Congenital Isolated Hyperinsulinism (Congenital Hyperinsulinism; CHI) — Disease Characteristics Research Report
Target Disease
- Disease name: Congenital isolated hyperinsulinism (commonly discussed as congenital hyperinsulinism, CHI) (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2)
- Category: Rare endocrine/metabolic disorder of dysregulated pancreatic insulin secretion causing recurrent hypoglycemia (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2)
- MONDO ID / OMIM / Orphanet / MeSH / ICD-10/ICD-11: Not retrievable with the available tool corpus in this run; identifiers should be filled from OMIM/Orphanet/MONDO/MeSH/ICD authoritative resources outside this evidence set (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2, banerjee2022congenitalhyperinsulinismin pages 1-2).
Executive overview (current understanding)
Congenital (isolated) hyperinsulinism (CHI) is characterized by inappropriate insulin secretion despite low blood glucose, and is widely described as the most common cause of persistent hypoglycemia in infancy/childhood (mittal2024molecularmechanismsunderlying pages 1-2, takasawa2024clinicalmanagementof pages 1-6). It is clinically, genetically, and histologically heterogeneous, with focal, diffuse, and atypical forms; correct subtype classification is critical because focal CHI can be surgically cured, while diffuse disease often requires long-term medical therapy and occasionally near-total pancreatectomy (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2, graca2023managingcongenitalhyperinsulinisma pages 33-37).
Table (click to expand)
| Domain | Key findings | Supporting citation IDs |
|---|---|---|
| Definition | Congenital isolated hyperinsulinism (CHI) is inappropriate insulin secretion despite hypoglycemia and is the most common cause of persistent hypoglycemia in infancy/childhood. Presentation is usually neonatal or early infancy and may be life-threatening because of recurrent neuroglycopenia. | (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2, banerjee2022congenitalhyperinsulinismin pages 1-2) |
| Histologic/clinical subtypes | Diffuse CHI: whole-pancreas β-cell involvement, often due to recessive or dominant KATP-channel defects; often medically difficult and may require near-total pancreatectomy. Focal CHI: localized lesion, classically from a paternally inherited ABCC8/KCNJ11 variant plus somatic loss of maternal 11p15; potentially curable by limited resection. Atypical CHI: less common mixed/nonclassic histology. | (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2, globa2024congenitalhyperinsulinismin pages 2-3, burroni2021earlydiagnosisof pages 1-2) |
| Major causal genes | Most common genes are ABCC8 and KCNJ11 (KATP channel; SUR1/Kir6.2). Other reported genes include GLUD1, GCK, HADH, SLC16A1, HNF4A, HNF1A, UCP2, CACNA1D, and less commonly syndromic/non-isolated causes in broader HI cohorts. KATP defects account for ~40–50% of persistent CHI in recent national data. | (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2, burroni2021earlydiagnosisof pages 1-2) |
| Gene-specific phenotype notes | ABCC8/KCNJ11: often diazoxide-unresponsive when inactivating; diffuse with biallelic/dominant forms, focal with single paternal recessive variant. GLUD1: hyperinsulinism-hyperammonemia, usually diazoxide responsive. GCK: activating variants can cause CHI. HADH, HNF4A, HNF1A: often diazoxide responsive in many cases. SLC16A1: exercise/protein-sensitive phenotypes reported in HI literature. | (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, takasawa2024clinicalmanagementof pages 1-6, globa2024congenitalhyperinsulinismin pages 2-3) |
| Typical inheritance | Autosomal recessive and autosomal dominant forms both occur; focal disease typically reflects paternal inheritance plus somatic maternal allele loss in the lesion. Consanguinity increases incidence in some populations. | (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, mittal2024molecularmechanismsunderlying pages 1-2, banerjee2022congenitalhyperinsulinismin pages 1-2) |
| Critical sample hallmarks | During hypoglycemia, typical findings are detectable/inappropriately unsuppressed insulin and C-peptide, suppressed ketones and free fatty acids, and high glucose infusion requirement often >8–10 mg/kg/min. Example review data include insulin 14.4 µIU/mL, C-peptide 1 ng/mL, ketones 0.5 mmol/L in a CHI case. | (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, mittal2024molecularmechanismsunderlying pages 1-2, graca2023managingcongenitalhyperinsulinism media 3dcd38ca, graca2023managingcongenitalhyperinsulinism media 1893ff72) |
