Congenital Sialidosis Type II (Hydropic/Congenital Severe Sialidosis): Comprehensive Disease Characteristics Report
Executive summary
Congenital sialidosis type II is the most severe prenatal-onset end of the NEU1-related lysosomal storage disorder spectrum (“sialidosis”), typically presenting in utero or at birth with nonimmune hydrops fetalis, ascites, and generalized edema and often resulting in stillbirth or death shortly after birth. It is caused by biallelic loss-of-function variants in NEU1 (autosomal recessive inheritance) leading to near-complete deficiency of lysosomal neuraminidase-1 (sialidase) activity and subsequent lysosomal accumulation and urinary excretion of sialylated oligosaccharides and glycopeptides (“sialyl-oligosacchariduria”). Recent 2023–2024 studies emphasize additional downstream mechanisms beyond “storage,” including dysregulated lysosomal exocytosis (via LAMP1 hypersialylation) and organ-specific consequences such as kidney disease (“nephrosialidosis”) linked to hypersialylation-dependent megalin trafficking defects. (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, kho2023severekidneydysfunction pages 1-2)
1. Disease information
1.1 Definition and overview
Sialidosis (mucolipidosis I) is an ultra-rare, autosomal recessive lysosomal storage disease caused by deficiency of lysosomal neuraminidase-1 (NEU1), leading to accumulation and excretion of sialylated metabolites. Clinical severity ranges from type I (attenuated) to type II (severe neuropathic). Type II is further subdivided into congenital/hydropic, infantile, and juvenile forms based on age at onset. (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, khan2018sialidosisareview pages 1-3)
The congenital/hydropic subtype is described as the severe prenatal form: “It manifests itself prenatally and is characterized by ascites and hydrops fetalis, hepatomegaly and stillbirths or death at a very early age.” (khan2018sialidosisareview pages 3-5)
1.2 Key identifiers
Table (click to expand)
| Identifier system | ID | Preferred name | Synonyms / notes | Source URL | Publication date |
|---|---|---|---|---|---|
| MONDO | MONDO_0009738 | sialidosis type 2 | Disease entity for severe early-onset NEU1-related sialidosis; congenital/hydropic, infantile, and juvenile forms are subtypes/clinical subdivisions of type II (OpenTargets Search: Sialidosis, khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5) | https://platform.opentargets.org/disease/MONDO_0009738 | — |
| Orphanet | Orphanet_87876 | sialidosis type II | Orphanet disease entry corresponding to type II sialidosis; congenital (hydropic) form is the prenatal/neonatal severe subtype (OpenTargets Search: Sialidosis, khan2018sialidosisareview pages 3-5) | https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=87876 | — |
| OMIM / MIM | 256550 | Sialidosis | Core inherited disorder caused by NEU1 deficiency; type II includes congenital/hydropic subtype; review explicitly cites “Sialidosis (MIM #256550)” (khan2018sialidosisareview pages 3-5, khan2018sialidosisareview pages 1-3) | https://omim.org/entry/256550 | 2018 (review citing MIM) |
| MeSH | Not clearly established for congenital type II as a distinct MeSH term | Sialidosis / Mucolipidosis I (broader indexing may vary) | Literature commonly indexes the broader disorder rather than a separate congenital type II term; use disease-level MeSH with subtype noted in free text (khan2018sialidosisareview pages 1-3, peng2025geneticinsightsand pages 1-2) | https://meshb.nlm.nih.gov/ | — |
| ICD-10 | No specific code identified for congenital type II | Sialidosis type II, congenital (hydropic) subtype | Usually classified under broader lysosomal storage/metabolic disorder categories rather than a unique congenital-type-specific ICD-10 code; verify locally in coding system implementation (khan2018sialidosisareview pages 3-5) | https://icd.who.int/browse10/2019/en | — |
| ICD-11 | No specific code confirmed from retrieved evidence | Sialidosis type II, congenital (hydropic) subtype | Retrieved evidence supports nomenclature but not a distinct ICD-11 identifier for the congenital subtype; map to broader sialidosis entry if needed after terminology validation (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5) | https://icd.who.int/ | — |
| Clinical subtype designation | — | congenital sialidosis type II | Also called congenital or hydropic subtype; manifests prenatally and is characterized by ascites, hydrops fetalis, hepatomegaly, and stillbirth or very early death (khan2018sialidosisareview pages 3-5) | https://doi.org/10.3390/diagnostics8020029 | 2018 |
| Clinical subtype designation | — | sialidosis type II, congenital/hydropic form | Severe neuropathic NEU1-deficiency phenotype within type II; recent preclinical gene-therapy paper uses the same classification framework (vlekkert2024aavmediatedgenetherapy pages 1-5) | https://doi.org/10.1101/2023.11.10.566667 | 2024 |
Table: This table summarizes the main identifiers and naming conventions for congenital sialidosis type II, linking the congenital/hydropic form to the broader type II disease entity. It is useful for harmonizing OMIM, MONDO, Orphanet, and coding terminology in a disease knowledge base.
