CHARGE Syndrome — Comprehensive Disease Characteristics Report
Target disease: CHARGE syndrome (Mendelian developmental disorder)
Primary causal gene: CHD7 (autosomal dominant, typically de novo)
1. Disease Information
1.1 Overview (current understanding)
CHARGE syndrome is a clinically defined multiple congenital anomaly disorder originally described as a non-random cluster of malformations. The CHARGE acronym denotes Coloboma, Heart defects, Atresia of choanae, Retarded growth/development, Genital hypoplasia, and Ear anomalies/deafness. (bergman2011chd7mutationsand pages 1-6, mcj2006chargesyndromethe pages 1-2)
A key consistent feature emphasized in early molecular-era cohorts is semicircular canal hypoplasia leading to vestibular areflexia, which helps explain balance and motor delay phenotypes. (mcj2006chargesyndromethe pages 1-2)
1.2 Key identifiers and cross-references
- OMIM: 214800 (CHARGE syndrome) (mcj2006chargesyndromethe pages 1-2, bergman2011chd7mutationsand pages 1-6)
- Orphanet/Orpha code: ORPHA:138 (wolanska2025analysisofthe pages 7-10)
- Synonyms / alternative names: “Hall–Hittner syndrome” (also referred to historically as CHARGE association) (wolanska2025analysisofthe pages 7-10)
- MONDO / ICD / MeSH: Not found explicitly in the retrieved full-text set; therefore not reported here.
1.3 Evidence type note
Most knowledge summarized here derives from aggregated disease-level resources (systematic reviews, cohort/genotype-phenotype studies, mechanistic studies) rather than EHR-derived real-world datasets. Examples include systematic review evidence for CHD epidemiology and outcomes (polito2024chargesyndromeand pages 1-2, polito2024chargesyndromeand pages 2-3) and mutation-positive cohort summaries (bergman2011chd7mutationsand pages 6-11).
2. Etiology
2.1 Primary causes
Genetic cause (dominant): Pathogenic variants in CHD7 are the major cause of CHARGE syndrome. CHARGE is described as autosomal dominant with variable expressivity; most pathogenic CHD7 variants arise de novo, but parent-to-child transmission occurs. (bergman2011chd7mutationsand pages 1-6, bergman2011chd7mutationsand pages 6-11)
CHD7 was identified as a major gene on chromosome 8q12.1. (mcj2006chargesyndromethe pages 1-2)
2.2 Risk factors
Genetic risk factor: Having a pathogenic CHD7 variant (typically heterozygous loss-of-function) is the dominant risk factor. Clinical genetic counseling must account for de novo predominance plus rare transmission and mosaicism. (mcj2006chargesyndromethe pages 1-2, bergman2011chd7mutationsand pages 24-29)
Non-genetic risk factors: No established environmental/toxic/infectious risk factors were identified in the retrieved sources.
2.3 Protective factors
No validated genetic or environmental protective factors were identified in the retrieved sources.
2.4 Gene–gene / oligogenic interactions (emerging)
A 2024 report proposes digenic inheritance/modifier effects involving CHD7 plus SMCHD1 in a family with variable hypogonadotropic hypogonadism and CHARGE-overlapping features, suggesting oligogenic contributions to penetrance/expressivity in some CHD7-related presentations. (wang2024digenicchd7and pages 1-2, wang2024digenicchd7and pages 2-4)
3. Phenotypes
3.1 Core phenotype spectrum (with frequencies)
Phenotypic variability is high, but several features are highly prevalent in mutation-positive cohorts.