| Dynamic testing | A positive glycemic response to glucagon during hypoglycemia supports excess insulin action and depleted hepatic glycogen stores in CHI. | (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, mittal2024molecularmechanismsunderlying pages 1-2) |
| Imaging/pathology | 18F-DOPA PET/CT is central for distinguishing focal from diffuse disease and localizing focal lesions preoperatively; reported performance in one cohort/review: sensitivity 88%, specificity 94%, accuracy 88–100%. | (globa2024congenitalhyperinsulinismin pages 2-3, burroni2021earlydiagnosisof pages 1-2) |
| First-line treatment | Diazoxide is the only approved first-line chronic drug; it opens SUR1-containing KATP channels. Effectiveness exceeds 70% overall in one 2024 single-center summary, but response strongly depends on genotype. | (mittal2024molecularmechanismsunderlying pages 1-2, takasawa2024clinicalmanagementof pages 1-6, graca2023managingcongenitalhyperinsulinisma pages 33-37) |
| Second-line/adjunct treatment | Octreotide is the common second-line therapy for diazoxide-unresponsive CHI; long-acting somatostatin analogs such as lanreotide are used in practice. Home CGM is increasingly used for management and feeding/treatment adjustment. | (takasawa2024clinicalmanagementof pages 1-6, globa2024congenitalhyperinsulinismin pages 2-3, graca2023managingcongenitalhyperinsulinisma pages 33-37) |
| Surgery | Focal lesionectomy/partial pancreatectomy can be curative. Near-total pancreatectomy is reserved for refractory diffuse CHI because of later diabetes/exocrine insufficiency risk. In the Ukrainian national cohort, complete recovery occurred in all 14 focal cases after surgery. | (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2, burroni2021earlydiagnosisof pages 1-2, graca2023managingcongenitalhyperinsulinisma pages 33-37) |
| Emerging/refractory therapies | Sirolimus and nifedipine are described as refractory/off-label options in reviews. GLP-1 receptor antagonist exendin(9-39) has been tested in pilot trials; NCT00571324 was an open-label randomized crossover phase 1/2 study (n=9), and NCT00835328 studied infants with diazoxide-refractory CHI. | (graca2023managingcongenitalhyperinsulinisma pages 33-37, NCT00571324 chunk 1, NCT00835328 chunk 2) |
| Epidemiology | Reported incidence is ~1:28,000–1:50,000 in Western populations, rising to ~1:2,500 where consanguinity is higher. Japanese estimates cited in a 2024 series were 1 in 13,600 for transient CHI and 1 in 31,600 for persistent CHI. | (banerjee2022congenitalhyperinsulinismin pages 1-2, takasawa2024clinicalmanagementof pages 1-6) |
| Genetic diagnosis rates | In the 2024 Ukrainian national study, a molecular diagnosis was made in 67.5% (27/40) overall, including 86.3% (19/22) of persistent CHI and 44.4% (8/18) of early-remission CHI. | (globa2024congenitalhyperinsulinismin pages 1-2) |
| Histology proportions | In 19 surgically characterized Ukrainian persistent CHI cases, histology was focal 73.7% (14/19), diffuse 10.5% (2/19), atypical 15.8% (3/19). | (globa2024congenitalhyperinsulinismin pages 1-2) |
| Clinical presentation stats | Hypoglycemia presents in the first week in 60–70% of cases; ~50% present with seizures; 20–30% are diagnosed in the first year and ~10% after age 1 year. | (banerjee2022congenitalhyperinsulinismin pages 1-2) |
| Neurodevelopment/QoL burden | Abnormal neurodevelopmental outcomes have been reported in 26–44% of children in the QoL review. HI Global Registry/family survey data showed 70% (36/51) of parents of children <5 years felt life was “ruled by HI,” 48% (59/123) reported physical health impact, and 67% (82/123) mental health impact. | (kristensen2021healthrelatedqualityof pages 1-2, banerjee2022congenitalhyperinsulinismin pages 9-10) |
| Economic burden | A UK cost-of-illness study estimated total annual CHI cost to the NHS at £3,408,398.59, average £2,124.95 per patient; 5.9% of patients (95 infants in first year of life) accounted for 61.8% of total costs. | (banerjee2022congenitalhyperinsulinismin pages 9-10, graca2023managingcongenitalhyperinsulinism pages 43-45) |
Table: This table condenses the main disease-definition, genetics, diagnostic, treatment, and burden-of-disease findings for congenital isolated hyperinsulinism. It is useful as a quick-reference evidence map with directly traceable context-ID citations.
1. Disease Information
1.1 Definition
- CHI is defined as inappropriate insulin secretion during hypoglycemia; “critical sample” biochemical testing during hypoglycemia initiates a diagnostic cascade (mittal2024molecularmechanismsunderlying pages 1-2, ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6).