Notes: In the retrieved evidence, identifiers are most consistently available at the disease-entity level (sialidosis; sialidosis type II) rather than as a separate MONDO/Orphanet entity specifically for the “congenital/hydropic” subtype. (OpenTargets Search: Sialidosis, khan2018sialidosisareview pages 3-5)
1.3 Synonyms and alternative names
Commonly used: - Sialidosis type II, congenital/hydropic subtype (or “congenital sialidosis”) (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5) - Mucolipidosis I / sialidosis (vlekkert2024aavmediatedgenetherapy pages 1-5)
1.4 Source type of disease information
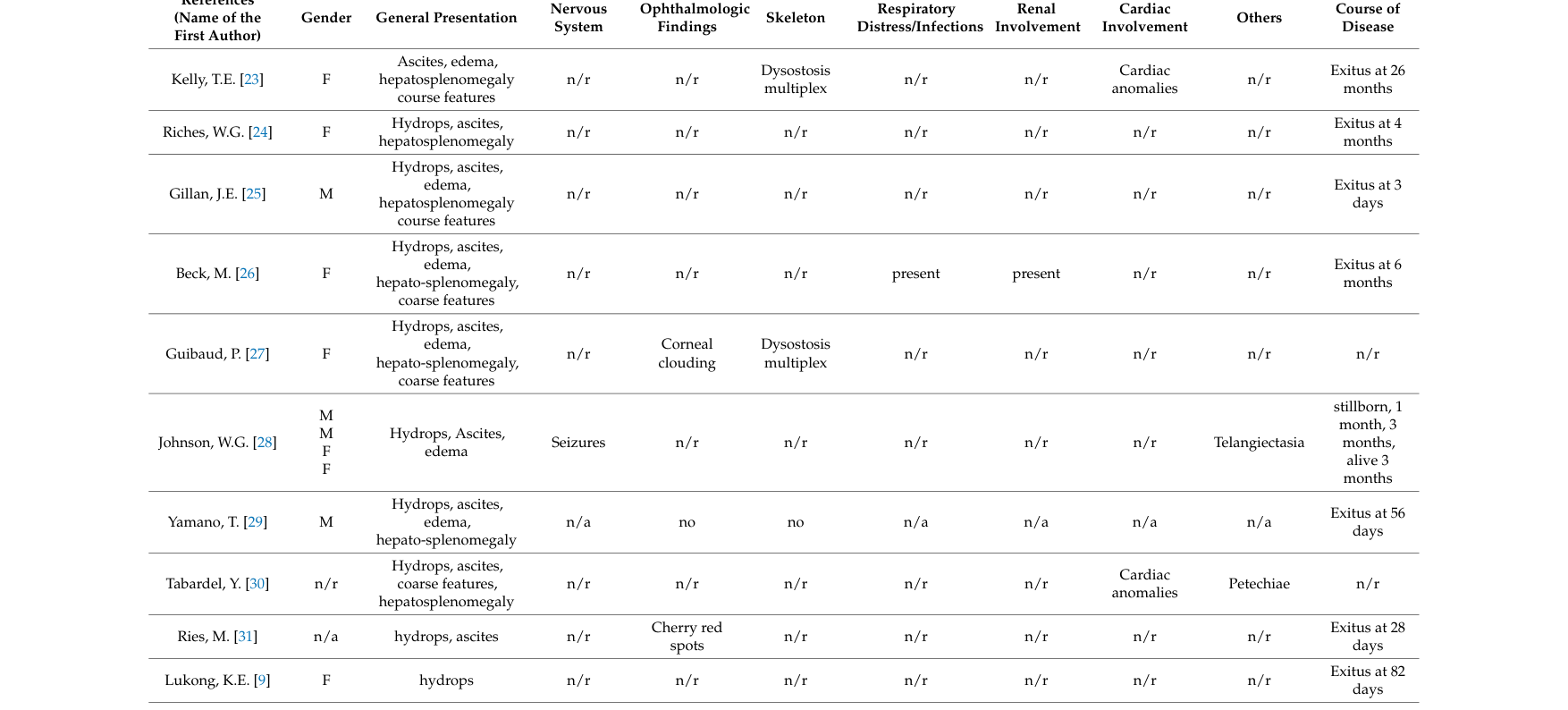
The congenital/hydropic phenotype is primarily derived from aggregated disease-level resources (reviews) that compile individual case reports and fetal pathology series (e.g., tabulated congenital cases). (khan2018sialidosisareview pages 3-5, khan2018sialidosisareview media 784849f6, khan2018sialidosisareview media 429fda1b, khan2018sialidosisareview media 29bee092)
2. Etiology
2.1 Disease causal factors
Genetic cause: biallelic pathogenic variants in NEU1 encoding lysosomal neuraminidase-1 (sialidase). (khan2018sialidosisareview pages 1-3, peng2025geneticinsightsand pages 1-2)
Primary mechanistic defect: deficient NEU1 activity prevents normal lysosomal catabolism of sialylated glycoconjugates, leading to lysosomal accumulation and abnormal excretion of sialylated oligosaccharides/glycopeptides. (vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3, kho2023severekidneydysfunction pages 1-2)
2.2 Risk factors
- Family history / parental carrier status for autosomal recessive NEU1 variants. (khan2018sialidosisareview pages 11-13)
- Consanguinity and population founder variants are plausible contributors in ultra-rare AR diseases, but explicit consanguinity/founder-effect quantification for congenital type II was not available in retrieved primary sources.
2.3 Protective factors
No validated genetic or environmental protective factors specific to congenital sialidosis type II were identified in retrieved sources.
2.4 Gene–environment interactions
No established gene–environment interactions specific to congenital sialidosis type II were found in the retrieved evidence.
3. Phenotypes
3.1 Phenotype spectrum and HPO mapping
The congenital/hydropic subtype is characterized by prenatal fluid overload and early lethality; additional findings vary across cases. In a comprehensive review, the distinguishing congenital group features were “hydrops, ascites, and edema … followed by coarse features, dysostosis multiplex, and hepatosplenomegaly.” (khan2018sialidosisareview pages 3-5)
Table (click to expand)
| Clinical feature | Suggested HPO term(s) | Typical onset | Notes / frequency info if available | Key supporting citations |
|---|---|---|---|---|
| Hydrops fetalis | Hydrops fetalis HP:0001789 | Prenatal | Hallmark of the congenital/hydropic subtype; described as a distinguishing feature of the severe congenital group and associated with stillbirth or death shortly after birth | (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3, khan2018sialidosisareview media 784849f6, khan2018sialidosisareview media 429fda1b, khan2018sialidosisareview media 29bee092) |
| Ascites / neonatal ascites | Ascites HP:0001541; Fetal ascites HP:0001791 | Prenatal / neonatal | Prominent in congenital type II; often reported together with hydrops fetalis; may be isolated or refractory congenital ascites in case literature summarized in the review | (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3, khan2018sialidosisareview media 784849f6, khan2018sialidosisareview media 429fda1b, khan2018sialidosisareview media 29bee092) |
| Generalized edema / facial edema | Edema HP:0000969; Generalized edema HP:0007430; Facial edema HP:0010669 | Prenatal / neonatal | Review states hydrops, ascites, and edema are distinguishing congenital features; gene-therapy review specifically notes facial edema in acute congenital cases | (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, khan2018sialidosisareview pages 11-13) |
| Hepatomegaly | Hepatomegaly HP:0002240 | Prenatal / neonatal / infantile | Included in congenital presentation and severe infantile type II; may be part of broader visceromegaly | (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3) |
| Hepatosplenomegaly | Hepatosplenomegaly HP:0001433; Splenomegaly HP:0001744 | Neonatal / infantile | Common severe type II somatic feature; congenital review notes hepatosplenomegaly among frequent findings after hydrops/ascites/edema | (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3, peng2025geneticinsightsand pages 1-2) |
| Coarse facial features | Coarse facial features HP:0000280 | Neonatal / infantile | Frequent in infantile/juvenile type II; also noted among congenital cases | (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3, peng2025geneticinsightsand pages 1-2) |
| Dysostosis multiplex / skeletal dysplasia | Dysostosis multiplex HP:0000943; Skeletal dysplasia HP:0002652; Abnormal vertebral morphology HP:0000925 | Prenatal / infantile | Congenital review highlights dysostosis multiplex; severe type II also described with skeletal dysplasia and vertebral deformities | (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3) |
| Inguinal hernia | Inguinal hernia HP:0000023 | Neonatal | Specifically mentioned in acute congenital form; review notes it as an infrequent congenital manifestation | (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5) |
| Cherry-red macula / cherry-red spot | Cherry red spot of the macula HP:0010729 | Infantile | More typical of infantile/juvenile severe type II than hydropic congenital cases; part of the classic ophthalmic phenotype | (vlekkert2024aavmediatedgenetherapy pages 1-5, peng2025geneticinsightsand pages 1-2, khan2018sialidosisareview pages 11-13) |
| Myoclonus | Myoclonus HP:0001336 | Infantile / juvenile | Common neurologic sign in severe type II beyond the congenital lethal presentation; used clinically with seizures for symptomatic management | (vlekkert2024aavmediatedgenetherapy pages 1-5, peng2025geneticinsightsand pages 1-2, khan2018sialidosisareview pages 11-13) |