Mutation-positive cohort frequencies (Bergman et al., 2011; CHD7+ cohort, n≈280): * Semicircular canal anomaly: 110/117 (~94%) (bergman2011chd7mutationsand pages 6-11) * Coloboma: 189/234 (~81%) (bergman2011chd7mutationsand pages 6-11) * Choanal atresia: 99/179 (~55%) (bergman2011chd7mutationsand pages 6-11) * Congenital heart defect: 191/252 (~76%) (bergman2011chd7mutationsand pages 6-11) * Feeding difficulties: 90/110, and tube feeding was frequently required (“necessitating tube feeding 82% (32–93%)”) (bergman2011chd7mutationsand pages 6-11) * Cranial nerve dysfunction: 173/174 (~99%) (bergman2011chd7mutationsand pages 6-11)
Broad phenotype frequency summary (Wieland et al., 2020; tabulated summary): * Developmental delay: 100% * Semicircular canal anomaly: 95% * External ear anomaly: 95% * Cranial nerve dysfunction: 95% * Coloboma: 80% * Congenital heart defect: 80% * Feeding difficulties: 80% * Choanal atresia: 50% * Tracheoesophageal anomaly: 25% (among other features) (wieland2020chargesyndrome pages 1-3)
Quality-of-life-related phenotype study (Wolańska, 2024/2025 thesis; 29 genetically confirmed): * Coloboma 100%, heart defects 82.8%, choanal atresia 35%, genital abnormalities 58.6%, hearing loss 86.2%; 76% had height below 3rd percentile. Family QoL measured by PedsQL Family Impact was described as intermediate/average, with higher QoL among parents who accept the child’s illness. (wolanska2025analysisofthe pages 76-79)
3.2 Phenotype characteristics
Age of onset: Predominantly congenital/neonatal with multi-organ malformations; neurodevelopmental features emerge in infancy/childhood. (mcj2006chargesyndromethe pages 1-2, wieland2020chargesyndrome pages 1-3)
Progression: Some domains may be progressive (e.g., mixed hearing loss reported as potentially progressive in clinical management guidance). (wieland2020chargesyndrome pages 10-11)
3.3 Suggested HPO terms (non-exhaustive, for KB mapping)
Below are commonly used HPO concepts aligned to the phenotypes reported in the cited sources: * Coloboma — HP:0000589 * Choanal atresia — HP:0000453 * Congenital heart defect — HP:0001627 * Abnormal semicircular canals / semicircular canal hypoplasia — HP:0008558 (or related vestibular/inner ear structure terms) * Sensorineural hearing impairment — HP:0000407 * Feeding difficulties — HP:0011968 * Facial palsy — HP:0007209 * Cleft lip/palate — HP:0000202 / HP:0000175 * Hypogonadotropic hypogonadism — HP:0000044 * Developmental delay — HP:0001263
(Exact HPO IDs may vary by knowledge base conventions; the above are intended as practical starting points for curation.)
4. Genetic / Molecular Information
4.1 Causal gene(s)
- CHD7 (chromodomain helicase DNA-binding protein 7) is the primary causal gene, encoding an ATP-dependent chromatin remodeler. (mcj2006chargesyndromethe pages 1-2, driesen2024chd7disorder—notcharge pages 1-2)
4.2 Pathogenic variant spectrum (human)
In a large clinical genetics review, CHD7 variant classes in clinically diagnosed CHARGE include a predominance of truncating variants (nonsense/frameshift), with additional splice-site and missense variants; haploinsufficiency is emphasized as the key mechanism. (bergman2011chd7mutationsand pages 6-11)
A 2023 case report illustrates challenges in interpreting non-canonical intronic variants and provides a workflow for functional classification. In two unrelated patients, an intronic CHD7 variant c.5607+17A>G was shown to induce aberrant splicing using minigene assays and patient cDNA validation, upgrading a VUS toward pathogenic. (rossi2023casereportfunctional pages 1-2, rossi2023casereportfunctional pages 2-4)
4.3 Inheritance, penetrance, expressivity, mosaicism
CHARGE is autosomal dominant with variable expressivity; most CHD7 mutations occur de novo, but inherited cases occur. (bergman2011chd7mutationsand pages 1-6, bergman2011chd7mutationsand pages 6-11)
Somatic mosaicism has been reported (e.g., in an unaffected mother in a sib pair), supporting germline mosaicism as a recurrence mechanism. (mcj2006chargesyndromethe pages 1-2)
Genetic counseling guidance: recurrence risk from parental mosaicism is estimated at ~2–3%, and transmission risk from an affected individual is 50%; prenatal molecular testing/ultrasound and preimplantation genetic diagnosis are recommended for discussion. (bergman2011chd7mutationsand pages 24-29)
4.4 Modifier genes / oligogenicity
Mouse model work proposes that foliation-related genes (e.g., Engrailed, FGF pathway genes, Zic genes) may modify neurodevelopmental phenotypes in CHARGE. (whittaker2017distinctcerebellarfoliation pages 9-10)
Human family report: co-inheritance of pathogenic CHD7 truncation and a SMCHD1 missense variant is proposed to contribute to intrafamilial variability (not definitive proof of causality but a notable 2024 development). (wang2024digenicchd7and pages 1-2, wang2024digenicchd7and pages 2-4)
4.5 Epigenetics / episignatures (emerging)
CHARGE is considered a “chromatinopathy” (chromatin remodeling disorder) conceptually, and clinical trials are now including DNA methylation episignature characterization for prenatal-onset disorders including CHD7-associated conditions. (NCT06475651 chunk 2)
5. Environmental Information
No consistent environmental, lifestyle, or infectious causal factors were identified in the retrieved evidence set. The condition is primarily genetic/developmental. (bergman2011chd7mutationsand pages 1-6, mcj2006chargesyndromethe pages 1-2)
6. Mechanism / Pathophysiology
6.1 CHD7 function and upstream mechanism
CHD7 encodes an ATP-dependent nucleosome remodeling factor involved in tissue-specific gene regulation during development. (driesen2024chd7disorder—notcharge pages 1-2)
A core mechanistic model is that CHD7 regulates enhancer activity and cell-type-specific transcriptional programs.