1.2 Common synonyms and alternative names
- Congenital hyperinsulinism (CHI) (common in contemporary literature) (globa2024congenitalhyperinsulinismin pages 1-2, takasawa2024clinicalmanagementof pages 1-6)
- Persistent hyperinsulinemic hypoglycemia of infancy (PHHI) (historical/alternative clinical term) (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, burroni2021earlydiagnosisof pages 1-2)
- Hyperinsulinemic hypoglycemia (HH) (broader umbrella used in some texts; CHI is a major cause in infancy) (globa2024congenitalhyperinsulinismin pages 1-2, banerjee2022congenitalhyperinsulinismin pages 1-2)
1.3 Source type (patient-level vs aggregated)
- Evidence in this report is derived largely from aggregated disease-level resources (reviews, national registry study) plus clinical trial registry records (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2, banerjee2022congenitalhyperinsulinismin pages 1-2, NCT00571324 chunk 1).
2. Etiology
2.1 Disease causal factors
Primary causal factor: genetic disruption of pancreatic β-cell insulin secretion regulation, particularly KATP-channel pathway genes ABCC8/KCNJ11 (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2).
2.2 Genetic risk factors (causal genes/variants)
- KATP channel genes dominate persistent CHI: In a 10-year Ukrainian national registry study (Frontiers in Endocrinology; publication date Dec 2024; URL https://doi.org/10.3389/fendo.2024.1497579), “Pathogenic variants in the K-ATP channel genes were the only identified genetic cause of p-CHI (ABCC8 (n=17) and KCNJ11 (n=2))” (globa2024congenitalhyperinsulinismin pages 1-2).
- A 2024 mechanistic review (Journal of Pediatric Endocrinology and Diabetes; Aug 2024; URL https://doi.org/10.25259/jped_25_2024) states that “The majority of the cases relate to defects in KATP channels … attributable to mutations in ABCC8 and KCNJ11” (mittal2024molecularmechanismsunderlying pages 1-2).
- Additional genes commonly cited in CHI/HH literature include GLUD1, GCK, HADH, SLC16A1, HNF4A, HNF1A, UCP2, CACNA1D (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, globa2024congenitalhyperinsulinismin pages 2-3, burroni2021earlydiagnosisof pages 1-2).
Genotype–histology correlations - Diffuse CHI: arises from dominant or recessive KATP mutations (recessive often more severe) (mittal2024molecularmechanismsunderlying pages 1-2). - Focal CHI: classically results from a paternally inherited germline KATP pathogenic variant plus post-zygotic loss of the maternal allele in the focal lesion (somatic UPD/unmasking), enabling curative lesionectomy (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 2-3).
2.3 Environmental risk factors / protective factors
- For isolated CHI, the retrieved evidence emphasizes genetic etiologies; no validated environmental protective factors were captured in the retrieved corpus (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2).
2.4 Gene–environment interactions
- Not specifically documented in the retrieved corpus for isolated CHI; syndromic/secondary hyperinsulinism contexts exist (e.g., Beckwith–Wiedemann), but constitute a distinct category from isolated CHI (globa2024congenitalhyperinsulinismin pages 1-2, ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6).
3. Phenotypes
3.1 Core phenotype spectrum
Onset and presentation - CHI commonly presents early: a 2022 Orphanet Journal of Rare Diseases review (Feb 2022; URL https://doi.org/10.1186/s13023-022-02214-y) reports hypoglycemia presents in the first week in 60–70% of cases; ~50% present with seizures; 20–30% diagnosed in the first year and ~10% after age 1 year (banerjee2022congenitalhyperinsulinismin pages 1-2). - Neonatal/infant presentations include severe non-ketotic hypoglycemia, lethargy, seizures, and other neuroglycopenic symptoms (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, banerjee2022congenitalhyperinsulinismin pages 1-2).
Laboratory phenotype - Non-ketotic hypoglycemia with suppressed ketones and free fatty acids plus detectable/inappropriately high insulin/C-peptide during hypoglycemia (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, mittal2024molecularmechanismsunderlying pages 1-2).
3.2 Neurodevelopmental and quality-of-life impact
- A 2021 scoping review on HRQoL in CHI (Frontiers in Endocrinology; Dec 2021; URL https://doi.org/10.3389/fendo.2021.784932) reports substantial neurodevelopmental morbidity: “incidence rates of abnormal neurodevelopmental outcomes have been reported between 26% and 44%” (kristensen2021healthrelatedqualityof pages 1-2).