| Seizures | Seizure HP:0001250 | Infantile / juvenile | Mentioned in severe type II spectrum and in supportive-care discussions; often grouped with myoclonus | (vlekkert2024aavmediatedgenetherapy pages 1-5) |
| Hearing loss | Hearing impairment HP:0000365 | Infantile / juvenile | Included in severe type II phenotype in the 2024 preclinical gene-therapy introduction; less emphasized for hydropic congenital cases | (vlekkert2024aavmediatedgenetherapy pages 1-5, khan2018sialidosisareview pages 5-7) |
| Cardiomyopathy | Cardiomyopathy HP:0001638 | Infantile | Listed among severe type II manifestations in recent review/preclinical summary; uncommon but clinically important | (vlekkert2024aavmediatedgenetherapy pages 1-5) |
| Nephrotic syndrome / nephrosialidosis | Nephrotic syndrome HP:0000100; Proteinuria HP:0000093 | Infantile / childhood | Subset of type II patients develop nephrosialidosis with abrupt fulminant glomerular nephropathy; renal involvement is infrequent in congenital review but important in severe type II | (kho2023severekidneydysfunction pages 1-2) |
| Developmental delay / severe intellectual disability | Global developmental delay HP:0001263; Intellectual disability, severe HP:0010864 | Infantile | Severe type II is associated with marked neurodevelopmental impairment/“mental retardation”; often not assessable in lethal prenatal congenital cases | (mosca2020conventionalandunconventional pages 1-3, peng2025geneticinsightsand pages 1-2, khan2018sialidosisareview pages 11-13) |
| Stillbirth / very early death | Stillbirth HP:0003826; Neonatal death HP:0003811 | Prenatal / neonatal | Congenital hydropic form is often lethal during fetal development or shortly after birth | (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3) |
Table: This table maps the major prenatal, neonatal, and infantile manifestations of congenital/severe sialidosis type II to suggested Human Phenotype Ontology terms. It is designed for knowledge-base curation and highlights which findings are characteristic of the hydropic congenital form versus the broader severe type II spectrum.
3.2 Age of onset, progression, and severity
- Congenital/hydropic: prenatal onset; frequently lethal in utero or shortly after birth due to near-complete NEU1 deficiency. (khan2018sialidosisareview pages 3-5)
- Infantile/juvenile type II: early-life onset with multisystem disease including coarse facies, skeletal dysplasia/dysostosis multiplex, hepatosplenomegaly, neurodevelopmental impairment, and (in some) renal disease. (vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3)
3.3 Frequency among affected individuals
Formal phenotype frequencies specifically for congenital/hydropic type II were not quantified in the retrieved evidence set. The congenital-case table compiled in the 2018 review provides case-by-case features rather than aggregate percentages. (khan2018sialidosisareview media 784849f6, khan2018sialidosisareview media 429fda1b, khan2018sialidosisareview media 29bee092)
3.4 Quality-of-life impact
For congenital/hydropic cases, quality-of-life measures are generally not applicable due to fetal/neonatal lethality. For infantile/juvenile type II survivors, major impacts include severe neurologic disability and multisystem complications; standardized HRQoL instruments (e.g., EQ-5D/SF-36) were not identified in retrieved sources.
4. Genetic / molecular information
4.1 Causal gene(s)
- NEU1 (neuraminidase 1), lysosomal sialidase. (peng2025geneticinsightsand pages 1-2)
NEU1 catalyzes lysosomal removal of terminal sialic acids; a recent genetics review states NEU1 “catalyzes the breakdown of sialic acid-containing substances within lysosomes by hydrolyzing terminal sialic acid residues.” (peng2025geneticinsightsand pages 1-2)
4.2 Inheritance
- Autosomal recessive (biallelic NEU1 variants). (khan2018sialidosisareview pages 1-3, peng2025geneticinsightsand pages 1-2)
4.3 Variant spectrum and genotype–phenotype
- A 2018 review reported “more than 40 mutations within the NEU1 gene” known at that time and emphasized genotype–phenotype correlation with residual neuraminidase activity. (khan2018sialidosisareview pages 3-5, khan2018sialidosisareview pages 1-3)
- A 2025 genetics review reported “Over 90 pathogenic NEU1 variants, predominantly missense mutations,” and another section noted ClinVar listing 94 probable pathogenic NEU1 variants. (peng2025geneticinsightsand pages 1-2, peng2025geneticinsightsand pages 4-6)
- Frameshift variants were described as “predominantly linked to type II” in the 2025 review, consistent with more severe loss-of-function. (peng2025geneticinsightsand pages 4-6)
Example genotype–phenotype detail (recent review): A reported patient with compound heterozygosity (exon3:c.544A>G and exon5:c.1021+1G>A) had neuraminidase activity “reduced to 6.4% of normal,” illustrating residual activity as a severity correlate. (peng2025geneticinsightsand pages 4-6)
4.4 NEU1 multienzyme complex and related disorders
NEU1 depends on a lysosomal multienzyme complex including PPCA/CTSA (protective protein/cathepsin A) and β-galactosidase (GLB1). In a 2024 preclinical gene-therapy study: “In mammalian tissues NEU1 is found in a high molecular weight complex with … PPCA … and … β-galactosidase” and “Interaction with PPCA is a prerequisite for the proper localization, stability and activation of NEU1. In absence of a functional PPCA, NEU1 is enzymatically silent.” (vlekkert2024aavmediatedgenetherapy pages 1-5)
This is central to the differential diagnosis with galactosialidosis (CTSA/PPCA deficiency) causing secondary NEU1 deficiency. (khan2018sialidosisareview pages 1-3, mosca2020conventionalandunconventional pages 1-3)
4.5 Epigenetics / chromosomal abnormalities
No disease-specific epigenetic alterations or recurrent chromosomal abnormalities were identified in the retrieved evidence.
5. Environmental information
Congenital sialidosis type II is a monogenic lysosomal disorder; no consistent environmental/lifestyle/infectious contributors were identified in retrieved sources.