6.2 Neural crest dysfunction (neurocristopathy framework)
A human iPSC model supports the long-standing hypothesis that CHARGE is a neurocristopathy: * “CHARGE syndrome modeling using patient-iPSCs reveals defective migration of neural crest cells harboring CHD7 mutations” with altered expression of migration-related genes and impaired delamination/migration/motility. (okuno2017chargesyndromemodeling pages 1-2, okuno2017chargesyndromemodeling pages 5-6)
Enhancer regulation in human neural crest cells: CHD7 binding is enriched at active enhancers, with TFAP2A motifs in hNCC-specific CHD7 peaks and enrichment near neural crest regulators (e.g., SOX9, MSX1/2). (sanosaka2022chromatinremodelerchd7 pages 2-3, sanosaka2022chromatinremodelerchd7 pages 1-2)
Causal chain (conceptual): CHD7 haploinsufficiency → altered enhancer accessibility / target-gene expression in neural crest lineages → impaired NCC migration/adhesion programs → malformations of NCC-derived/populated structures (craniofacial, heart outflow tract, ear, eye). (okuno2017chargesyndromemodeling pages 1-2, sanosaka2022chromatinremodelerchd7 pages 2-3)
6.3 Inner ear / hair cell differentiation mechanisms
Human inner ear organoids show that CHD7 is required for otic lineage specification and sensory epithelium formation: * Loss of CHD7 (or its chromatin remodeling activity) leads to “complete absence of hair cells and supporting cells,” and transcriptome profiling suggests “disruption of deafness gene expression” as a mechanism for CHARGE-associated sensorineural hearing loss. (nie2022chd7regulatesotic pages 1-2)
6.4 p53 pathway contribution (mouse genetics)
A high-impact mouse genetics study provides evidence that inappropriate p53 activation contributes to CHARGE-like phenotypes: * CHD7 binds the p53 promoter and negatively regulates p53; CHD7 loss activates p53 in mouse neural crest cells and patient samples, and p53 reduction partially rescues Chd7-null phenotypes. (nostrand2014inappropriatep53activation pages 1-2)
6.5 Cerebellar developmental defects and modifier pathways
In Chd7 haploinsufficient mice, cerebellar hypoplasia and foliation anomalies show incomplete penetrance (e.g., 67% overall penetrance for specific foliation phenotypes in combined analyses) and may be modified by developmental patterning genes (Engrailed/FGF/Zic pathways). (whittaker2017distinctcerebellarfoliation pages 3-6, whittaker2017distinctcerebellarfoliation pages 9-10)
6.6 Multi-omics (zebrafish; emerging target discovery)
A zebrafish CHARGE model used transcriptomics + proteomics integration to identify dysregulated pathways and candidate downstream mediators; CRISPR knockdown of candidate genes (capgb, nefla, rdh5) phenocopied behavioral defects seen in chd7 mutants, supporting a pipeline for therapeutic target nomination. (hancock2026multiomicanalysesidentify pages 1-3, hancock2026multiomicanalysesidentify pages 11-13)
6.7 Suggested ontology terms for mechanisms
GO Biological Process (examples): * Chromatin remodeling — GO:0006338 * Regulation of transcription, DNA-templated — GO:0006355 * Neural crest cell migration — GO:0001755 * Inner ear development — GO:0048839 * Sensory perception of sound — GO:0007605
Cell Ontology (CL) (examples): * Neural crest cell — CL:0000135 * Otic progenitor / hair cell / supporting cell (use lineage-appropriate CL terms)
GO Cellular Component (examples): * Nucleus — GO:0005634 * Chromatin — GO:0000785
7. Anatomical Structures Affected
7.1 Organ and system level (primary)
- Eye (coloboma) (bergman2011chd7mutationsand pages 6-11, wieland2020chargesyndrome pages 1-3)
- Heart / great vessels (multiple CHDs) (polito2024chargesyndromeand pages 1-2, bergman2011chd7mutationsand pages 6-11)
- Nasal choanae / upper airway (choanal atresia/stenosis) (bergman2011chd7mutationsand pages 6-11, wieland2020chargesyndrome pages 1-3)
- Ear (external/middle/inner ear), vestibular apparatus, cochleovestibular nerve (bergman2011chd7mutationsand pages 6-11, wieland2020chargesyndrome pages 10-11)
- CNS including cerebellum (neurodevelopmental delay; cerebellar anomalies in models) (whittaker2017distinctcerebellarfoliation pages 1-2, wolanska2025analysisofthe pages 76-79)
- Cranial nerves (feeding/swallowing, facial palsy) (bergman2011chd7mutationsand pages 6-11, webb2021aframeworkfor pages 8-10)