- A 2022 challenges/unmet-needs review emphasizes psychosocial burden. Reported family/parent impacts include: “70% (36/51) of respondents with children below 5 years of age feel that their lives were ‘ruled by HI’,” “48% (59/123) … physical health has suffered,” and “over two-thirds … (67% [82/123]) … mental health has suffered” (banerjee2022congenitalhyperinsulinismin pages 9-10).
3.3 Suggested HPO terms (examples; non-exhaustive)
- Hypoglycemia (HP:0001943)
- Hyperinsulinemia (HP:0000842)
- Seizure (HP:0001250)
- Lethargy (HP:0001254)
- Abnormality of ketone body metabolism / low ketones during hypoglycemia (map to laboratory phenotype; often encoded via metabolic phenotype terms)
- Developmental delay (HP:0001263)
- Abnormality of motor development (HP:0001270)
- Abnormality of language development (HP:0000750)
(HPO IDs are provided as standard ontology suggestions; not directly asserted by the retrieved papers, which describe the underlying clinical features.)
4. Genetic / Molecular Information
4.1 Causal genes (high-confidence in retrieved evidence)
- ABCC8 and KCNJ11 (KATP channel subunits SUR1 and Kir6.2) are repeatedly identified as major causal genes and are the only persistent-CHI causes found in the Ukrainian national study cohort (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2, burroni2021earlydiagnosisof pages 1-2).
4.2 Pathogenic variant types and consequences (qualitative)
- Mechanistically, many ABCC8/KCNJ11 CHI variants are described as inactivating / loss-of-function at the KATP channel level, leading to constitutive β-cell depolarization and insulin secretion (globa2024congenitalhyperinsulinismin pages 1-2, takasawa2024clinicalmanagementof pages 1-6).
4.3 Modifier genes / epigenetic information
- Not specifically resolved in retrieved evidence for isolated CHI; focal CHI mechanism involves somatic loss of the maternal allele (mosaic, tissue-specific genetic event) (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, globa2024congenitalhyperinsulinismin pages 2-3).
5. Environmental Information
- No CHI-specific environmental exposures were identified in the retrieved corpus for isolated CHI; disease causality is predominantly genetic in this evidence set (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2).
6. Mechanism / Pathophysiology
6.1 Core causal chain (KATP-centered model)
- Genetic lesion (often ABCC8/KCNJ11) impairs β-cell KATP channel function (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2).
- β-cell membrane becomes inappropriately depolarized, promoting calcium influx and insulin granule exocytosis even at low glucose (review-level mechanism summary) (mittal2024molecularmechanismsunderlying pages 1-2).
- Excess insulin action suppresses ketogenesis and lipolysis, leading to low ketones and free fatty acids during hypoglycemia and increased glucose infusion requirements (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, mittal2024molecularmechanismsunderlying pages 1-2).
- Recurrent neuroglycopenia increases risk of seizures and neurodevelopmental impairment (banerjee2022congenitalhyperinsulinismin pages 1-2, kristensen2021healthrelatedqualityof pages 1-2).
6.2 Focal lesion mechanism
- Focal CHI is described as paternally inherited germline mutation “together with post-zygotic loss of normal maternal allele,” creating a localized hyperfunctional β-cell population that can be surgically cured (mittal2024molecularmechanismsunderlying pages 1-2, ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, globa2024congenitalhyperinsulinismin pages 2-3).
6.3 Suggested ontology terms
GO biological process (examples): - Regulation of insulin secretion - Glucose homeostasis - Potassium ion transmembrane transport
Cell type (CL) suggestions: - Pancreatic β cell (endocrine pancreas)
(These are standard mechanistic ontology mappings; the retrieved evidence supports β-cell involvement and insulin secretion dysregulation.)
7. Anatomical Structures Affected
- Primary organ: pancreas (endocrine pancreas/islets; β-cells) (mittal2024molecularmechanismsunderlying pages 1-2, burroni2021earlydiagnosisof pages 1-2).
- Secondary organ system impacts: central nervous system (hypoglycemia-related seizures and neurodevelopmental sequelae) (banerjee2022congenitalhyperinsulinismin pages 1-2, kristensen2021healthrelatedqualityof pages 1-2).
UBERON suggestions: pancreas; pancreatic islet of Langerhans.
8. Temporal Development
- Typically congenital/neonatal onset, frequently within the first days/week of life (banerjee2022congenitalhyperinsulinismin pages 1-2, ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6).
