affected: walking, handwriting, falling, swallowing, speech; SF-36v2 physical component mean 34.3 ± 11.0 in a landmark cohort; substantial diagnostic delay (~2 years to first medical attention plus ~3 more years to diagnosis) (rhodes2009clinicalfeaturesof pages 6-8) |
| Key diagnostic tests | Genetic confirmation of AR CAG expansion is definitive; supportive tests include EMG/neurophysiology, quantitative muscle assessment, timed walk tests, SBMAFRS/ALSFRS-R, tongue pressure, pulmonary metrics (%FVC, %PEF), muscle MRI with Dixon fat quantification, and cardiac ECG/CMR when indicated (NCT06169046 chunk 1, NCT00303446 chunk 1, inagaki2022developmentofa pages 6-7, hashizume2020diseasemechanismbiomarker pages 5-8, steinmetz2022jwavesyndromes pages 1-2) |
| Biomarkers / outcome measures | Prominent biomarkers: serum creatinine (declines before overt weakness), CK/liver enzymes, MRI muscle fat fraction, MUNE, tongue pressure; trial/research outcomes include 6MWT, 2MWD, AMAT, QMA, SBMAFRS, ALSFRS-R, %FVC, %PEF, neurophysiology, and mutant AR in muscle/skin (NCT05517603 chunk 1, NCT06169046 chunk 1, NCT00303446 chunk 1, inagaki2022developmentofa pages 6-7, hashizume2020diseasemechanismbiomarker pages 1-5, hashizume2020diseasemechanismbiomarker pages 5-8) |
| Notable recent mechanism (2023) | LSD1/PRMT6 axis: androgen-dependent overexpression of AR co-regulators LSD1 and PRMT6 specifically in SBMA skeletal muscle; they synergistically enhance AR transactivation, and silencing them suppresses toxic gain-of-function and improves phenotypes in flies and mice (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, prakasam2023lsd1prmt6targetinggenetherapy pages 9-12, prakasam2023lsd1prmt6targetinggenetherapy pages 14-15) |
| Notable recent mechanism (2023) | Early skeletal-muscle pathology: defective excitation-contraction coupling and impaired mitochondrial respiration occur before denervation; early events are androgen-dependent and reversible with castration or AR silencing; patient biopsies and mouse models support muscle as a primary toxicity site (hashizume2020diseasemechanismbiomarker pages 8-12, hashizume2020diseasemechanismbiomarker pages 12-15) |
| Notable recent mechanism (2024) | Energy metabolism / NAD+ biology: SBMA muscle shows reduced NAD+ and ATP, altered nicotinamide/NAD+ salvage pathways, and decreased Nmrk2/NRK2, helping explain why nicotinamide riboside failed to restore muscle NAD+ or improve disease in mice; integrated metabolomics/proteomics implicated glycolysis, PPP, pyruvate, glutathione, and amino-acid pathways (debartolo2024differentiallydisruptedspinal pages 10-12, debartolo2024differentiallydisruptedspinal pages 1-2, debartolo2024differentiallydisruptedspinal pages 8-10) |
| Notable recent mechanism (2024 preclinical) | Synaptic dysregulation / hyperexcitability: early postnatal nuclear accumulation of polyQ-AR in motor neurons dysregulates glutamatergic synaptic genes via Rest/Rest4; iPSC-derived motor neurons are hyperexcitable; antisense correction rescued pathology in mice (preprint evidence) (prakasam2023lsd1prmt6targetinggenetherapy pages 9-12) |
| Anatomy / cell types chiefly affected | Lower motor neurons in spinal cord and brainstem; skeletal muscle is a major primary toxicity site; neuromuscular junction and muscle fiber-type composition are altered; cardiac involvement may occur in a subset (ECG/CMR abnormalities, fibrosis) (hashizume2020diseasemechanismbiomarker pages 1-5, steinmetz2022jwavesyndromes pages 1-2, hashizume2020diseasemechanismbiomarker pages 8-12, hashizume2020diseasemechanismbiomarker pages 12-15) |
| Cardiac / systemic comorbidity signals | In a 30-patient SBMA cohort, 70% had abnormal ECGs; diffuse myocardial fibrosis on T1 mapping in 73.9% vs 9.1% of controls; metabolic comorbidities include glucose intolerance/insulin resistance and dyslipidemia in some patients (debartolo2024differentiallydisruptedspinal pages 1-2, steinmetz2022jwavesyndromes pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5) |
| Supportive / real-world implementation | Long-term gait rehabilitation using wearable HAL in one 68-year-old patient: 9 courses over ~5 years, 3 sessions/week for 3 weeks each; 2MWD improved from 94 m to 101.8 m, gait item on ALSFRS-R remained stable at 3, and independent walking was maintained (iijima2023longtermeffectsof pages 1-2, iijima2023longtermeffectsof pages 9-10, iijima2023longtermeffectsof pages 2-4) |
| Historical / completed interventional trials | NCT00303446 dutasteride 0.5 mg/day vs placebo for 24 months, phase 2; primary endpoint QMA; failed primary outcome in review summary (NCT00303446 chunk 1, hashizume2020diseasemechanismbiomarker pages 5-8). NCT00004771 leuprolide + testosterone, phase 2; genotype-confirmed by AR exon-1 mutation (NCT00004771 chunk 1). NCT00851461 goserelin (listed as completed) (clinical-trial search context in conversation). NCT02024932 BVS857 / IGF-1 mimetic, phase 2, showed muscle-volume signal in review summary (hashizume2020diseasemechanismbiomarker pages 5-8) |
| Active / recent trials (2023-2025 records) | NCT06169046 clenbuterol 0.04 mg/day for 48 weeks, phase 2, primary endpoint 6MWT, enrollment 90, requires AR CAG ≥38 (NCT06169046 chunk 1); NCT05517603 AJ201 600 mg/day for 12 weeks, phase 1/2a, enrollment 25, pharmacodynamic endpoint mutant AR in skeletal muscle, requires AR CAG ≥36 (NCT05517603 chunk 1); NCT06411912 NIDO-361 in genetically confirmed SBMA, phase 2, enrollment 54 (NCT06411912 chunk 2); NCT06862596 mexiletine hydrochloride phase 2/3 recruiting (clinical-trial search context in conversation) |
| Expert/consensus interpretation | Current expert view is that SBMA combines androgen-dependent toxic gain-of-function of expanded AR with elements of partial AR loss-of-function; skeletal muscle is not merely secondary but a therapeutically relevant primary target, supporting both endocrine modulation and AR-lowering / co-regulator-targeting strategies (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, hashizume2020diseasemechanismbiomarker pages 12-15, hashizume2020diseasemechanismbiomarker pages 1-5) |
Table: This table condenses the main evidence-backed facts about Kennedy disease / SBMA, including identifiers, genetics, epidemiology, phenotypes, biomarkers, mechanisms, and clinical trials. It is designed as a quick-reference summary for knowledge-base curation or report drafting.