6. Mechanism / pathophysiology
6.1 Core causal chain (current understanding)
NEU1 loss-of-function → lysosomal failure to desialylate glycoconjugates → storage of sialylated glycoproteins/oligosaccharides and systemic dysfunction. (vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3)
A 2024 preprint abstract describes the foundational mechanism: “Sialidosis is a glycoprotein storage disease caused by deficiency of the lysosomal sialidase NEU1, which leads to pathogenic accumulation of sialylated glycoproteins and oligosaccharides in tissues and body fluids.” (vlekkert2024aavmediatedgenetherapy pages 1-5)
6.2 Dysregulated lysosomal exocytosis (LAMP1 axis)
Beyond storage, NEU1 has been characterized as a negative regulator of lysosomal exocytosis. In Neu1−/− mice, NEU1 deficiency increases LAMP1+ lysosomes docked at the plasma membrane, promoting extracellular release of lysosomal contents, with downstream tissue remodeling and fibrosis. (vlekkert2024aavmediatedgenetherapy pages 1-5, khan2018sialidosisareview pages 1-3)
6.3 Kidney involvement (nephrosialidosis) and megalin trafficking
A 2023 JCI Insight study focused on the severe renal presentation in a subset of type II patients (“nephrosialidosis”) and modeled it in Neu1-deficient mice. Its abstract states that mice exhibited “elevated urinary albumin levels, loss of nephrons, renal fibrosis, presence of storage vacuoles,” and that glycoprotein sialylation was increased, including megalin; “Megalin levels were severely reduced, and the protein was directed to lysosomes instead of the apical membrane,” supporting impaired proximal tubular protein reabsorption as a mechanism for proteinuria. (kho2023severekidneydysfunction pages 1-2)
6.4 Suggested pathway/ontology annotations
GO Biological Process (suggested): - Lysosomal catabolic process; glycoprotein catabolic process; lysosomal exocytosis; protein glycosylation/desialylation; endocytosis and receptor-mediated endocytosis; extracellular matrix organization/fibrosis; autophagy (renal findings include autophagy markers in mechanistic work). (vlekkert2024aavmediatedgenetherapy pages 1-5, kho2023severekidneydysfunction pages 9-11, kho2023severekidneydysfunction pages 1-2)
GO Cellular Component (suggested): - Lysosome; plasma membrane; endosome; extracellular space (lysosomal content release); glomerulus-related compartments (renal). (vlekkert2024aavmediatedgenetherapy pages 1-5, kho2023severekidneydysfunction pages 1-2)
Cell Ontology terms (suggested key cell types): - Kidney proximal tubule epithelial cell; podocyte; glomerular endothelial cell; macrophage/microglia (neuroinflammation/microgliosis in models); connective tissue fibroblast/myocyte (fibrosis/atrophy models). (khan2018sialidosisareview pages 5-7, kho2023severekidneydysfunction pages 9-11, vlekkert2024aavmediatedgenetherapy pages 1-5)
7. Anatomical structures affected
Organ-level involvement (severe type II spectrum): - Fetal/placental and systemic prenatal involvement with multiorgan vacuolation in congenital cases (liver, bone marrow, kidney, brain) and placental vacuolation indicating early fetal storage. (khan2018sialidosisareview pages 3-5, khan2018sialidosisareview pages 5-7) - Liver/spleen: hepatomegaly/hepatosplenomegaly. (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5) - Skeletal system: dysostosis multiplex/skeletal dysplasia and vertebral deformities. (vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3) - Central nervous system: severe neuropathology in type II; neurodegeneration mechanisms described in mouse models. (kho2023severekidneydysfunction pages 1-2, vlekkert2024aavmediatedgenetherapy pages 1-5) - Kidney: nephrosialidosis with glomerular nephropathy/nephrotic syndrome subset; megalin-mediated reabsorption defects in models. (kho2023severekidneydysfunction pages 1-2)
Subcellular localization: Lysosome (primary); plasma membrane docking/fusion events are emphasized in lysosomal exocytosis model; cell-surface hypersialylation affects trafficking receptors such as megalin. (vlekkert2024aavmediatedgenetherapy pages 1-5, kho2023severekidneydysfunction pages 1-2)
UBERON suggestions (high-level): fetal placenta, liver, spleen, kidney glomerulus, bone, brain.
8. Temporal development
- Onset: congenital/hydropic type II begins prenatally; infantile severe type II begins 0–12 months; juvenile 2–20 years (classification). (khan2018sialidosisareview pages 3-5, vlekkert2024aavmediatedgenetherapy pages 1-5)
- Progression: congenital cases are often lethal; Neu1−/− mice show neonatal onset and rapid progression with premature death (4–6 months in a commonly used model), supporting a rapidly progressive systemic-neurodegenerative course. (vlekkert2024aavmediatedgenetherapy pages 1-5)
9. Inheritance and population
9.1 Epidemiology
- A 2020 review reported sialidosis incidence estimates of “1:250,000 to 1:2,000,000 live births.” (mosca2020conventionalandunconventional pages 1-3)
- A 2023 mechanistic paper stated prevalence “less than 1/1,000,000 live births.” (kho2023severekidneydysfunction pages 1-2)
These values are for sialidosis overall; congenital/hydropic type II is expected to be rarer, but subtype-specific rates were not found in retrieved sources.
9.2 Population/variant distribution and phenotype statistics
A 2025 genetics review summarized population differences across sialidosis cohorts (not limited to congenital type II): - “Asian patients exhibit a lower incidence of impaired cognition (21.7%) … [vs] Caucasian patients (50.0%)” and “the occurrence of cherry-red patches is also lower in Asians (40.7% compared to 61.1% in Caucasians, P = 0.02).” (peng2025geneticinsightsand pages 2-4, peng2025geneticinsightsand pages 4-6)
A Taiwan-associated variant (c.544A>G; p.Ser182Gly) was highlighted as common in Asians and absent in Caucasians in the same review. (peng2025geneticinsightsand pages 4-6)
9.3 Penetrance/expressivity
Congenital type II is associated with profound loss of enzyme function and high severity; explicit penetrance estimates were not identified.