- Course may be persistent or remit early in some patients; Ukrainian study defined early remission as spontaneous remission by age 2 with 24 months free of hypoglycemia (globa2024congenitalhyperinsulinismin pages 2-3).
9. Inheritance and Population
9.1 Epidemiology (recent quantitative summaries)
- Incidence reported in a 2022 review: ~1:28,000–1:50,000 in Western populations and up to ~1:2,500 in populations with higher consanguinity (banerjee2022congenitalhyperinsulinismin pages 1-2).
- A 2024 single-center experience cites Japanese incidence estimates (2017–2018 survey): transient CHI 1 in 13,600 and persistent CHI 1 in 31,600 births (takasawa2024clinicalmanagementof pages 1-6).
9.2 Population genetics and heterogeneity
- In the Ukrainian registry, genetic diagnoses were made in 67.5% (27/40) overall; 86.3% (19/22) in persistent CHI; 44.4% (8/18) in early-remission CHI (globa2024congenitalhyperinsulinismin pages 1-2).
- Persistent CHI in that registry was exclusively ABCC8/KCNJ11, while early-remission CHI showed broader heterogeneity including syndromic causes (globa2024congenitalhyperinsulinismin pages 1-2).
10. Diagnostics
10.1 Biochemical diagnosis (“critical sample”)
Biochemical characterization during hypoglycemia is central. - Typical critical sample features described include inappropriate insulin and detectable C-peptide, hypoketonemia, low free fatty acids, and positive glycemic response to glucagon, often with high glucose infusion needs (>8–10 mg/kg/min) (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, mittal2024molecularmechanismsunderlying pages 1-2).
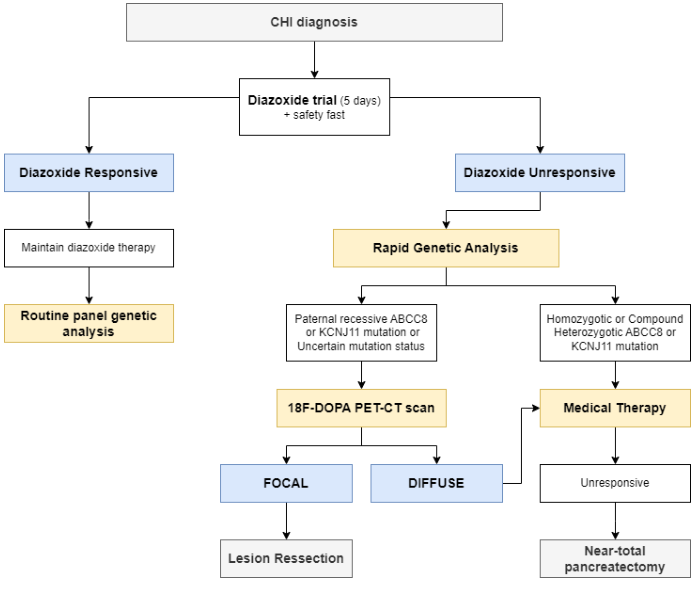
The 2023 review includes a diagnostic biochemical criteria table (graca2023managingcongenitalhyperinsulinism media 1893ff72) and a management flowchart incorporating biochemical and genetic steps (graca2023managingcongenitalhyperinsulinism media 3dcd38ca).
10.2 Imaging and subtype classification
- 18F-DOPA PET/CT is emphasized as the key modality for localizing focal lesions and differentiating focal vs diffuse disease to guide surgery (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 2-3, burroni2021earlydiagnosisof pages 1-2).
- Performance metrics reported in the Ukrainian study excerpt: sensitivity 88%, specificity 94%, accuracy 88–100%; reported superior to 68Ga-DOTANOC PET/CT for predicting focal lesions (globa2024congenitalhyperinsulinismin pages 2-3).
10.3 Histopathology
- Histologic types: focal, diffuse, atypical (burroni2021earlydiagnosisof pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2).
- In the Ukrainian p-CHI surgical subset with histology (n=19): focal 73.7% (14/19), diffuse 10.5% (2/19), atypical 15.8% (3/19) (globa2024congenitalhyperinsulinismin pages 1-2).
10.4 Genetic testing strategy
- Ukrainian national study used tiered testing: Sanger sequencing of ABCC8/KCNJ11 followed by targeted NGS panel covering at least ABCC8, KCNJ11, GLUD1, GCK, CACNA1D, HADH, HNF1A, HNF4A, INSR, PMM2, SLC16A1, TRMT10A (globa2024congenitalhyperinsulinismin pages 2-3).