1. Disease Information
1.1 Definition and overview (current understanding)
SBMA is described as a hereditary neuromuscular disorder caused by CAG trinucleotide expansion in the gene encoding the androgen receptor (AR), with selective involvement of lower motor neurons and clear skeletal muscle pathology. (hashizume2020diseasemechanismbiomarker pages 1-5)
Recent review wording (abstract quote): “Spinal and bulbar muscular atrophy (SBMA) is a hereditary neuromuscular disorder caused by CAG trinucleotide expansion in the gene encoding the androgen receptor (AR).” (Hashizume et al., 2020-09; JNNP; https://doi.org/10.1136/jnnp-2020-322949) (hashizume2020diseasemechanismbiomarker pages 1-5)
A 2024 review similarly defines it as “an X-linked neuromuscular disorder characterized by the progressive loss of motor neurons in the spinal cord and brainstem.” (Chang & Chen, 2024-05; Antioxidants; https://doi.org/10.3390/antiox13060649) (chang2024theroleof pages 11-12)
1.2 Key identifiers (as available from retrieved evidence)
- MONDO: MONDO_0010735 (“Kennedy disease”) from Open Targets disease entity metadata. (OpenTargets Search: Spinal and bulbar muscular atrophy,Kennedy disease-AR)
- OMIM: 313200 (“X-linked spinal and bulbar muscular atrophy (SMAX1, Kennedy disease, OMIM 313200)”). (wang2020apathogenicmissense pages 1-2)
- MeSH: “Bulbo-Spinal Atrophy, X-Linked” (MeSH ID D055534) as used in multiple ClinicalTrials.gov condition mappings. (NCT06411912 chunk 2, NCT06169046 chunk 1)
Not found in retrieved full text: Orphanet identifier and ICD-10/ICD-11 codes were not explicitly present in the gathered sources, so they cannot be cited from this tool run. (wang2020apathogenicmissense pages 1-2, NCT06169046 chunk 1)
1.3 Synonyms / alternative names
Common synonyms in the retrieved literature include Kennedy disease/Kennedy’s disease, spinal and bulbar muscular atrophy (SBMA), X-linked spinal and bulbar muscular atrophy, and “X-linked recessive bulbospinal neuronopathy.” (lee2024morethanautophony pages 1-2, debartolo2024differentiallydisruptedspinal pages 14-15, wang2020apathogenicmissense pages 1-2, cantara2024antisenseoligonucleotides(asos) pages 14-16)
1.4 Evidence-source type
The evidence used here is predominantly aggregated disease-level resources (reviews, clinical trial registries) plus human clinical cohorts/case reports and model organism + in vitro research, rather than EHR-derived analyses. (rhodes2009clinicalfeaturesof pages 6-8, hashizume2020diseasemechanismbiomarker pages 1-5, NCT06169046 chunk 1)
2. Etiology
2.1 Disease causal factors
Primary cause (genetic): CAG repeat expansion in AR exon 1 producing polyglutamine-expanded AR. (cantara2024antisenseoligonucleotides(asos) pages 14-16, prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5)
A landmark clinical cohort explicitly frames the disease as a “ligand-dependent toxic gain of function in the mutant androgen receptor,” linking causality to androgen binding. (Rhodes et al., 2009-10; Brain; https://doi.org/10.1093/brain/awp258) (rhodes2009clinicalfeaturesof pages 2-3)
2.2 Risk factors
- Sex: adult males are primarily affected due to androgen dependence of toxicity. (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5)
- Repeat length: longer CAG repeat length correlates with earlier onset and greater severity in reviewed evidence. (cantara2024antisenseoligonucleotides(asos) pages 14-16)
- Androgen exposure: toxicity requires ligand binding to mutant AR (testosterone/DHT), making androgenic signaling a mechanistic risk factor. (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, hashizume2020diseasemechanismbiomarker pages 8-12)
2.3 Protective factors
Mechanistic protective interventions (preclinical): androgen deprivation and AR silencing are repeatedly reported to prevent or reverse early pathological processes in SBMA models. (debartolo2024differentiallydisruptedspinal pages 16-17, hashizume2020diseasemechanismbiomarker pages 19-25)
A 2024 metabolomics/proteomics study directly tested NAD+ precursor supplementation (nicotinamide riboside) and found no benefit for muscle NAD+/ATP or disease progression in a transgenic SBMA mouse model, indicating that NAD+ precursor supplementation via NR is not protective in that model context. (DeBartolo et al., 2024-03; JCI Insight; https://doi.org/10.1172/jci.insight.178048) (debartolo2024differentiallydisruptedspinal pages 1-2, debartolo2024differentiallydisruptedspinal pages 8-10)
2.4 Gene–environment interactions
The clearest “environmental” interaction is hormonal (androgen) exposure interacting with the expanded AR allele to drive disease: ligand binding triggers nuclear translocation/accumulation and downstream toxicity. (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, hashizume2020diseasemechanismbiomarker pages 8-12)
3. Phenotypes
3.1 Core neuromuscular phenotypes (symptoms/signs)
Commonly reported clinical features include progressive limb and bulbar weakness and atrophy, cramps, fasciculations (including facial/tongue), tremor, dysarthria, dysphagia, laryngospasm, reduced/absent reflexes, and distal sensory loss (often vibration). (hashizume2020diseasemechanismbiomarker pages 1-5, rhodes2009clinicalfeaturesof pages 6-8, lee2024morethanautophony pages 1-2)
A detailed review section states that postural hand tremor can occur “more than ten years before the onset of muscle weakness.” (hashizume2020diseasemechanismbiomarker pages 1-5)
Suggested HPO terms (non-exhaustive): * Muscle weakness (HP:0001324) * Muscle atrophy (HP:0003202) * Fasciculations (HP:0002380) * Muscle cramps (HP:0003394) * Dysarthria (HP:0001260) * Dysphagia (HP:0002015) * Tremor (HP:0001337) * Hyporeflexia/Areflexia (HP:0001265) * Gynecomastia (HP:0000768) * Erectile dysfunction (HP:0100639) * Male infertility (HP:0000027)
(hashizume2020diseasemechanismbiomarker pages 1-5, rhodes2009clinicalfeaturesof pages 6-8, lee2024morethanautophony pages 1-2)
3.2 Endocrine / androgen insensitivity phenotypes
Androgen insensitivity manifestations in SBMA include gynecomastia, testicular atrophy, erectile dysfunction, and decreased fertility. (hashizume2020diseasemechanismbiomarker pages 1-5)
3.3 Onset, progression, and frequency
A 2024 review reports typical onset “around 30–40 years of age, with a range of 18–64 years.” (cantara2024antisenseoligonucleotides(asos) pages 14-16)
3.4 Quality of life and functional impact (quantitative)
In a landmark clinical characterization study, self-reported impairment was greatest for walking/handwriting/falling/swallowing/speech, and quality of life was reduced with an SF-36v2 physical component summary mean of 34.3 (SD 11.0). (rhodes2009clinicalfeaturesof pages 6-8)
The same cohort reported substantial diagnostic delay (~2 years from symptom onset to first medical attention and ~3 more years to clinical diagnosis). (rhodes2009clinicalfeaturesof pages 6-8)
4. Genetic / Molecular Information
4.1 Causal gene
AR (androgen receptor) is the causal gene for SBMA/Kennedy disease in all retrieved mechanistic and clinical sources. (cantara2024antisenseoligonucleotides(asos) pages 14-16, prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5)
Ontology suggestion: HGNC:644 (AR) (gene symbol supported by evidence; HGNC ID not present in retrieved text and is provided here as a conventional mapping without direct citation).