10. Diagnostics
Table (click to expand)
| Diagnostic modality | Sample type | Expected finding in sialidosis type II | Distinguishes from | Notes / pitfalls | Key citations | URL / publication date |
|---|---|---|---|---|---|---|
| Urine oligosaccharide screening by thin-layer chromatography (TLC) | Urine | Abnormal urinary oligosaccharide pattern; increased urinary excretion of sialylated oligosaccharides / glycopeptides is a diagnostic hallmark of NEU1 deficiency | Supports separation from non-oligosaccharidosis causes of hydrops/ascites; helps prioritize sialidosis or galactosialidosis among lysosomal storage diseases | Screening test rather than standalone confirmation; congenital cases may also show oligosaccharides in ascitic fluid in historical reports summarized by review literature | (khan2018sialidosisareview pages 3-5, khan2018sialidosisareview pages 1-3, khan2018sialidosisareview pages 13-14, kho2023severekidneydysfunction pages 1-2) | https://doi.org/10.3390/diagnostics8020029 (2018-04-20); https://doi.org/10.1172/jci.insight.166470 (2023-10-23) |
| Neuraminidase (alpha-N-acetyl neuraminidase / NEU1) enzyme assay | Cultured skin fibroblasts from biopsy (gold standard), also leukocytes in some reports | Deficient lysosomal sialidase activity / neuraminidase deficiency | Confirms primary neuraminidase deficiency and narrows differential within lysosomal storage diseases | Fibroblasts are emphasized as the preferred material for sialidosis/galactosialidosis enzymology; specialized metabolic laboratories are often required | (khan2018sialidosisareview pages 3-5, serrano2024hepatomegalyandsplenomegaly pages 7-8, khan2018sialidosisareview pages 1-3, khan2018sialidosisareview pages 11-13) | https://doi.org/10.3390/diagnostics8020029 (2018-04-20); https://doi.org/10.3390/jcm13051465 (2024-03-01) |
| Paired beta-galactosidase assay with neuraminidase testing | Cultured fibroblasts or other enzymology sample used for lysosomal enzyme workup | Beta-galactosidase activity should be normal in primary sialidosis type II | Galactosialidosis (secondary combined NEU1 and beta-galactosidase deficiency due to CTSA/PPCA defects) | Essential differential step because galactosialidosis can phenocopy sialidosis; PPCA deficiency causes secondary NEU1 deficiency | (khan2018sialidosisareview pages 3-5, khan2018sialidosisareview pages 1-3, mosca2020conventionalandunconventional pages 1-3) | https://doi.org/10.3390/diagnostics8020029 (2018-04-20); https://doi.org/10.3390/jcm9030695 (2020-03-04) |
| Molecular genetic confirmation: targeted NEU1 sequencing | Blood, saliva, buccal swab, or DNA extracted from clinical specimen | Biallelic pathogenic/likely pathogenic NEU1 variants confirming autosomal recessive sialidosis | Distinguishes primary NEU1-related sialidosis from CTSA-related galactosialidosis and from other lysosomal storage disorders with overlapping hydrops/hepatosplenomegaly phenotypes | Definitive diagnosis requires variant interpretation in clinical context; useful for prenatal testing, carrier testing, and family counseling | (khan2018sialidosisareview pages 3-5, khan2018sialidosisareview pages 1-3, khan2018sialidosisareview pages 11-13, peng2025geneticinsightsand pages 1-2) | https://doi.org/10.3390/diagnostics8020029 (2018-04-20); https://doi.org/10.3390/genes16020151 (2025-01) |
| Exome / genome sequencing (WES/WGS) | Blood-derived DNA or family trio DNA | Identification of biallelic NEU1 variants, including novel missense, splice-site, frameshift, or compound-heterozygous variants | Useful when biochemical phenotype overlaps with other lysosomal disorders or when prenatal/neonatal presentations are nonspecific | Particularly valuable in atypical presentations or when single-gene testing is unrevealing; sequencing results may still require biochemical validation | (khan2018sialidosisareview pages 3-5, serrano2024hepatomegalyandsplenomegaly pages 7-8, peng2025geneticinsightsand pages 6-8) | https://doi.org/10.3390/diagnostics8020029 (2018-04-20); https://doi.org/10.3390/jcm13051465 (2024-03-01); https://doi.org/10.3390/genes16020151 (2025-01) |
| Prenatal / family-based molecular diagnosis | Fetal DNA, parental DNA, or prenatal specimen in at-risk pregnancies | Detection of familial NEU1 pathogenic variants before birth | Helps distinguish congenital hydropic sialidosis from other genetic causes of nonimmune hydrops fetalis | Particularly relevant because congenital type II may be lethal in utero or shortly after birth; enables recurrence-risk counseling | (khan2018sialidosisareview pages 11-13, khan2018sialidosisareview pages 13-14) | https://doi.org/10.3390/diagnostics8020029 (2018-04-20) |
| Peripheral blood smear / bone marrow smear | Peripheral blood or bone marrow | Storage granules in lymphocytes may be present | Supports lysosomal storage disease rather than isolated renal/hepatic disease | Ancillary, not disease-specific; should not replace enzyme or molecular confirmation | (khan2018sialidosisareview pages 3-5) | https://doi.org/10.3390/diagnostics8020029 (2018-04-20) |
| Integrated LSD second-line workup | Plasma, leukocytes, fibroblasts, urine, DNA | Combination of low neuraminidase activity, abnormal urine oligosaccharides, and biallelic NEU1 variants | Helps distinguish from mucolipidoses, multiple sulfatase deficiency, and other hepatosplenomegaly-associated LSDs | Serrano 2024 recommends molecular testing accompanied by enzymatic testing when feasible; biopsy should generally be last resort | (serrano2024hepatomegalyandsplenomegaly pages 7-8, serrano2024hepatomegalyandsplenomegaly pages 2-3) | https://doi.org/10.3390/jcm13051465 (2024-03-01) |
| Interpretation safeguard for VUS / pseudodeficiency | DNA plus matched biochemical testing | Variants of uncertain significance should be corroborated by enzymology and/or biomarkers; pseudodeficiency may show low in vitro activity without true disease | Prevents overcalling sialidosis when lab abnormalities are non-pathogenic or inconclusive | Important pitfall from LSD diagnostics: VUS often need targeted biochemical confirmation; specialized labs improve interpretation | (lange2024systematicdiagnosisof pages 4-6, serrano2024hepatomegalyandsplenomegaly pages 7-8) | https://doi.org/10.51847/isa3beaurx (2024-01); https://doi.org/10.3390/jcm13051465 (2024-03-01) |
Table: This table summarizes the core diagnostic workflow and major differential-diagnostic issues for congenital sialidosis type II. It highlights the complementary roles of urine oligosaccharide screening, fibroblast neuraminidase assays, beta-galactosidase testing, and molecular confirmation, including key pitfalls such as VUS interpretation and pseudodeficiency.
Key diagnostic concepts (with direct quotes)
- Screening: “thin-layer chromatography … useful screening test to find an abnormal urinary oligosaccharide pattern.” (khan2018sialidosisareview pages 3-5)
- Enzymology: “Deficiency of the lysosomal sialidase activity (neuraminidase) can be demonstrated in cultured skin fibroblasts …” (khan2018sialidosisareview pages 3-5)
- Differential from galactosialidosis: “β-galactosidase … should be normal” in sialidosis. (khan2018sialidosisareview pages 3-5)
- Confirmation: “The final diagnosis is made after whole genome sequencing.” (khan2018sialidosisareview pages 3-5)
Differential diagnosis (non-exhaustive)
- Galactosialidosis (CTSA/PPCA deficiency) with secondary NEU1 deficiency. (khan2018sialidosisareview pages 1-3)
- Other lysosomal storage diseases presenting with hepatosplenomegaly, dysostosis multiplex, or hydrops fetalis (general LSD diagnostic frameworks emphasize combined molecular + enzyme testing). (serrano2024hepatomegalyandsplenomegaly pages 7-8)
11. Outcome / prognosis
- Congenital/hydropic type II is often lethal: “Profoundly severe mutational alterations may lead to a complete absence of lysosomal neuraminidase and are lethal during fetal development or at birth.” (khan2018sialidosisareview pages 3-5)
- Severe type II is associated with multisystem morbidity and premature mortality; NIH natural-history protocol describes glycoproteinoses (including sialidosis) as “fatal or severely debilitating,” and states “No effective therapy … has yet been demonstrated.” (NCT00029965 chunk 1)
Robust survival curves, median survival, or subtype-stratified mortality statistics were not identified in the retrieved evidence.