- Genetic diagnosis informs likelihood of diazoxide responsiveness and focal vs diffuse pathways (ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6, takasawa2024clinicalmanagementof pages 1-6).
10.5 Differential diagnosis (evidence-limited in retrieved corpus)
- The evidence set focuses on CHI; comprehensive differential diagnosis lists (e.g., endocrine deficiencies, inborn errors of metabolism) are not fully enumerated in retrieved excerpts.
11. Outcome / Prognosis
11.1 Surgical outcomes
- In the Ukrainian registry, “complete recovery was observed in all 14 with focal disease” after surgery, while relapse occurred in some diffuse/atypical cases (globa2024congenitalhyperinsulinismin pages 1-2).
11.2 Neurodevelopmental outcomes
- Abnormal neurodevelopmental outcomes reported in the literature summarized by the 2021 scoping review: 26–44% (kristensen2021healthrelatedqualityof pages 1-2).
- Ongoing concerns that long-term developmental outcomes “have not significantly improved” are emphasized in the 2022 review, supporting the need for early recognition and specialized multidisciplinary care (banerjee2022congenitalhyperinsulinismin pages 1-2).
12. Treatment
12.1 Current standard-of-care ladder and real-world implementation
A 2023 review presents a management algorithm (graca2023managingcongenitalhyperinsulinism media 3dcd38ca) and discusses that: - Diazoxide is the only approved first-line chronic drug (graca2023managingcongenitalhyperinsulinism pages 33-37, graca2023managingcongenitalhyperinsulinisma pages 33-37). - Octreotide and long-acting somatostatin analogs are used as second-line therapies; nifedipine and sirolimus are reserved for refractory cases in some settings (graca2023managingcongenitalhyperinsulinisma pages 33-37).
A 2024 single-center experience provides pragmatic implementation details for diazoxide-unresponsive CHI, including: - Emphasis on early genetic diagnosis guiding therapy decisions; - Continuous subcutaneous octreotide as a common second-line approach to avoid subtotal pancreatectomy; - Switching to long-acting somatostatin analogs such as lanreotide and using home continuous glucose monitoring (CGM) for management (takasawa2024clinicalmanagementof pages 1-6).
12.2 Surgery
- Focal CHI: partial pancreatectomy/lesionectomy is curative in many cases when guided by imaging and histology (mittal2024molecularmechanismsunderlying pages 1-2, globa2024congenitalhyperinsulinismin pages 1-2, burroni2021earlydiagnosisof pages 1-2).
- Diffuse CHI: near-total pancreatectomy is reserved for refractory disease due to high risks of postoperative diabetes/exocrine insufficiency (mittal2024molecularmechanismsunderlying pages 1-2, graca2023managingcongenitalhyperinsulinisma pages 33-37).
12.3 Emerging/experimental: GLP-1 receptor antagonism (exendin(9-39); avexitide)
ClinicalTrials.gov evidence (trial registry; provides dates and protocol design): - NCT00571324 (“Effect of Exendin-(9-39) on Glycemic Control in Subjects With Congenital Hyperinsulinism”) is a randomized crossover, open-label Phase 1/2 pilot study (enrollment 9) evaluating whether exendin(9-39) increases fasting glucose and characterizing pharmacokinetics; study start 2007-08; completion 2014-12; results posted 2016-11-09; last update 2017-12-11 (NCT00571324 chunk 1). - NCT00835328 (“Effect of Exendin (9-39) on Glucose Requirements to Maintain Euglycemia”) targets infants <12 months with diazoxide-refractory CHI and includes PK endpoints and metabolic measures (insulin/glucose and beta-hydroxybutyrate sampling) (NCT00835328 chunk 2).
URL (ClinicalTrials.gov): https://clinicaltrials.gov/study/NCT00571324 ; https://clinicaltrials.gov/study/NCT00835328 (NCT00571324 chunk 1, NCT00835328 chunk 2).
12.4 Suggested MAXO terms (examples)
- Diazoxide therapy (pharmacotherapy)
- Somatostatin analog therapy (octreotide/lanreotide)
- Partial pancreatectomy / lesionectomy
- Near-total pancreatectomy
- Continuous glucose monitoring
- Glucagon administration (acute management)
(MAXO IDs not retrievable within this run; terms are provided as controlled-action suggestions.)
13. Prevention
- Primary prevention: not established for isolated genetic CHI.
- Secondary/tertiary prevention: prevention of neurodevelopmental injury relies on early recognition, rapid stabilization of glucose, and specialist referral (banerjee2022congenitalhyperinsulinismin pages 1-2, kristensen2021healthrelatedqualityof pages 1-2).