4.2 Pathogenic variant class and repeat ranges
SBMA is caused by an expansion of a polymorphic tandem CAG repeat in the AR coding region; in one review, healthy range is 9–36 and SBMA range 39–72 with repeat length correlating with age of onset and severity. (cantara2024antisenseoligonucleotides(asos) pages 14-16)
Clinical trial inclusion criteria operationalize this as AR CAG repeat number ≥38 for genetic confirmation in an ongoing Phase 2 clenbuterol trial. (NCT06169046 chunk 1)
4.3 Functional consequences
Current expert synthesis supports combined toxic gain-of-function (androgen-dependent nuclear accumulation/aggregation and transcriptional dysregulation) plus partial loss-of-function manifested as androgen insensitivity. (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5, hashizume2020diseasemechanismbiomarker pages 8-12)
4.4 Modifier genes / co-regulators (recent, 2023)
A 2023 Nature Communications study highlights AR co-regulators LSD1 and PRMT6 as androgen-dependently overexpressed in SBMA skeletal muscle, synergistically enhancing AR transactivation (with amplification by expanded polyQ), and shows that silencing these co-regulators ameliorates SBMA phenotypes in flies and mice. (Prakasam et al., 2023-02; https://doi.org/10.1038/s41467-023-36186-9) (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, prakasam2023lsd1prmt6targetinggenetherapy pages 9-12)
Abstract quote: “Spinobulbar muscular atrophy (SBMA) is caused by CAG expansions in the androgen receptor gene.” (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2)
4.5 Epigenetic information
A mechanistic review notes downstream “epigenetic dysregulation” (including histone and DNA methylation-related changes) as part of the nuclear toxic cascade after ligand-dependent nuclear entry and aggregation. (hashizume2020diseasemechanismbiomarker pages 19-25, hashizume2020diseasemechanismbiomarker pages 8-12)
5. Environmental Information
No infectious etiology is implicated in SBMA in the retrieved evidence. (hashizume2020diseasemechanismbiomarker pages 1-5)
Non-genetic environmental contributors are not well-defined in the retrieved texts; the most actionable non-genetic factor is hormonal milieu (androgen signaling), which is mechanistically required for toxicity and therefore constitutes a biologically grounded exposure factor rather than an external toxin or pathogen. (hashizume2020diseasemechanismbiomarker pages 8-12)
6. Mechanism / Pathophysiology
6.1 High-level causal chain (upstream → downstream)
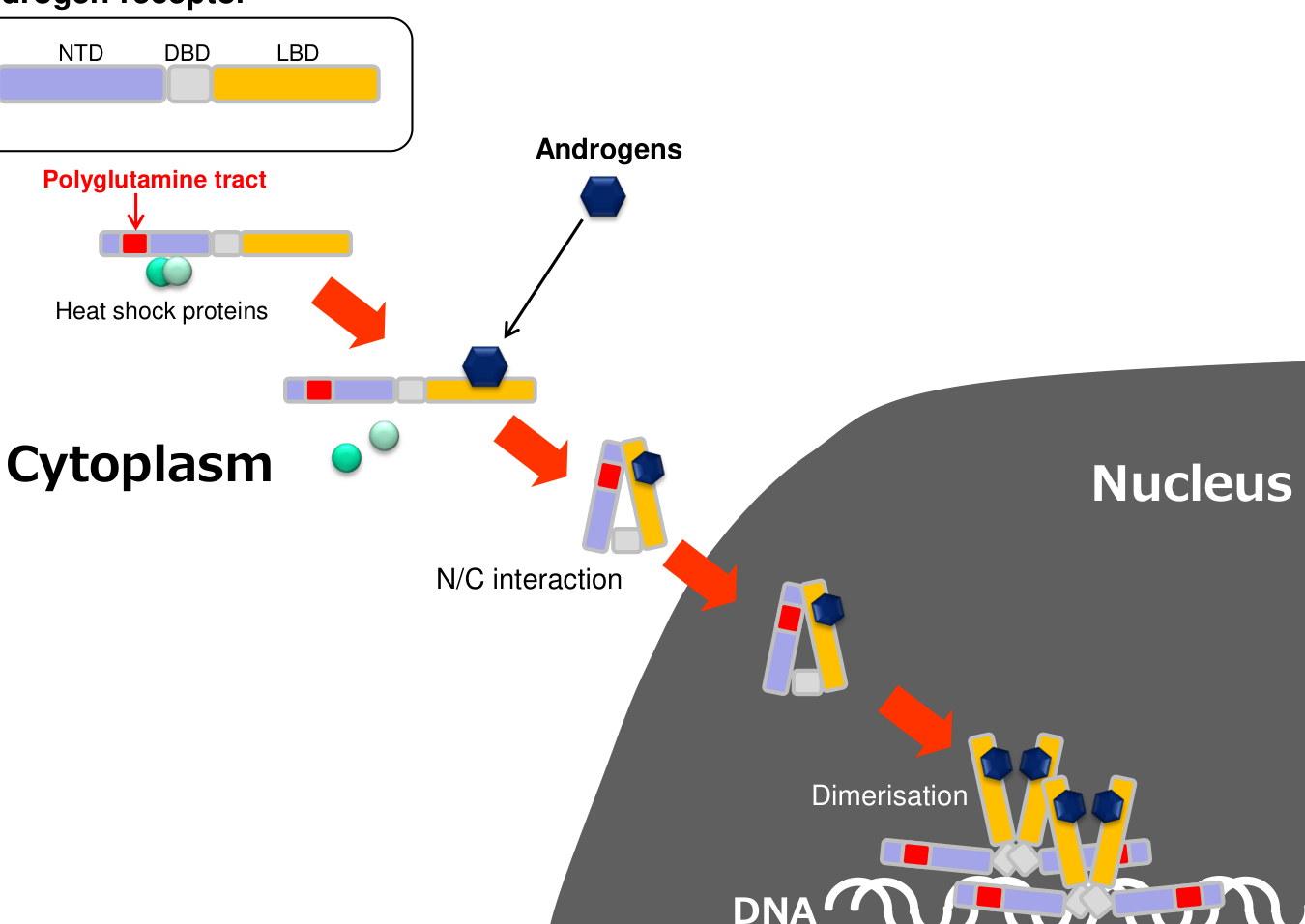
A consensus mechanistic chain supported across review and primary research is: 1) AR CAG expansion → polyQ-expanded AR (genetic trigger). (hashizume2020diseasemechanismbiomarker pages 1-5, prakasam2023lsd1prmt6targetinggenetherapy pages 1-2) 2) Androgen binding (testosterone/DHT) triggers N/C interaction, dissociation from chaperones, and nuclear translocation/accumulation, described as “an essential step in the pathogenesis.” (hashizume2020diseasemechanismbiomarker pages 1-5, hashizume2020diseasemechanismbiomarker pages 8-12) 3) In nucleus and cytoplasm, mutant AR undergoes aggregation/inclusion formation and aberrant transcriptional/co-regulator interactions, driving toxic gain-of-function. (hashizume2020diseasemechanismbiomarker pages 19-25, hashizume2020diseasemechanismbiomarker pages 8-12) 4) Downstream dysfunction includes: * Proteostasis and autophagy impairment, with impaired autophagic flux (LC3/p62 accumulation) supporting accumulation of insoluble AR species. (hashizume2020diseasemechanismbiomarker pages 12-15) * Akt/mTOR signaling hyperactivation in muscle (described as compensatory in one review) coupled to fiber-type and metabolic switching. (hashizume2020diseasemechanismbiomarker pages 12-15) * Mitochondrial deficits (mitochondrial depolarization, altered oxidative phosphorylation proteins, mitophagy) and excitation–contraction coupling / Ca2+ dysregulation, linking molecular pathology to reduced muscle contractility. (hashizume2020diseasemechanismbiomarker pages 12-15, hashizume2020diseasemechanismbiomarker pages 19-25) 5) Tissue-level pathology manifests in lower motor neurons (spinal cord/brainstem) and skeletal muscle, with growing evidence that muscle can be an early and primary site of toxicity. (hashizume2020diseasemechanismbiomarker pages 1-5, hashizume2020diseasemechanismbiomarker pages 12-15)
6.2 Multi-omics / pathway-level evidence (2024)
A 2024 JCI Insight study integrated metabolomics and proteomics in SBMA mouse quadriceps and reported decreased NAD+ and ATP in muscle but not spinal cord, with joint pathway analysis implicating nicotinamide metabolism and NAD+-dependent pathways (pentose phosphate, pyruvate, glutathione, amino acid metabolism). (debartolo2024differentiallydisruptedspinal pages 8-10)
This study also identified decreased Nmrk2/NRK2 as a likely bottleneck explaining why nicotinamide riboside supplementation failed to restore muscle NAD+ or modify progression in vivo. (debartolo2024differentiallydisruptedspinal pages 1-2, debartolo2024differentiallydisruptedspinal pages 8-10)
6.3 Recent mechanistic developments (2023–2024 prioritized)