12. Treatment
12.1 Current real-world management
No approved disease-modifying therapy is established; current care is primarily supportive. In the 2024 gene-therapy preprint: “Currently, there is no target treatment or cure available for type I or II sialidosis. Patients receive supportive care and symptomatic relief, primarily for the management of myoclonus and seizures.” (vlekkert2024aavmediatedgenetherapy pages 1-5)
12.2 Experimental and emerging therapeutics (preclinical emphasis)
Table (click to expand)
| Intervention type | Specific intervention | Mechanism / rationale | Evidence type | Key findings / outcomes | Limitations / notes | Key citations | URL and publication date |
|---|---|---|---|---|---|---|---|
| Supportive | Symptomatic management of myoclonus and seizures | No disease-modifying therapy is established; care focuses on controlling neurologic symptoms in surviving type II patients | Human clinical practice / review | Recent preclinical review states patients with sialidosis currently receive “supportive care and symptomatic relief, primarily for the management of myoclonus and seizures” | Evidence is descriptive rather than protocolized; especially relevant to infantile/juvenile type II rather than lethal hydropic congenital presentations | (vlekkert2024aavmediatedgenetherapy pages 1-5) | https://doi.org/10.1101/2023.11.10.566667 ; posted 2023-11-13 / cited as 2024 preprint |
| Supportive | Multidisciplinary monitoring in natural history protocols | Longitudinal neurologic, ophthalmologic, imaging, electrophysiologic, biochemical, and functional assessments help define progression and future trial endpoints | Observational trial / real-world implementation | NIH natural history protocol enrolls enzyme- or DNA-confirmed sialidosis/galactosialidosis patients and uses MRI/MRS, hearing testing, EEG, EMG/NCV, echocardiogram, abdominal ultrasound, ophthalmology, rehabilitation, speech, neurology, psychology, and biomarker sampling | Observational only; not a treatment; includes all sialidosis forms rather than congenital type II specifically | (NCT00029965 chunk 1) | https://clinicaltrials.gov/study/NCT00029965 ; first posted 2002-01-28, recruiting update 2026-05-29 |
| Experimental | Recombinant NEU1 enzyme replacement therapy (ERT) | Replaces deficient lysosomal neuraminidase-1 to reduce storage of sialylated glycoproteins / oligosaccharides | Mouse preclinical | Review summarizes “short-term ERT” in Neu1−/− mice using recombinant Neu1 from insect cells; corrected systemic pathology in mice | Strong immunogenicity limited durability/translation; no approved human ERT for sialidosis | (mosca2020conventionalandunconventional pages 1-3, khan2018sialidosisareview pages 11-13, khan2018sialidosisareview pages 14-16, peng2025geneticinsightsand pages 1-2) | https://doi.org/10.3390/jcm9030695 ; 2020-03-04 |
| Experimental | PPCA-based pharmacologic chaperone approach | NEU1 requires protective protein/cathepsin A (PPCA) for folding, lysosomal localization, stability, and activation; augmenting PPCA can rescue residual mutant NEU1 | In vitro and mouse preclinical | Recombinant PPCA and related chaperone strategies produced small but consistent increases in residual NEU1 activity in patient fibroblasts; prior AAV-PPCA work improved residual activity and prevented kidney pathology / oligosacchariduria in a type I model | Likely most applicable where residual NEU1 is present; evidence strongest for type I or attenuated variants, not fully null congenital cases | (mosca2020conventionalandunconventional pages 1-3, vlekkert2024aavmediatedgenetherapy pages 1-5, khan2022neu1—auniquetherapeutic pages 2-3) | https://doi.org/10.3390/jcm9030695 ; 2020-03-04 |
| Experimental | AAV-PPCA chaperone-mediated gene therapy | Liver-directed PPCA expression acts as a systemic chaperone to enhance residual NEU1 function | Mouse preclinical | Prior work cited in the 2024 preprint showed a single liver-specific AAV-PPCA dose “enhanced residual Neu1 activity and prevented kidney pathology and oligosacchariduria” | Mutation-dependent; mainly relevant to residual-activity alleles rather than severe congenital null alleles | (vlekkert2024aavmediatedgenetherapy pages 1-5, khan2018sialidosisareview pages 11-13) | https://doi.org/10.1101/2023.11.10.566667 ; posted 2023-11-13 / cited as 2024 preprint |
| Experimental | Dual AAV gene therapy (NEU1 + PPCA; scAAV2/8 co-injection) | Directly restores NEU1 plus its required chaperone PPCA, aiming to correct enzyme deficiency, storage, lysosomal exocytosis, fibrosis, and neurovisceral pathology | Mouse preclinical | In Neu1−/− mice, treated animals were “phenotypically indistinguishable from their WT controls,” with restored NEU1 activity in most tissues, reversal of sialyl-oligosacchariduria, diminished/absent vacuolization in visceral organs and brain, normalization of lysosomal exocytosis, and prevention of generalized fibrosis | Preclinical bioRxiv study; not yet a human trial; long-term durability, dosing, immunity, and CNS translation remain to be established | (vlekkert2024aavmediatedgenetherapy pages 1-5) | https://doi.org/10.1101/2023.11.10.566667 ; posted 2023-11-13 / cited as 2024 preprint |
| Experimental | Pharmacologic proteostasis / proteasome inhibition (e.g., MG132; celastrol discussed in review) | May improve folding, trafficking, and lysosomal localization of defective NEU1 proteins | In vitro / preclinical | Review notes MG132 “enhances enzyme activity and its localization in cells expressing defective sialidase”; celastrol and other compounds were explored as adjunctive approaches | Early-stage only; toxicity and translational feasibility uncertain; not specific to congenital type II | (khan2018sialidosisareview pages 11-13, khan2018sialidosisareview pages 14-16) | https://doi.org/10.3390/diagnostics8020029 ; 2018-04-20 |
| Experimental / adjunctive | Dietary betaine supplementation | Proposed to stabilize residual mutant NEU1 and improve oligosaccharide handling | Mouse preclinical and patient fibroblasts | In residual-activity mouse models, betaine increased mutant NEU1 levels and resolved oligosacchariduria; patient fibroblasts showed small activity gains with several compounds | Data are primarily for type I / residual-activity disease, not severe congenital type II | (mosca2020conventionalandunconventional pages 1-3) | https://doi.org/10.3390/jcm9030695 ; 2020-03-04 |
| Supportive / preventive | Carrier detection, prenatal molecular diagnosis, genetic counseling | Because congenital hydropic type II can be lethal prenatally or neonatally, family-based molecular diagnosis supports reproductive planning and early diagnosis | Human clinical genetics | Review recommends “carrier detection in affected families, prenatal molecular diagnosis, and improved genetic counseling”; congenital cases have been diagnosed prenatally in reported literature | Prevents recurrence risk rather than treating affected fetus/newborn; requires known familial variants or robust molecular testing | (khan2018sialidosisareview pages 11-13, khan2018sialidosisareview pages 13-14) | https://doi.org/10.3390/diagnostics8020029 ; 2018-04-20 |
| Real-world research infrastructure | Biomarker and cell-model development within natural history studies | Builds outcome measures and translational platforms for future therapy testing | Observational trial / translational implementation | NIH protocol collects CSF, blood, urine biomarkers; establishes fibroblast cultures for “testing potential therapeutic agents” and creates iPSC-derived neural tissues for mechanistic and preclinical work | Indirect therapeutic value; no interventional efficacy results yet | (NCT00029965 chunk 1) | https://clinicaltrials.gov/study/NCT00029965 ; first posted 2002-01-28, recruiting update 2026-05-29 |
Table: This table summarizes the current management landscape for sialidosis type II, from symptomatic care to preclinical gene therapy and enzyme/chaperone approaches. It also includes natural-history study infrastructure that is already being used in practice to support biomarker discovery and future interventional trials.