- Genetic counseling: family planning is impacted; in one reported family survey, “36% chose not to have any additional children and 19% were delaying having additional children,” supporting the importance of genetic diagnosis and counseling (banerjee2022congenitalhyperinsulinismin pages 9-10).
14. Other Species / Natural Disease
- No direct evidence on naturally occurring CHI analogs in non-human species was retrieved in this corpus.
15. Model Organisms
- No model organism papers were retrieved in this evidence set. Mechanistic work in the field commonly uses β-cell models (islets/cell lines) and genetically engineered animals for KATP pathway genes, but specific models cannot be cited here due to corpus limitations.
Recent developments (2023–2024 emphasis) and expert analysis
- Genotype-informed, subtype-targeted management is increasingly operationalized (genetic testing + 18F-DOPA PET/CT to decide focal surgery vs chronic medical management), supported by a 2024 national registry study that combined genetic testing with PET/CT and achieved surgical cure in all focal cases (globa2024congenitalhyperinsulinismin pages 1-2, globa2024congenitalhyperinsulinismin pages 2-3).
- Real-world implementation improvements reported in 2024 include switching from short-acting octreotide to long-acting somatostatin analogs and integration of home CGM for ongoing management and developmental follow-up (takasawa2024clinicalmanagementof pages 1-6).
- Unmet need remains high: a major 2022 review emphasizes that “long-term developmental outcomes have not significantly improved,” and families report substantial psychosocial burden, supporting ongoing need for new therapies that reduce monitoring and hypoglycemia risk (banerjee2022congenitalhyperinsulinismin pages 1-2, banerjee2022congenitalhyperinsulinismin pages 9-10).
Key visual evidence (diagnostic/treatment pathway)
- CHI management algorithm / flowchart: (graca2023managingcongenitalhyperinsulinism media 3dcd38ca)
- Biochemical criteria table for CHI diagnosis: (graca2023managingcongenitalhyperinsulinism media 1893ff72)
Evidence limitations (important for knowledge base curation)
- Database identifiers (OMIM/Orphanet/MONDO/MeSH/ICD codes) were not retrievable from the current tool run; they should be filled from authoritative resources directly.
- 2023–2024 primary literature on sirolimus and next-generation GLP-1R antagonists (avexitide) outcomes was not captured in the retrieved corpus beyond reviews and ClinicalTrials.gov protocol records; additional targeted retrieval would be required for up-to-date efficacy/safety statistics.
References
-
(mittal2024molecularmechanismsunderlying pages 1-2): Medha Mittal, Amit Kumar Gupta, and Seema Kapoor. Molecular mechanisms underlying congenital hyperinsulinemia of infancy and its relevance to management – a review. Journal of Pediatric Endocrinology and Diabetes, 4:9-20, Aug 2024. URL: https://doi.org/10.25259/jped_25_2024, doi:10.25259/jped_25_2024. This article has 1 citations.
-
(globa2024congenitalhyperinsulinismin pages 1-2): Evgenia Globa, Henrik Thybo Christesen, Michael Bau Mortensen, Jayne A. L. Houghton, Anne Lerberg Nielsen, Sönke Detlefsen, and Sarah E. Flanagan. Congenital hyperinsulinism in the ukraine: a 10-year national study. Frontiers in Endocrinology, Dec 2024. URL: https://doi.org/10.3389/fendo.2024.1497579, doi:10.3389/fendo.2024.1497579. This article has 4 citations.
-
(banerjee2022congenitalhyperinsulinismin pages 1-2): Indraneel Banerjee, Julie Raskin, Jean-Baptiste Arnoux, Diva D. De Leon, Stuart A. Weinzimer, Mette Hammer, David M. Kendall, and Paul S. Thornton. Congenital hyperinsulinism in infancy and childhood: challenges, unmet needs and the perspective of patients and families. Orphanet Journal of Rare Diseases, Feb 2022. URL: https://doi.org/10.1186/s13023-022-02214-y, doi:10.1186/s13023-022-02214-y. This article has 101 citations and is from a peer-reviewed journal.
-
(takasawa2024clinicalmanagementof pages 1-6): Kei Takasawa, Ryosei Iemura, Ryuta Orimoto, Haruki Yamano, Shizuka Kirino, Eriko Adachi, Yoko Saito, Kurara Yamamoto, Nozomi Matsuda, Shigeru Takishima, Kumi Shuno, Hanako Tajima, Manabu Sugie, Yuki Mizuno, Akito Sutani, Kentaro Okamoto, Michiya Masue, Tomohiro Morio, and Kenichi Kashimada. Clinical management of diazoxide-unresponsive congenital hyperinsulinism: a single-center experience. Clinical Pediatric Endocrinology, 33:187-194, Jun 2024. URL: https://doi.org/10.1297/cpe.2024-0004, doi:10.1297/cpe.2024-0004. This article has 4 citations and is from a peer-reviewed journal.