- Endocrine/reproductive axis (hypogonadotropic hypogonadism; genital hypoplasia) (driesen2024chd7disorder—notcharge pages 8-9, wang2024digenicchd7and pages 2-4)
- Esophagus/trachea (tracheoesophageal anomalies; feeding/aspiration risk) (wieland2020chargesyndrome pages 1-3, polito2024chargesyndromeand pages 7-8)
7.2 Suggested UBERON terms (examples)
- Eye — UBERON:0000970
- Heart — UBERON:0000948
- Choana — UBERON:0000467
- Inner ear — UBERON:0001768
- Semicircular canal — UBERON:0001786
- Cerebellum — UBERON:0002037
- Cranial nerve — UBERON:0001021
- Pituitary gland / hypothalamus — UBERON:0000007 / UBERON:0001898
8. Temporal Development
- Typical onset: congenital/neonatal (multi-organ malformations) (mcj2006chargesyndromethe pages 1-2, wieland2020chargesyndrome pages 1-3)
- Course: lifelong, with major early-life morbidity driven by airway/feeding and cardiac anomalies; neurodevelopmental and sensory impairments require long-term supports. (meisner2020congenitalheartdefects pages 5-6, wieland2020chargesyndrome pages 10-11)
9. Inheritance and Population
9.1 Epidemiology
Incidence/prevalence estimates vary by study and ascertainment. * Estimated birth prevalence in early cohort reports: 1/10,000 to 1/15,000; a regional estimate reported 1/8,500 in Atlantic Canada. (mcj2006chargesyndromethe pages 1-2) * A 2024 clinical review estimated CHARGE incidence 1/15,000–1/17,000 live births, and separately estimated CHD7-mutation birth incidence 1/18,400. (driesen2024chd7disorder—notcharge pages 1-2) * A 2024 systematic review states incidence 1–3 per 10,000 births. (polito2024chargesyndromeand pages 1-2)
9.2 Inheritance and counseling-relevant points
- Autosomal dominant, variable expressivity; mostly de novo variants. (bergman2011chd7mutationsand pages 1-6, bergman2011chd7mutationsand pages 6-11)
- Mosaicism can occur; recurrence risk and prenatal testing options should be discussed. (mcj2006chargesyndromethe pages 1-2, bergman2011chd7mutationsand pages 24-29)
10. Diagnostics
10.1 Clinical criteria
Two widely used clinical criteria frameworks are Blake (1998) and Verloes (2005). A key Verloes contribution was emphasizing semicircular canal defects as a major criterion. (bergman2011chd7mutationsand pages 1-6, bergman2011chd7mutationsand pages 6-11)
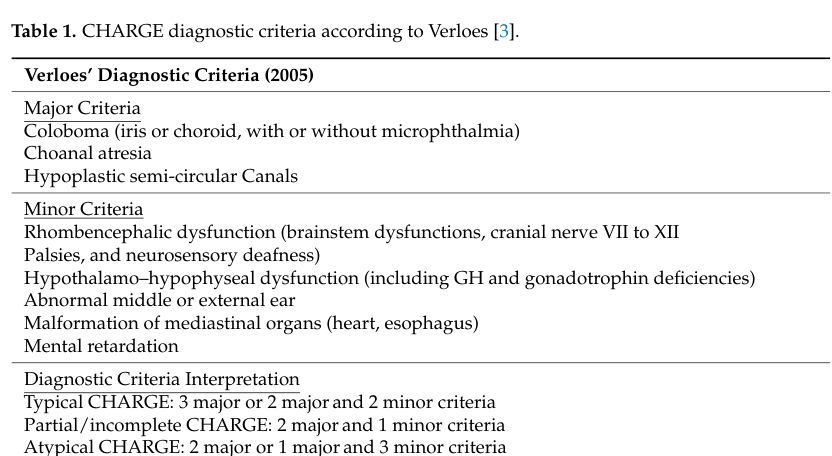
Verloes (2005) criteria (image evidence): Major criteria include coloboma, choanal atresia, and hypoplastic semicircular canals, with typical/partial/atypical categories defined by combinations of major/minor criteria. (driesen2024chd7disorder—notcharge media 9654fd32)
Text-form criteria are also reproduced in primary literature. (mcj2006chargesyndromethe pages 1-2, driesen2024chd7disorder—notcharge pages 8-9)
10.2 Genetic testing strategy
CHD7 testing is recommended broadly (not only those meeting strict criteria), because clinical criteria can miss mutation-positive individuals. (bergman2011chd7mutationsand pages 15-19)
Bergman et al. provide a pragmatic threshold for CHD7 testing (“3 cardinal or 2 cardinal + 1 supportive”) and emphasize semicircular canal imaging and cranial nerve evaluation in the diagnostic workup. (bergman2011chd7mutationsand pages 44-44)
10.3 Imaging and functional tests
- Temporal bone CT/MRI to detect semicircular canal abnormalities and nerve anatomy. (bergman2011chd7mutationsand pages 44-44, wieland2020chargesyndrome pages 10-11)
- Cardiac evaluation: standardized transthoracic echocardiography (TTE) first-line; CTA/cardiac MRI for complex extracardiac anatomy. (polito2024chargesyndromeand pages 7-8)
10.4 Differential diagnosis (examples)
Differential diagnoses in overlapping phenotypes include Kabuki syndrome and other craniofacial/multiple anomaly syndromes; genetic testing is emphasized as decisive when phenotypes overlap. (ouassifi2025chargesyndromein pages 1-4)
10.5 Emerging molecular diagnostics
Functional testing for splicing VUS: Minigene assays plus patient RNA/cDNA validation can resolve intronic CHD7 splicing variants that are otherwise difficult to classify by in silico prediction alone. (rossi2023casereportfunctional pages 2-4, rossi2023casereportfunctional pages 5-6)
Episignatures: DNA methylation episignature studies are being operationalized in observational protocols involving CHD7. (NCT06475651 chunk 2)
11. Outcomes / Prognosis
11.1 Cardiac burden and mortality (recent quantitative evidence)