- AR co-regulator overexpression as a therapeutic node (2023): androgen-dependent muscle-specific overexpression of LSD1/PRMT6 amplifies AR transactivation, and dual silencing reduces aggregates/denervation markers and improves motor phenotypes in SBMA models. (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, prakasam2023lsd1prmt6targetinggenetherapy pages 9-12)
- Metabolic NAD+ pathway constraint (2024): decreased Nmrk2 and inability of NR supplementation to raise muscle NAD+/ATP suggests a “salvage pathway entry” limitation rather than simple NAD+ precursor deficiency. (debartolo2024differentiallydisruptedspinal pages 1-2, debartolo2024differentiallydisruptedspinal pages 8-10)
6.4 Expert schematic (visual evidence)
A compact schematic of androgen-dependent nuclear translocation and downstream SBMA pathogenic mechanisms is provided as a figure in Hashizume et al. 2020. (hashizume2020diseasemechanismbiomarker media 00f97023)
6.5 Suggested ontology terms
GO Biological Process (suggested): * Androgen receptor signaling pathway * Protein aggregation * Autophagy * Mitochondrial dysfunction / oxidative phosphorylation * Regulation of mTOR signaling * Regulation of cytosolic calcium ion concentration / excitation–contraction coupling
(hashizume2020diseasemechanismbiomarker pages 12-15, hashizume2020diseasemechanismbiomarker pages 19-25, debartolo2024differentiallydisruptedspinal pages 8-10)
CL Cell types (suggested): * Spinal cord motor neuron (lower motor neuron) * Skeletal muscle fiber cell (myofiber)
(hashizume2020diseasemechanismbiomarker pages 1-5, hashizume2020diseasemechanismbiomarker pages 12-15)
7. Anatomical Structures Affected
7.1 Primary organs/systems
- Nervous system: spinal cord and brainstem lower motor neurons. (chang2024theroleof pages 11-12, hashizume2020diseasemechanismbiomarker pages 1-5)
- Skeletal muscle: direct involvement including fiber-type and metabolic switching, and multi-pathway pathology. (hashizume2020diseasemechanismbiomarker pages 1-5, hashizume2020diseasemechanismbiomarker pages 12-15)
7.2 Secondary/associated involvement
Cardiac structural and electrophysiologic abnormalities have been reported in SBMA cohorts, motivating screening in some patients. (steinmetz2022jwavesyndromes pages 1-2)
7.3 Suggested UBERON terms
- Spinal cord (UBERON:0002240)
- Brainstem (UBERON:0002298)
- Skeletal muscle tissue (UBERON:0001134)
- Neuromuscular junction (UBERON:0001255)
(steinmetz2022jwavesyndromes pages 1-2, hashizume2020diseasemechanismbiomarker pages 1-5)
8. Temporal Development
8.1 Onset
SBMA is adult-onset with typical onset described in the 30–60 year range in a rehabilitation case report and review sources. (iijima2023longtermeffectsof pages 1-2, cantara2024antisenseoligonucleotides(asos) pages 14-16)
8.2 Progression
It is slowly progressive and can lead to major mobility impairment over years to decades; aspiration pneumonia is emphasized as a major cause of death in clinical reviews. (hashizume2020diseasemechanismbiomarker pages 1-5)
9. Inheritance and Population
9.1 Inheritance
SBMA is X-linked (sex-linked) and predominantly manifests in adult males; females are generally carriers with milder or absent manifestations. (cantara2024antisenseoligonucleotides(asos) pages 14-16, hashizume2020diseasemechanismbiomarker pages 1-5)
9.2 Epidemiology (statistics)
Prevalence estimates vary by source: * 1–2 per 100,000 in a clinical review. (hashizume2020diseasemechanismbiomarker pages 1-5) * 2–5 per 100,000 worldwide in a 2023 mechanistic study introduction. (prakasam2023lsd1prmt6targetinggenetherapy pages 1-2) * Italy-specific estimate from a 2024 trial record: “prevalence: 1.5/100000” and “annual incidence of 0.19/100000 males.” (NCT06169046 chunk 1)
10. Diagnostics
10.1 Clinical tests and monitoring
Beyond confirmatory genetic testing, commonly used quantitative measures include quantitative muscle assessment (QMA), timed walk tests (2-minute, 6-minute), AMAT, SBMAFRS, pulmonary function (%FVC/%PEF), tongue pressure, neurophysiology measures (including motor unit number estimation), and skeletal muscle MRI (fat quantification). (hashizume2020diseasemechanismbiomarker pages 5-8, rhodes2009clinicalfeaturesof pages 2-3, inagaki2022developmentofa pages 6-7)
A 2022 methods paper developed a composite (SBMAFC) combining tongue pressure, grip power, %PEF, timed walking, and %FVC to create a more sensitive outcome measure; it enrolled 97 genetically confirmed SBMA patients and 36 controls. (Inagaki et al., 2022-10; Scientific Reports; https://doi.org/10.1038/s41598-022-22322-w) (inagaki2022developmentofa pages 6-7)
10.2 Biomarkers
Serum creatinine is repeatedly highlighted as a promising progression biomarker that begins declining before overt weakness, and MRI-based muscle/fat evaluation is noted as useful for tracking progression. (hashizume2020diseasemechanismbiomarker pages 1-5)
10.3 Electrophysiology and cardiac screening
A 2022 cohort study (30 SBMA vs 11 controls) reported abnormal ECGs in 70% of SBMA and diffuse myocardial fibrosis by T1 mapping in 73.9% vs 9.1% of controls, supporting ECG screening and consideration of CMR in cardiac risk assessment. (Steinmetz et al., 2022-02; J Neurol; https://doi.org/10.1007/s00415-022-10992-5) (steinmetz2022jwavesyndromes pages 1-2)