-
(graca2023managingcongenitalhyperinsulinisma pages 33-37): IMCG Graça. Managing congenital hyperinsulinism: a review of current diagnostic and therapeutic methods. Unknown journal, 2023.
-
(globa2024congenitalhyperinsulinismin pages 2-3): Evgenia Globa, Henrik Thybo Christesen, Michael Bau Mortensen, Jayne A. L. Houghton, Anne Lerberg Nielsen, Sönke Detlefsen, and Sarah E. Flanagan. Congenital hyperinsulinism in the ukraine: a 10-year national study. Frontiers in Endocrinology, Dec 2024. URL: https://doi.org/10.3389/fendo.2024.1497579, doi:10.3389/fendo.2024.1497579. This article has 4 citations.
-
(burroni2021earlydiagnosisof pages 1-2): Luca Burroni, Andrea Palucci, Giuseppina Biscontini, and Valentino Cherubini. Early diagnosis of focal congenital hyperinsulinism: a fluorine-18-labeled l-dihydroxyphenylalanine positron emission tomography/computed tomography study. World Journal of Nuclear Medicine, 20:395-397, Oct 2021. URL: https://doi.org/10.4103/wjnm.wjnm_159_20, doi:10.4103/wjnm.wjnm_159_20. This article has 4 citations and is from a peer-reviewed journal.
-
(ouadghiri2025neonatalcongenitalhyperinsulinism pages 5-6): Fouad Khalil El Ouadghiri, Anass Ayyad, Sahar Messaoudi, and Rim Amrani. Neonatal congenital hyperinsulinism: a case-based contribution to the understanding of a rare disorder. Cureus, Aug 2025. URL: https://doi.org/10.7759/cureus.89272, doi:10.7759/cureus.89272. This article has 0 citations.
-
(graca2023managingcongenitalhyperinsulinism media 3dcd38ca): IMCG Graça. Managing congenital hyperinsulinism: a review of current diagnostic and therapeutic methods. Unknown journal, 2023.
-
(graca2023managingcongenitalhyperinsulinism media 1893ff72): IMCG Graça. Managing congenital hyperinsulinism: a review of current diagnostic and therapeutic methods. Unknown journal, 2023.
-
(NCT00571324 chunk 1): Diva De Leon. Effect of Exendin-(9-39) on Glycemic Control in Subjects With Congenital Hyperinsulinism. Diva De Leon. 2007. ClinicalTrials.gov Identifier: NCT00571324
-
(NCT00835328 chunk 2): Diva De Leon. Effect of Exendin (9-39) on Glucose Requirements to Maintain Euglycemia. Diva De Leon. 2009. ClinicalTrials.gov Identifier: NCT00835328
-
(kristensen2021healthrelatedqualityof pages 1-2): Kaja Kristensen, Julia Quitmann, and Stefanie Witt. Health-related quality of life of children and adolescents with congenital hyperinsulinism – a scoping review. Frontiers in Endocrinology, Dec 2021. URL: https://doi.org/10.3389/fendo.2021.784932, doi:10.3389/fendo.2021.784932. This article has 3 citations.
-
(banerjee2022congenitalhyperinsulinismin pages 9-10): Indraneel Banerjee, Julie Raskin, Jean-Baptiste Arnoux, Diva D. De Leon, Stuart A. Weinzimer, Mette Hammer, David M. Kendall, and Paul S. Thornton. Congenital hyperinsulinism in infancy and childhood: challenges, unmet needs and the perspective of patients and families. Orphanet Journal of Rare Diseases, Feb 2022. URL: https://doi.org/10.1186/s13023-022-02214-y, doi:10.1186/s13023-022-02214-y. This article has 101 citations and is from a peer-reviewed journal.
-
(graca2023managingcongenitalhyperinsulinism pages 43-45): IMCG Graça. Managing congenital hyperinsulinism: a review of current diagnostic and therapeutic methods. Unknown journal, 2023.
-
(graca2023managingcongenitalhyperinsulinism pages 33-37): IMCG Graça. Managing congenital hyperinsulinism: a review of current diagnostic and therapeutic methods. Unknown journal, 2023.