A 2024 systematic review (68 studies; n=943 reported CHARGE patients) found a 76.6% prevalence of congenital heart defects, with common lesions including PDA (26%), VSD (21%), ASD (18%), TOF (11%), and aortic abnormalities (24%). Cardiac surgery was performed in 62% of reported patients (150/242), and in-hospital mortality in the literature was ~9.5% in case series (and ~12% in case reports). (polito2024chargesyndromeand pages 1-2, polito2024chargesyndromeand pages 2-3)
Aspiration related to feeding problems was a major non-cardiovascular cause of death (“aspiration of secretions due to feeding problems was the most common cause of non-CV death in about 50%”). (polito2024chargesyndromeand pages 7-8)
11.2 Neurodevelopment and QoL
Cognitive outcomes are variable and can be confounded by dual sensory impairment. Prognostic indicators for worse cognitive outcomes include extensive colobomas and brain malformations, but improvement over time is possible with support. (wieland2020chargesyndrome pages 7-8)
Family QoL (29 genetically confirmed children) was described as intermediate/average with parental acceptance associated with higher QoL scores. (wolanska2025analysisofthe pages 76-79)
12. Treatment
There is no disease-modifying therapy for CHARGE; management is multidisciplinary and targeted to organ system complications.
12.1 Multidisciplinary care (real-world implementation)
CHARGE care is repeatedly emphasized as best delivered through specialized multidisciplinary teams, including genetics, ENT/audiology, ophthalmology, cardiology, endocrinology, speech/OT/PT, and others. (wieland2020chargesyndrome pages 6-7, bergman2011chd7mutationsand pages 19-21)
12.2 Cardiac treatment
Corrective cardiac surgery is frequently required; risk is amplified by noncardiac issues (airway/feeding/aspiration), and perioperative management should prioritize aspiration prevention. (meisner2020congenitalheartdefects pages 5-6, polito2024chargesyndromeand pages 7-8)
12.3 Airway and feeding support
Feeding difficulties are common and can require nasogastric feeding and/or gastrostomy; reflux management and dysphagia clinic referral are recommended. (wieland2020chargesyndrome pages 6-7)
Choanal atresia requires acute airway management and surgical repair; endoscopic transnasal approaches and stenting are commonly used, with higher reoperation rates reported in CHARGE. (wieland2020chargesyndrome pages 8-10)
12.4 Hearing interventions
Audiologic evaluation at diagnosis (including ABR and imaging) and ongoing follow-up is recommended. Cochlear implantation can improve outcomes but requires careful assessment due to temporal bone and nerve anomalies; ABI may be considered when cochlear nerve aplasia limits benefit. (wieland2020chargesyndrome pages 10-11)
12.5 Vision and developmental therapies
Early ophthalmology assessment and management (amblyopia screening, low-vision aids, strabismus treatment) plus early developmental therapies (speech/language, OT/PT) are emphasized to maximize function. (wieland2020chargesyndrome pages 7-8, wieland2020chargesyndrome pages 6-7)
12.6 MAXO suggestions (examples for KB annotation)
- Cardiac surgical repair — MAXO term for congenital heart defect surgery
- Choanal atresia repair — MAXO term for nasal/airway reconstructive surgery
- Gastrostomy tube placement — MAXO term for enteral feeding support
- Fundoplication — MAXO term for anti-reflux surgery
- Cochlear implantation — MAXO term for cochlear implant procedure
- Hearing aid fitting — MAXO term for amplification device use
- Speech therapy / occupational therapy / physical therapy — MAXO therapy terms
13. Prevention
Primary prevention is generally not applicable because CHARGE is primarily genetic and typically de novo. Prevention focuses on: * Genetic counseling (recurrence risk with mosaicism; options for prenatal diagnosis/PGD). (bergman2011chd7mutationsand pages 24-29) * Secondary/tertiary prevention: early detection and management of airway/feeding/cardiac issues to reduce morbidity and early mortality, especially aspiration prevention. (meisner2020congenitalheartdefects pages 5-6, polito2024chargesyndromeand pages 7-8)
14. Other Species / Natural Disease
No naturally occurring veterinary CHARGE syndrome cases were identified in the retrieved sources. The comparative biology evidence base in this report therefore relies on experimental models.