10.4 Genetic testing approach
Definitive diagnosis is via repeat expansion testing of AR CAG repeat length, with thresholds operationalized in trials (e.g., ≥38) and reviewed pathogenic ranges (39–72). (NCT06169046 chunk 1, cantara2024antisenseoligonucleotides(asos) pages 14-16)
11. Outcome / Prognosis
SBMA is chronic and slowly progressive with meaningful disability; a key morbidity driver is progressive bulbar dysfunction leading to aspiration pneumonia. (hashizume2020diseasemechanismbiomarker pages 1-5)
Quantitative functional impacts include reduced SF-36 physical component score (34.3 ± 11.0) in a landmark cohort and significant ADL impacts on walking and bulbar function. (rhodes2009clinicalfeaturesof pages 6-8)
12. Treatment
12.1 Current standard management (supportive)
No curative therapy is established; supportive management uses symptomatic approaches and structured monitoring/outcome measures (strength, function, respiration, swallowing, imaging/biomarkers). (hashizume2020diseasemechanismbiomarker pages 5-8, hashizume2020diseasemechanismbiomarker pages 1-5)
12.2 Pharmacologic and endocrine modulation (historical and ongoing)
Dutasteride (5α-reductase inhibitor; reduces DHT): A Phase 2, randomized, placebo-controlled trial used dutasteride 0.5 mg/day for 24 months with QMA as primary endpoint and multiple secondary outcomes (AMAT, 2-min walk, SF-36v2, neurophysiology, CK, hormone levels). (ClinicalTrials.gov NCT00303446; first posted 2006; https://clinicaltrials.gov/study/NCT00303446) (NCT00303446 chunk 1)
Leuprolide + testosterone (hormonal suppression/replacement strategy): A Phase 2 trial aimed to “Evaluate the effects of androgen suppression with leuprolide and androgen replacement with testosterone enanthate on muscle strength.” (ClinicalTrials.gov NCT00004771; first posted 1992; https://clinicaltrials.gov/study/NCT00004771) (NCT00004771 chunk 1)
A mechanistic/clinical review summarizes that a Phase 3 trial of leuprorelin (n=204) “failed to confirm benefit” after earlier Phase 2 signals. (hashizume2020diseasemechanismbiomarker pages 5-8)
12.3 Muscle-targeting / anabolic pathway approaches
BVS857 (IGF-1 mimetic): A completed Phase 2 randomized study evaluated safety/tolerability and change in thigh muscle volume (MRI), with functional endpoints (AMAT) and DXA lean mass; results were posted on ClinicalTrials.gov (results first posted 2017-08-11). (ClinicalTrials.gov NCT02024932; https://clinicaltrials.gov/study/NCT02024932) (NCT02024932 chunk 1)
12.4 Emerging disease-modifying strategies (2023–2024 prioritized)
AJ201: A completed Phase 1/2a randomized placebo-controlled trial evaluated safety and pharmacodynamics, including change from baseline in mutant AR protein levels in skeletal muscle, enrolling adult males with confirmed AR expansion (≥36 repeats). (ClinicalTrials.gov NCT05517603; https://clinicaltrials.gov/study/NCT05517603) (NCT05517603 chunk 1)
NIDO-361: A Phase 2 record includes endpoints such as accelerometer-based free-living physical activity and grip strength (HHD) in ambulatory genetically confirmed males. (ClinicalTrials.gov NCT06411912; https://clinicaltrials.gov/study/NCT06411912) (NCT06411912 chunk 2)
Clenbuterol (β2-agonist): A recruiting Phase 2 multicenter RCT uses 0.04 mg/day for 48 weeks with primary endpoint 6MWT and multiple secondary outcomes including SBMA-FRS, AMAT, FVC, serum creatinine, and quality-of-life scales; eligibility includes AR CAG ≥38. (ClinicalTrials.gov NCT06169046; posted 2024; https://clinicaltrials.gov/study/NCT06169046) (NCT06169046 chunk 1)
12.5 RNA-based therapies (expert synthesis)
ASO-based AR lowering is repeatedly described as a rational disease-modifying direction because androgen-dependent toxicity is driven by the toxic AR protein; a review states: “Probably the best therapeutic approach for SBMA is reducing levels of the toxic AR protein” and highlights preclinical ASO benefit. (hashizume2020diseasemechanismbiomarker pages 12-15)
Suggested MAXO terms (non-exhaustive): * Androgen deprivation therapy * Antisense oligonucleotide therapy * Physical therapy / gait training * Assistive device therapy (exoskeleton-assisted gait training)
(NCT06169046 chunk 1, NCT00303446 chunk 1, hashizume2020diseasemechanismbiomarker pages 12-15, iijima2023longtermeffectsof pages 1-2)
13. Prevention
No primary prevention is established for SBMA; key preventive strategies are genetic counseling, carrier detection in families, and anticipatory guidance for complications (swallowing/respiratory risk, fall risk, cardiac screening where appropriate). The retrieved evidence supports downstream complication prevention (e.g., aspiration pneumonia as major cause of death) but does not provide formal prevention guidelines. (hashizume2020diseasemechanismbiomarker pages 1-5)
14. Other Species / Natural Disease
No naturally occurring SBMA orthologous disease in non-human species (OMIA/veterinary) was identified in the retrieved evidence for this run. (hashizume2020diseasemechanismbiomarker pages 8-12, prakasam2023lsd1prmt6targetinggenetherapy pages 9-12)