Key 2023–2024 development: dual AAV gene therapy (NEU1 + PPCA) in Neu1−/− mice. The abstract reports treated animals “phenotypically indistinguishable from their WT controls,” with “restoration of NEU1 activity in most tissues, reversal of sialyl-oligosacchariduria, and normalization of lysosomal exocytosis,” and prevention of generalized fibrosis. (vlekkert2024aavmediatedgenetherapy pages 1-5)
MAXO suggestions (high-level): antiseizure medication therapy; supportive care; genetic counseling; enzyme replacement therapy (experimental); gene therapy (AAV-based) (preclinical).
12.3 Clinical trials and real-world implementations
No interventional NEU1 gene therapy or ERT trials for sialidosis type II were identified in the retrieved trial list. However, natural history and biomarker studies relevant to future trials exist, including: - NCT00029965 (NIH): Natural History of Glycosphingolipid Storage Disorders and Glycoprotein Disorders (includes enzyme/DNA-confirmed sialidosis). (NCT00029965 chunk 1)
13. Prevention
Because this is a Mendelian autosomal recessive disorder, prevention focuses on recurrence-risk management: - Carrier detection and prenatal molecular diagnosis with genetic counseling are explicitly recommended in the congenital/type II context. (khan2018sialidosisareview pages 11-13)
Primary prevention via lifestyle or vaccination is not applicable.
14. Other species / natural disease
No naturally occurring (non-model) animal cases were identified in the retrieved evidence.
15. Model organisms and model systems
15.1 Mouse models (Neu1−/−)
Neu1−/− mice are widely used and closely resemble severe type II disease. The 2024 preprint states: “Neu1 knockout mice (Neu1−/−) closely resemble type II sialidosis … [with] severe growth retardation … premature death (4–6 months).” (vlekkert2024aavmediatedgenetherapy pages 1-5)
Kidney-specific mechanistic modeling of nephrosialidosis: Neu1-deficient mice show albuminuria, nephron loss, and renal fibrosis, and hypersialylation-driven megalin mistrafficking to lysosomes rather than the apical membrane. (kho2023severekidneydysfunction pages 1-2)
15.2 Cellular/iPSC models and technology applications
The NIH natural history protocol notes: “Some fibroblast lines will be used to create induced pluripotent stem cells (iPSC) for differentiation into neural tissues,” enabling CNS-relevant modeling and biomarker studies. (NCT00029965 chunk 1)
Evidence gaps and limitations of this report
- Subtype-specific epidemiology (congenital/hydropic type II) and formal phenotype frequency distributions were not available from the retrieved sources; most numbers are for sialidosis overall. (kho2023severekidneydysfunction pages 1-2, mosca2020conventionalandunconventional pages 1-3)
- Several clinical details for congenital cases are derived from aggregated historical case reports in reviews rather than contemporary multicenter registries. (khan2018sialidosisareview media 784849f6, khan2018sialidosisareview media 429fda1b, khan2018sialidosisareview media 29bee092)
- Most disease-modifying therapy evidence remains preclinical (mouse models) and includes at least one preprint (bioRxiv), so clinical translation remains uncertain. (vlekkert2024aavmediatedgenetherapy pages 1-5, mosca2020conventionalandunconventional pages 1-3)
Key URLs (selected)
- OMIM Sialidosis (MIM 256550): https://omim.org/entry/256550 (cited via review) (khan2018sialidosisareview pages 3-5)
- Open Targets disease entity (MONDO_0009738, sialidosis type 2): https://platform.opentargets.org/disease/MONDO_0009738 (OpenTargets Search: Sialidosis)
- 2023 JCI Insight (kidney dysfunction in sialidosis mice): https://doi.org/10.1172/jci.insight.166470 (Published 2023-10-23) (kho2023severekidneydysfunction pages 1-2)
- 2024 bioRxiv preprint (AAV-mediated gene therapy for sialidosis): https://doi.org/10.1101/2023.11.10.566667 (Posted 2023-11-13; indexed 2024) (vlekkert2024aavmediatedgenetherapy pages 1-5)
- 2024 J Clin Med diagnostic approach to LSDs with hepatosplenomegaly: https://doi.org/10.3390/jcm13051465 (Published 2024-03) (serrano2024hepatomegalyandsplenomegaly pages 7-8)
- ClinicalTrials.gov natural history (NCT00029965): https://clinicaltrials.gov/study/NCT00029965 (First posted 2002-01-28; updated 2026-05-29) (NCT00029965 chunk 1)
References
-
(khan2018sialidosisareview pages 3-5): Aiza Khan and Consolato Sergi. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics, 8:29, Apr 2018. URL: https://doi.org/10.3390/diagnostics8020029, doi:10.3390/diagnostics8020029. This article has 73 citations.
-
(vlekkert2024aavmediatedgenetherapy pages 1-5): Diantha van de Vlekkert, Huimin Hu, Leigh E. Fremuth, Scott A. Brown, Jason A. Weesner, Elida Gomero, Yvan Campos, and Alessandra d’Azzo. Aav-mediated gene therapy for sialidosis. bioRxiv, Nov 2024. URL: https://doi.org/10.1101/2023.11.10.566667, doi:10.1101/2023.11.10.566667. This article has 11 citations.
-
(kho2023severekidneydysfunction pages 1-2): Ikhui Kho, Ekaterina P. Demina, Xuefang Pan, Irene Londono, Christopher W. Cairo, Luisa Sturiale, Angelo Palmigiano, Angela Messina, Domenico Garozzo, Roth-Visal Ung, Fabrice Mac-Way, Éric Bonneil, Pierre Thibault, Mathieu Lemaire, Carlos R. Morales, and Alexey V. Pshezhetsky. Severe kidney dysfunction in sialidosis mice reveals an essential role for neuraminidase 1 in reabsorption. JCI Insight, Oct 2023. URL: https://doi.org/10.1172/jci.insight.166470, doi:10.1172/jci.insight.166470. This article has 14 citations and is from a domain leading peer-reviewed journal.
-
(khan2018sialidosisareview pages 1-3): Aiza Khan and Consolato Sergi. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics, 8:29, Apr 2018. URL: https://doi.org/10.3390/diagnostics8020029, doi:10.3390/diagnostics8020029. This article has 73 citations.
-
(OpenTargets Search: Sialidosis): Open Targets Query (Sialidosis, 5 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(peng2025geneticinsightsand pages 1-2): Mei-Ling Peng, Siu-Fung Chau, Jia-Ying Chien, Peng-Yeong Woon, Yu-Chen Chen, Wai-Man Cheang, Hsien-Yang Tsai, and Shun-Ping Huang. Genetic insights and clinical implications of neu1 mutations in sialidosis. Genes, 16:151, Jan 2025. URL: https://doi.org/10.3390/genes16020151, doi:10.3390/genes16020151. This article has 10 citations.