15. Model Organisms and Experimental Models
15.1 Mouse models
- p53 activation model: p53 hyperactivation induces CHARGE-like developmental defects; CHD7 negatively regulates p53, and p53 reduction partially rescues phenotypes in Chd7-null mice. (nostrand2014inappropriatep53activation pages 1-2)
- Cerebellar foliation model: Chd7 haploinsufficient mice exhibit mild cerebellar hypoplasia and foliation anomalies with incomplete penetrance (e.g., ~67% for specific patterns) and suggest modifier genes/pathways (Engrailed/FGF/Zic). (whittaker2017distinctcerebellarfoliation pages 3-6, whittaker2017distinctcerebellarfoliation pages 9-10)
15.2 Zebrafish models
Multi-omics datasets from larval zebrafish head tissue in a CHARGE model were integrated to identify candidate downstream effectors; functional CRISPR knockdown of candidate genes phenocopied behavioral defects. (hancock2026multiomicanalysesidentify pages 1-3, hancock2026multiomicanalysesidentify pages 11-13)
15.3 Human iPSC / organoid models
- Patient iPSC-derived neural crest cells demonstrate defective migration. (okuno2017chargesyndromemodeling pages 1-2)
- Human inner ear organoids show CHD7 dependence of hair cell/supporting cell differentiation and disruption of deafness gene expression in mutants. (nie2022chd7regulatesotic pages 1-2)
2023–2024 Highlights (recent developments prioritized)
- “CHD7 disorder” spectrum framing (2024): Adoption of the term “CHD7 disorder” to capture presentations that do not meet classic CHARGE criteria, including isolated cochleovestibular dysfunction; emphasizes CHD7 testing in nonsyndromic hearing loss with inner ear malformations. (driesen2024chd7disorder—notcharge pages 1-2, driesen2024chd7disorder—notcharge pages 8-9)
- Systematic review of congenital heart disease in CHARGE (2024): Quantifies CHD lesion spectrum, surgery rates, and in-hospital mortality; highlights aspiration from feeding problems as a major cause of death. (polito2024chargesyndromeand pages 1-2, polito2024chargesyndromeand pages 7-8)
- Digenic/oligogenic inheritance hypothesis (2024): CHD7+SMCHD1 co-inheritance proposed to underlie intrafamilial variability (hypogonadotropic hypogonadism / CHARGE-overlap). (wang2024digenicchd7and pages 1-2)
- Functional resolution of intronic splicing VUS (2023): Minigene + patient cDNA assays demonstrate a CHD7 intronic variant causes aberrant splicing, illustrating a path to improve molecular diagnostic yield. (rossi2023casereportfunctional pages 2-4, rossi2023casereportfunctional pages 1-2)
Key URLs (from retrieved sources)
- Driesen et al., Genes (Published 2024-05-19): https://doi.org/10.3390/genes15050643 (driesen2024chd7disorder—notcharge pages 1-2)
- Polito et al., Monaldi Archives for Chest Disease (Published 2024-09): https://doi.org/10.4081/monaldi.2023.2661 (polito2024chargesyndromeand pages 1-2)
- Wang et al., Heliyon (Published 2024-01): https://doi.org/10.1016/j.heliyon.2023.e23272 (wang2024digenicchd7and pages 1-2)
- Rossi et al., Frontiers in Genetics (Published 2023-02): https://doi.org/10.3389/fgene.2023.1082100 (rossi2023casereportfunctional pages 1-2)
- Nie et al., Nature Communications (Published 2022-11): https://doi.org/10.1038/s41467-022-34759-8 (nie2022chd7regulatesotic pages 1-2)
- Okuno et al., eLife (Published 2017-11): https://doi.org/10.7554/eLife.21114 (okuno2017chargesyndromemodeling pages 1-2)
Limitations of this tool-based synthesis
- ICD-10/ICD-11, MeSH, and MONDO identifiers were not explicitly present in the retrieved full-text set; therefore they are not asserted here.