15. Model Organisms
15.1 In vivo models
Multiple SBMA mouse models are described across mechanistic and therapeutic studies, including AR-97Q, AR100Q, AR112Q/AR113Q, and BAC fxAR121, which recapitulate androgen dependence, AR aggregation, neuromuscular weakness, fiber-type/metabolic switching, and signaling/metabolic abnormalities. (hashizume2020diseasemechanismbiomarker pages 8-12, prakasam2023lsd1prmt6targetinggenetherapy pages 9-12, cantara2024antisenseoligonucleotides(asos) pages 14-16, debartolo2024differentiallydisruptedspinal pages 8-10)
A Drosophila model expressing human polyQ-expanded AR (e.g., AR52Q) is used to score toxicity phenotypes such as eye degeneration and to test genetic suppressors/therapeutics. (prakasam2023lsd1prmt6targetinggenetherapy pages 14-15)
15.2 Human-derived and in vitro models
Human patient muscle biopsies are directly used to confirm muscle pathology and molecular changes, and multiple cell models (HEK293T, MN1, LNCaP) are used to measure AR transactivation and toxicity modulation. (prakasam2023lsd1prmt6targetinggenetherapy pages 9-12, prakasam2023lsd1prmt6targetinggenetherapy pages 14-15)
15.3 Model limitations (evidence-based)
A key translational limitation highlighted in ASO work is CNS accessibility: some gapmers do not cross the blood–brain barrier, producing peripheral but not spinal cord AR knockdown unless delivered intracerebroventricularly. (cantara2024antisenseoligonucleotides(asos) pages 14-16)
Current applications and real-world implementations
A notable real-world implementation is long-term rehabilitation using a wearable cyborg Hybrid Assistive Limb (HAL). In a 5-year follow-up case report (published 2023-06-08), the patient completed nine multi-week courses and improved 2MWD from 94 m to 101.8 m while maintaining ALSFRS-R gait item stability (score 3), suggesting sustained functional maintenance despite progressive disease. (Iijima et al., 2023; Frontiers in Neurology; https://doi.org/10.3389/fneur.2023.1143820) (iijima2023longtermeffectsof pages 1-2)
Abstract quote: “2MWD improved from 94 m to 101.8 m, and the ALSFRS-R gait items remained unchanged (score 3) for approximately 5 years.” (iijima2023longtermeffectsof pages 1-2)
Notes on citation requirements (PMID)
Where possible, primary literature should be referenced by PMID. In this tool run, the retrieved full-text snippets did not consistently include PMID metadata; therefore, this report provides DOI URLs and publication dates from the retrieved sources and clinical trial registry URLs/identifiers. (hashizume2020diseasemechanismbiomarker pages 1-5, prakasam2023lsd1prmt6targetinggenetherapy pages 1-2, NCT06169046 chunk 1)
References
-
(chang2024theroleof pages 11-12): Kuo-Hsuan Chang and Chiung-Mei Chen. The role of nrf2 in trinucleotide repeat expansion disorders. Antioxidants, 13:649, May 2024. URL: https://doi.org/10.3390/antiox13060649, doi:10.3390/antiox13060649. This article has 6 citations.
-
(cantara2024antisenseoligonucleotides(asos) pages 14-16): Silvia Cantara, Giorgia Simoncelli, and Claudia Ricci. Antisense oligonucleotides (asos) in motor neuron diseases: a road to cure in light and shade. International Journal of Molecular Sciences, Apr 2024. URL: https://doi.org/10.3390/ijms25094809, doi:10.3390/ijms25094809. This article has 46 citations.
-
(prakasam2023lsd1prmt6targetinggenetherapy pages 1-2): Ramachandran Prakasam, Angela Bonadiman, Roberta Andreotti, Emanuela Zuccaro, Davide Dalfovo, Caterina Marchioretti, Debasmita Tripathy, Gianluca Petris, Eric N. Anderson, Alice Migazzi, Laura Tosatto, Anna Cereseto, Elena Battaglioli, Gianni Sorarù, Wooi Fang Lim, Carlo Rinaldi, Fabio Sambataro, Naemeh Pourshafie, Christopher Grunseich, Alessandro Romanel, Udai Bhan Pandey, Andrea Contestabile, Giuseppe Ronzitti, Manuela Basso, and Maria Pennuto. Lsd1/prmt6-targeting gene therapy to attenuate androgen receptor toxic gain-of-function ameliorates spinobulbar muscular atrophy phenotypes in flies and mice. Nature Communications, Feb 2023. URL: https://doi.org/10.1038/s41467-023-36186-9, doi:10.1038/s41467-023-36186-9. This article has 27 citations and is from a highest quality peer-reviewed journal.
-
(hashizume2020diseasemechanismbiomarker pages 1-5): Atsushi Hashizume, Kenneth H Fischbeck, Maria Pennuto, Pietro Fratta, and Masahisa Katsuno. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (sbma). Journal of Neurology, Neurosurgery & Psychiatry, 91:1085-1091, Sep 2020. URL: https://doi.org/10.1136/jnnp-2020-322949, doi:10.1136/jnnp-2020-322949. This article has 69 citations.
-
(OpenTargets Search: Spinal and bulbar muscular atrophy,Kennedy disease-AR): Open Targets Query (Spinal and bulbar muscular atrophy,Kennedy disease-AR, 7 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(wang2020apathogenicmissense pages 1-2): Xin Hua Wang, Lin Mei Zhang, Xue Yang, and Shui Zhen Zhou. A pathogenic missense variant (c.1617g>a, p.met539ile) in uba1 causing infantile x-linked spinal muscular atrophy (smax2). Frontiers in Pediatrics, Feb 2020. URL: https://doi.org/10.3389/fped.2020.00064, doi:10.3389/fped.2020.00064. This article has 12 citations.