-
(khan2018sialidosisareview media 784849f6): Aiza Khan and Consolato Sergi. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics, 8:29, Apr 2018. URL: https://doi.org/10.3390/diagnostics8020029, doi:10.3390/diagnostics8020029. This article has 73 citations.
-
(khan2018sialidosisareview media 429fda1b): Aiza Khan and Consolato Sergi. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics, 8:29, Apr 2018. URL: https://doi.org/10.3390/diagnostics8020029, doi:10.3390/diagnostics8020029. This article has 73 citations.
-
(khan2018sialidosisareview media 29bee092): Aiza Khan and Consolato Sergi. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics, 8:29, Apr 2018. URL: https://doi.org/10.3390/diagnostics8020029, doi:10.3390/diagnostics8020029. This article has 73 citations.
-
(mosca2020conventionalandunconventional pages 1-3): Rosario Mosca, Diantha van de Vlekkert, Yvan Campos, Leigh E. Fremuth, Jaclyn Cadaoas, Vish Koppaka, Emil Kakkis, Cynthia Tifft, Camilo Toro, Simona Allievi, Cinzia Gellera, Laura Canafoglia, Gepke Visser, Ida Annunziata, and Alessandra d’Azzo. Conventional and unconventional therapeutic strategies for sialidosis type i. Journal of Clinical Medicine, 9:695, Mar 2020. URL: https://doi.org/10.3390/jcm9030695, doi:10.3390/jcm9030695. This article has 29 citations.
-
(khan2018sialidosisareview pages 11-13): Aiza Khan and Consolato Sergi. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics, 8:29, Apr 2018. URL: https://doi.org/10.3390/diagnostics8020029, doi:10.3390/diagnostics8020029. This article has 73 citations.
-
(khan2018sialidosisareview pages 5-7): Aiza Khan and Consolato Sergi. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics, 8:29, Apr 2018. URL: https://doi.org/10.3390/diagnostics8020029, doi:10.3390/diagnostics8020029. This article has 73 citations.
-
(peng2025geneticinsightsand pages 4-6): Mei-Ling Peng, Siu-Fung Chau, Jia-Ying Chien, Peng-Yeong Woon, Yu-Chen Chen, Wai-Man Cheang, Hsien-Yang Tsai, and Shun-Ping Huang. Genetic insights and clinical implications of neu1 mutations in sialidosis. Genes, 16:151, Jan 2025. URL: https://doi.org/10.3390/genes16020151, doi:10.3390/genes16020151. This article has 10 citations.
-
(kho2023severekidneydysfunction pages 9-11): Ikhui Kho, Ekaterina P. Demina, Xuefang Pan, Irene Londono, Christopher W. Cairo, Luisa Sturiale, Angelo Palmigiano, Angela Messina, Domenico Garozzo, Roth-Visal Ung, Fabrice Mac-Way, Éric Bonneil, Pierre Thibault, Mathieu Lemaire, Carlos R. Morales, and Alexey V. Pshezhetsky. Severe kidney dysfunction in sialidosis mice reveals an essential role for neuraminidase 1 in reabsorption. JCI Insight, Oct 2023. URL: https://doi.org/10.1172/jci.insight.166470, doi:10.1172/jci.insight.166470. This article has 14 citations and is from a domain leading peer-reviewed journal.
-
(peng2025geneticinsightsand pages 2-4): Mei-Ling Peng, Siu-Fung Chau, Jia-Ying Chien, Peng-Yeong Woon, Yu-Chen Chen, Wai-Man Cheang, Hsien-Yang Tsai, and Shun-Ping Huang. Genetic insights and clinical implications of neu1 mutations in sialidosis. Genes, 16:151, Jan 2025. URL: https://doi.org/10.3390/genes16020151, doi:10.3390/genes16020151. This article has 10 citations.
-
(khan2018sialidosisareview pages 13-14): Aiza Khan and Consolato Sergi. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics, 8:29, Apr 2018. URL: https://doi.org/10.3390/diagnostics8020029, doi:10.3390/diagnostics8020029. This article has 73 citations.
-
(serrano2024hepatomegalyandsplenomegaly pages 7-8): Teodoro Jerves Serrano, Jessica Gold, James A. Cooper, Heather J. Church, Karen L. Tylee, Hoi Yee Wu, Sun Young Kim, and Karolina M. Stepien. Hepatomegaly and splenomegaly: an approach to the diagnosis of lysosomal storage diseases. Journal of Clinical Medicine, 13:1465, Mar 2024. URL: https://doi.org/10.3390/jcm13051465, doi:10.3390/jcm13051465. This article has 17 citations.
-
(peng2025geneticinsightsand pages 6-8): Mei-Ling Peng, Siu-Fung Chau, Jia-Ying Chien, Peng-Yeong Woon, Yu-Chen Chen, Wai-Man Cheang, Hsien-Yang Tsai, and Shun-Ping Huang. Genetic insights and clinical implications of neu1 mutations in sialidosis. Genes, 16:151, Jan 2025. URL: https://doi.org/10.3390/genes16020151, doi:10.3390/genes16020151. This article has 10 citations.
-
(serrano2024hepatomegalyandsplenomegaly pages 2-3): Teodoro Jerves Serrano, Jessica Gold, James A. Cooper, Heather J. Church, Karen L. Tylee, Hoi Yee Wu, Sun Young Kim, and Karolina M. Stepien. Hepatomegaly and splenomegaly: an approach to the diagnosis of lysosomal storage diseases. Journal of Clinical Medicine, 13:1465, Mar 2024. URL: https://doi.org/10.3390/jcm13051465, doi:10.3390/jcm13051465. This article has 17 citations.
-
(lange2024systematicdiagnosisof pages 4-6): Martin Lange, Nicoleta Corcodel, and Falk Schwendicke. Systematic diagnosis of lysosomal storage diseases in patients with hepatosplenomegaly. Bulletin of Pioneering Researches of Medical and Clinical Science, 4:66-80, Jan 2024. URL: https://doi.org/10.51847/isa3beaurx, doi:10.51847/isa3beaurx. This article has 0 citations.
-
(NCT00029965 chunk 1): Natural History of Glycosphingolipid Storage Disorders and Glycoprotein Disorders. National Human Genome Research Institute (NHGRI). 2002. ClinicalTrials.gov Identifier: NCT00029965
-
(khan2018sialidosisareview pages 14-16): Aiza Khan and Consolato Sergi. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics, 8:29, Apr 2018. URL: https://doi.org/10.3390/diagnostics8020029, doi:10.3390/diagnostics8020029. This article has 73 citations.
-
(khan2022neu1—auniquetherapeutic pages 2-3): Aiza Khan and Consolato M. Sergi. Neu1—a unique therapeutic target for alzheimer’s disease. Frontiers in Pharmacology, Jun 2022. URL: https://doi.org/10.3389/fphar.2022.902259, doi:10.3389/fphar.2022.902259. This article has 20 citations.