- Some epidemiologic values are ascertainment-sensitive (clinical criteria vs molecular confirmation; regional differences) and thus reported as ranges with source-specific values.
References
-
(bergman2011chd7mutationsand pages 1-6): J. E. H. Bergman, N. Janssen, L. H. Hoefsloot, M. C. J. Jongmans, R. M. W. Hofstra, and C. M. A. van Ravenswaaij-Arts. Chd7 mutations and charge syndrome: the clinical implications of an expanding phenotype. Journal of Medical Genetics, 48:334-342, Mar 2011. URL: https://doi.org/10.1136/jmg.2010.087106, doi:10.1136/jmg.2010.087106. This article has 379 citations and is from a domain leading peer-reviewed journal.
-
(mcj2006chargesyndromethe pages 1-2): Jongmans Mcj, RJ Admiraal, KP vanderDonk, Vissers Lelm, AF Baas, L. Kapusta, JM vanHagen, D. Donnai, TJ deRavel, J. Veltman, A. GeurtsvanKessel, De Vries, Bba, H. Brunner, L. Hoefsloot, and van Ravenswaaij Cma. Charge syndrome: the phenotypic spectrum of mutations in the chd7 gene. Journal of Medical Genetics, 43:306-314, Sep 2006. URL: https://doi.org/10.1136/jmg.2005.036061, doi:10.1136/jmg.2005.036061. This article has 531 citations and is from a domain leading peer-reviewed journal.
-
(wolanska2025analysisofthe pages 7-10): E Wolańska. Analysis of the genotype and phenotype of children with charge syndrome and assessment of their families' quality of life. Unknown journal, 2025.
-
(polito2024chargesyndromeand pages 1-2): Maria Vincenza Polito, Mario Ferraioli, Alessandra Nocilla, Guido Coppola, Federica D'Auria, Antonio Marzano, Luca Barnabei, Marisa Malinconico, Eduardo Bossone, and Francesco Ferrara. Charge syndrome and congenital heart diseases: systematic review of literature. Monaldi archives for chest disease = Archivio Monaldi per le malattie del torace, Sep 2024. URL: https://doi.org/10.4081/monaldi.2023.2661, doi:10.4081/monaldi.2023.2661. This article has 10 citations.
-
(polito2024chargesyndromeand pages 2-3): Maria Vincenza Polito, Mario Ferraioli, Alessandra Nocilla, Guido Coppola, Federica D'Auria, Antonio Marzano, Luca Barnabei, Marisa Malinconico, Eduardo Bossone, and Francesco Ferrara. Charge syndrome and congenital heart diseases: systematic review of literature. Monaldi archives for chest disease = Archivio Monaldi per le malattie del torace, Sep 2024. URL: https://doi.org/10.4081/monaldi.2023.2661, doi:10.4081/monaldi.2023.2661. This article has 10 citations.
-
(bergman2011chd7mutationsand pages 6-11): J. E. H. Bergman, N. Janssen, L. H. Hoefsloot, M. C. J. Jongmans, R. M. W. Hofstra, and C. M. A. van Ravenswaaij-Arts. Chd7 mutations and charge syndrome: the clinical implications of an expanding phenotype. Journal of Medical Genetics, 48:334-342, Mar 2011. URL: https://doi.org/10.1136/jmg.2010.087106, doi:10.1136/jmg.2010.087106. This article has 379 citations and is from a domain leading peer-reviewed journal.
-
(bergman2011chd7mutationsand pages 24-29): J. E. H. Bergman, N. Janssen, L. H. Hoefsloot, M. C. J. Jongmans, R. M. W. Hofstra, and C. M. A. van Ravenswaaij-Arts. Chd7 mutations and charge syndrome: the clinical implications of an expanding phenotype. Journal of Medical Genetics, 48:334-342, Mar 2011. URL: https://doi.org/10.1136/jmg.2010.087106, doi:10.1136/jmg.2010.087106. This article has 379 citations and is from a domain leading peer-reviewed journal.
-
(wang2024digenicchd7and pages 1-2): Tian Wang, Wu Ren, Fangfang Fu, Hairong Wang, Yan Li, and Jie Duan. Digenic chd7 and smchd1 inheritance unveils phenotypic variability in a family mainly presenting with hypogonadotropic hypogonadism. Heliyon, 10:e23272, Jan 2024. URL: https://doi.org/10.1016/j.heliyon.2023.e23272, doi:10.1016/j.heliyon.2023.e23272. This article has 4 citations.