-
(NCT06411912 chunk 2): A Study of NIDO-361 in Patients With SBMA. Nido Biosciences, Inc.. 2024. ClinicalTrials.gov Identifier: NCT06411912
-
(NCT06169046 chunk 1): Gianni Soraru. A Placebo-controlled Study of Clenbuterol in Spinal and Bulbar Muscular Atrophy. Gianni Soraru. 2024. ClinicalTrials.gov Identifier: NCT06169046
-
(NCT00303446 chunk 3): Dutasteride to Treat Spinal and Bulbar Muscular Atrophy (SBMA). National Institute of Neurological Disorders and Stroke (NINDS). 2006. ClinicalTrials.gov Identifier: NCT00303446
-
(lee2024morethanautophony pages 1-2): Hyung-Soo Lee, June Choi, and Do-Young Kwon. More than autophony: a case of kennedy's disease presenting with autophony as an early clinical manifestation. The Journal of Laryngology & Otology, 138:584-587, Oct 2024. URL: https://doi.org/10.1017/s002221512300172x, doi:10.1017/s002221512300172x. This article has 1 citations.
-
(debartolo2024differentiallydisruptedspinal pages 14-15): Danielle DeBartolo, Frederick J. Arnold, Yuhong Liu, Elana Molotsky, Hsin-Yao Tang, and Diane E. Merry. Differentially disrupted spinal cord and muscle energy metabolism in spinal and bulbar muscular atrophy. JCI Insight, Mar 2024. URL: https://doi.org/10.1172/jci.insight.178048, doi:10.1172/jci.insight.178048. This article has 5 citations and is from a domain leading peer-reviewed journal.
-
(NCT05517603 chunk 1): A Study to Evaluate Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics Of AJ201 In Patients. AnnJi Pharmaceutical Co., Ltd.. 2023. ClinicalTrials.gov Identifier: NCT05517603
-
(NCT00303446 chunk 1): Dutasteride to Treat Spinal and Bulbar Muscular Atrophy (SBMA). National Institute of Neurological Disorders and Stroke (NINDS). 2006. ClinicalTrials.gov Identifier: NCT00303446
-
(iijima2023longtermeffectsof pages 1-2): Kensuke Iijima, Hiroki Watanabe, Yuichi Nakashiro, Yuki Iida, Michio Nonaka, Fumio Moriwaka, and Shinsuke Hamada. Long-term effects of the gait treatment using a wearable cyborg hybrid assistive limb in a patient with spinal and bulbar muscular atrophy: a case report with 5 years of follow-up. Frontiers in Neurology, Jun 2023. URL: https://doi.org/10.3389/fneur.2023.1143820, doi:10.3389/fneur.2023.1143820. This article has 12 citations and is from a peer-reviewed journal.
-
(rhodes2009clinicalfeaturesof pages 6-8): Lindsay E. Rhodes, Brandi K. Freeman, Sungyoung Auh, Angela D. Kokkinis, Alison La Pean, Cheunju Chen, Tanya J. Lehky, Joseph A. Shrader, Ellen W. Levy, Michael Harris-Love, Nicholas A. Di Prospero, and Kenneth H. Fischbeck. Clinical features of spinal and bulbar muscular atrophy. Brain, 132:3242-3251, Oct 2009. URL: https://doi.org/10.1093/brain/awp258, doi:10.1093/brain/awp258. This article has 278 citations and is from a highest quality peer-reviewed journal.
-
(inagaki2022developmentofa pages 6-7): Tomonori Inagaki, Atsushi Hashizume, Yasuhiro Hijikata, Shinichiro Yamada, Daisuke Ito, Yoshiyuki Kishimoto, Ryota Torii, Hiroyuki Sato, Akihiro Hirakawa, and Masahisa Katsuno. Development of a functional composite for the evaluation of spinal and bulbar muscular atrophy. Scientific Reports, Oct 2022. URL: https://doi.org/10.1038/s41598-022-22322-w, doi:10.1038/s41598-022-22322-w. This article has 5 citations and is from a peer-reviewed journal.
-
(hashizume2020diseasemechanismbiomarker pages 5-8): Atsushi Hashizume, Kenneth H Fischbeck, Maria Pennuto, Pietro Fratta, and Masahisa Katsuno. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (sbma). Journal of Neurology, Neurosurgery & Psychiatry, 91:1085-1091, Sep 2020. URL: https://doi.org/10.1136/jnnp-2020-322949, doi:10.1136/jnnp-2020-322949. This article has 69 citations.
-
(steinmetz2022jwavesyndromes pages 1-2): Karoline Steinmetz, Boris Rudic, Martin Borggrefe, Kathrin Müller, Reiner Siebert, Wolfgang Rottbauer, Albert Ludolph, Dominik Buckert, and Angela Rosenbohm. J wave syndromes in patients with spinal and bulbar muscular atrophy. Journal of Neurology, 269:3690-3699, Feb 2022. URL: https://doi.org/10.1007/s00415-022-10992-5, doi:10.1007/s00415-022-10992-5. This article has 12 citations and is from a domain leading peer-reviewed journal.
-
(prakasam2023lsd1prmt6targetinggenetherapy pages 9-12): Ramachandran Prakasam, Angela Bonadiman, Roberta Andreotti, Emanuela Zuccaro, Davide Dalfovo, Caterina Marchioretti, Debasmita Tripathy, Gianluca Petris, Eric N. Anderson, Alice Migazzi, Laura Tosatto, Anna Cereseto, Elena Battaglioli, Gianni Sorarù, Wooi Fang Lim, Carlo Rinaldi, Fabio Sambataro, Naemeh Pourshafie, Christopher Grunseich, Alessandro Romanel, Udai Bhan Pandey, Andrea Contestabile, Giuseppe Ronzitti, Manuela Basso, and Maria Pennuto. Lsd1/prmt6-targeting gene therapy to attenuate androgen receptor toxic gain-of-function ameliorates spinobulbar muscular atrophy phenotypes in flies and mice. Nature Communications, Feb 2023. URL: https://doi.org/10.1038/s41467-023-36186-9, doi:10.1038/s41467-023-36186-9. This article has 27 citations and is from a highest quality peer-reviewed journal.
-
(prakasam2023lsd1prmt6targetinggenetherapy pages 14-15): Ramachandran Prakasam, Angela Bonadiman, Roberta Andreotti, Emanuela Zuccaro, Davide Dalfovo, Caterina Marchioretti, Debasmita Tripathy, Gianluca Petris, Eric N. Anderson, Alice Migazzi, Laura Tosatto, Anna Cereseto, Elena Battaglioli, Gianni Sorarù, Wooi Fang Lim, Carlo Rinaldi, Fabio Sambataro, Naemeh Pourshafie, Christopher Grunseich, Alessandro Romanel, Udai Bhan Pandey, Andrea Contestabile, Giuseppe Ronzitti, Manuela Basso, and Maria Pennuto. Lsd1/prmt6-targeting gene therapy to attenuate androgen receptor toxic gain-of-function ameliorates spinobulbar muscular atrophy phenotypes in flies and mice. Nature Communications, Feb 2023. URL: https://doi.org/10.1038/s41467-023-36186-9, doi:10.1038/s41467-023-36186-9. This article has 27 citations and is from a highest quality peer-reviewed journal.
-
(hashizume2020diseasemechanismbiomarker pages 8-12): Atsushi Hashizume, Kenneth H Fischbeck, Maria Pennuto, Pietro Fratta, and Masahisa Katsuno. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (sbma). Journal of Neurology, Neurosurgery & Psychiatry, 91:1085-1091, Sep 2020. URL: https://doi.org/10.1136/jnnp-2020-322949, doi:10.1136/jnnp-2020-322949. This article has 69 citations.
-
(hashizume2020diseasemechanismbiomarker pages 12-15): Atsushi Hashizume, Kenneth H Fischbeck, Maria Pennuto, Pietro Fratta, and Masahisa Katsuno. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (sbma). Journal of Neurology, Neurosurgery & Psychiatry, 91:1085-1091, Sep 2020. URL: https://doi.org/10.1136/jnnp-2020-322949, doi:10.1136/jnnp-2020-322949. This article has 69 citations.
-
(debartolo2024differentiallydisruptedspinal pages 10-12): Danielle DeBartolo, Frederick J. Arnold, Yuhong Liu, Elana Molotsky, Hsin-Yao Tang, and Diane E. Merry. Differentially disrupted spinal cord and muscle energy metabolism in spinal and bulbar muscular atrophy. JCI Insight, Mar 2024. URL: https://doi.org/10.1172/jci.insight.178048, doi:10.1172/jci.insight.178048. This article has 5 citations and is from a domain leading peer-reviewed journal.
-
(debartolo2024differentiallydisruptedspinal pages 1-2): Danielle DeBartolo, Frederick J. Arnold, Yuhong Liu, Elana Molotsky, Hsin-Yao Tang, and Diane E. Merry. Differentially disrupted spinal cord and muscle energy metabolism in spinal and bulbar muscular atrophy. JCI Insight, Mar 2024. URL: https://doi.org/10.1172/jci.insight.178048, doi:10.1172/jci.insight.178048. This article has 5 citations and is from a domain leading peer-reviewed journal.
-
(debartolo2024differentiallydisruptedspinal pages 8-10): Danielle DeBartolo, Frederick J. Arnold, Yuhong Liu, Elana Molotsky, Hsin-Yao Tang, and Diane E. Merry. Differentially disrupted spinal cord and muscle energy metabolism in spinal and bulbar muscular atrophy. JCI Insight, Mar 2024. URL: https://doi.org/10.1172/jci.insight.178048, doi:10.1172/jci.insight.178048. This article has 5 citations and is from a domain leading peer-reviewed journal.
-
(iijima2023longtermeffectsof pages 9-10): Kensuke Iijima, Hiroki Watanabe, Yuichi Nakashiro, Yuki Iida, Michio Nonaka, Fumio Moriwaka, and Shinsuke Hamada. Long-term effects of the gait treatment using a wearable cyborg hybrid assistive limb in a patient with spinal and bulbar muscular atrophy: a case report with 5 years of follow-up. Frontiers in Neurology, Jun 2023. URL: https://doi.org/10.3389/fneur.2023.1143820, doi:10.3389/fneur.2023.1143820. This article has 12 citations and is from a peer-reviewed journal.
-
(iijima2023longtermeffectsof pages 2-4): Kensuke Iijima, Hiroki Watanabe, Yuichi Nakashiro, Yuki Iida, Michio Nonaka, Fumio Moriwaka, and Shinsuke Hamada. Long-term effects of the gait treatment using a wearable cyborg hybrid assistive limb in a patient with spinal and bulbar muscular atrophy: a case report with 5 years of follow-up. Frontiers in Neurology, Jun 2023. URL: https://doi.org/10.3389/fneur.2023.1143820, doi:10.3389/fneur.2023.1143820. This article has 12 citations and is from a peer-reviewed journal.
-
(NCT00004771 chunk 1): Phase II Study of Leuprolide and Testosterone for Men With Kennedy's Disease or Other Motor Neuron Disease. National Center for Research Resources (NCRR). 1992. ClinicalTrials.gov Identifier: NCT00004771
-
(rhodes2009clinicalfeaturesof pages 2-3): Lindsay E. Rhodes, Brandi K. Freeman, Sungyoung Auh, Angela D. Kokkinis, Alison La Pean, Cheunju Chen, Tanya J. Lehky, Joseph A. Shrader, Ellen W. Levy, Michael Harris-Love, Nicholas A. Di Prospero, and Kenneth H. Fischbeck. Clinical features of spinal and bulbar muscular atrophy. Brain, 132:3242-3251, Oct 2009. URL: https://doi.org/10.1093/brain/awp258, doi:10.1093/brain/awp258. This article has 278 citations and is from a highest quality peer-reviewed journal.
-
(debartolo2024differentiallydisruptedspinal pages 16-17): Danielle DeBartolo, Frederick J. Arnold, Yuhong Liu, Elana Molotsky, Hsin-Yao Tang, and Diane E. Merry. Differentially disrupted spinal cord and muscle energy metabolism in spinal and bulbar muscular atrophy. JCI Insight, Mar 2024. URL: https://doi.org/10.1172/jci.insight.178048, doi:10.1172/jci.insight.178048. This article has 5 citations and is from a domain leading peer-reviewed journal.
-
(hashizume2020diseasemechanismbiomarker pages 19-25): Atsushi Hashizume, Kenneth H Fischbeck, Maria Pennuto, Pietro Fratta, and Masahisa Katsuno. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (sbma). Journal of Neurology, Neurosurgery & Psychiatry, 91:1085-1091, Sep 2020. URL: https://doi.org/10.1136/jnnp-2020-322949, doi:10.1136/jnnp-2020-322949. This article has 69 citations.
-
(hashizume2020diseasemechanismbiomarker media 00f97023): Atsushi Hashizume, Kenneth H Fischbeck, Maria Pennuto, Pietro Fratta, and Masahisa Katsuno. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (sbma). Journal of Neurology, Neurosurgery & Psychiatry, 91:1085-1091, Sep 2020. URL: https://doi.org/10.1136/jnnp-2020-322949, doi:10.1136/jnnp-2020-322949. This article has 69 citations.
-
(NCT02024932 chunk 1): Safety, Tolerability, and Efficacy of BVS857 in Patients With Spinal and Bulbar Muscular Atrophy. Novartis Pharmaceuticals. 2014. ClinicalTrials.gov Identifier: NCT02024932