-
(wang2024digenicchd7and pages 2-4): Tian Wang, Wu Ren, Fangfang Fu, Hairong Wang, Yan Li, and Jie Duan. Digenic chd7 and smchd1 inheritance unveils phenotypic variability in a family mainly presenting with hypogonadotropic hypogonadism. Heliyon, 10:e23272, Jan 2024. URL: https://doi.org/10.1016/j.heliyon.2023.e23272, doi:10.1016/j.heliyon.2023.e23272. This article has 4 citations.
-
(wieland2020chargesyndrome pages 1-3): J Wieland, D Wieland, T Estiphan, and B Weakley. Charge syndrome. Encyclopedia of Autism Spectrum Disorders, Feb 2020. URL: https://doi.org/10.1007/978-1-4614-6430-3_38-2, doi:10.1007/978-1-4614-6430-3_38-2. This article has 140 citations.
-
(wolanska2025analysisofthe pages 76-79): E Wolańska. Analysis of the genotype and phenotype of children with charge syndrome and assessment of their families' quality of life. Unknown journal, 2025.
-
(wieland2020chargesyndrome pages 10-11): J Wieland, D Wieland, T Estiphan, and B Weakley. Charge syndrome. Encyclopedia of Autism Spectrum Disorders, Feb 2020. URL: https://doi.org/10.1007/978-1-4614-6430-3_38-2, doi:10.1007/978-1-4614-6430-3_38-2. This article has 140 citations.
-
(driesen2024chd7disorder—notcharge pages 1-2): Jef Driesen, Helen Van Hoecke, Leen Maes, Sandra Janssens, Frederic Acke, and Els De Leenheer. Chd7 disorder—not charge syndrome—presenting as isolated cochleovestibular dysfunction. Genes, 15:643, May 2024. URL: https://doi.org/10.3390/genes15050643, doi:10.3390/genes15050643. This article has 3 citations.
-
(rossi2023casereportfunctional pages 1-2): Cesare Rossi, Sherin Ramadan, Cecilia Evangelisti, Simona Ferrari, Maria Accadia, Reha M. Toydemir, and Emanuele Panza. Case report: functional characterization of a novel chd7 intronic variant in patients with charge syndrome. Frontiers in Genetics, Feb 2023. URL: https://doi.org/10.3389/fgene.2023.1082100, doi:10.3389/fgene.2023.1082100. This article has 5 citations and is from a peer-reviewed journal.
-
(rossi2023casereportfunctional pages 2-4): Cesare Rossi, Sherin Ramadan, Cecilia Evangelisti, Simona Ferrari, Maria Accadia, Reha M. Toydemir, and Emanuele Panza. Case report: functional characterization of a novel chd7 intronic variant in patients with charge syndrome. Frontiers in Genetics, Feb 2023. URL: https://doi.org/10.3389/fgene.2023.1082100, doi:10.3389/fgene.2023.1082100. This article has 5 citations and is from a peer-reviewed journal.
-
(whittaker2017distinctcerebellarfoliation pages 9-10): Danielle E. Whittaker, Sahrunizam Kasah, Alex P. A. Donovan, Jacob Ellegood, Kimberley L. H. Riegman, Holger A. Volk, Imelda McGonnell, Jason P. Lerch, and M. Albert Basson. Distinct cerebellar foliation anomalies in a chd7 haploinsufficient mouse model of charge syndrome. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 175:n/a-n/a, Nov 2017. URL: https://doi.org/10.1002/ajmg.c.31595, doi:10.1002/ajmg.c.31595. This article has 34 citations.
-
(NCT06475651 chunk 2): Characterization and Contribution of Genome-wide DNA Methylation (DNA Methylation Episignatures) in Rare Diseases With Prenatal Onset. Assistance Publique - Hôpitaux de Paris. 2026. ClinicalTrials.gov Identifier: NCT06475651
-
(okuno2017chargesyndromemodeling pages 1-2): Hironobu Okuno, Francois Renault Mihara, Shigeki Ohta, Kimiko Fukuda, Kenji Kurosawa, Wado Akamatsu, Tsukasa Sanosaka, Jun Kohyama, Kanehiro Hayashi, Kazunori Nakajima, Takao Takahashi, Joanna Wysocka, Kenjiro Kosaki, and Hideyuki Okano. Charge syndrome modeling using patient-ipscs reveals defective migration of neural crest cells harboring chd7 mutations. eLife, Nov 2017. URL: https://doi.org/10.7554/elife.21114, doi:10.7554/elife.21114. This article has 74 citations and is from a domain leading peer-reviewed journal.
-
(okuno2017chargesyndromemodeling pages 5-6): Hironobu Okuno, Francois Renault Mihara, Shigeki Ohta, Kimiko Fukuda, Kenji Kurosawa, Wado Akamatsu, Tsukasa Sanosaka, Jun Kohyama, Kanehiro Hayashi, Kazunori Nakajima, Takao Takahashi, Joanna Wysocka, Kenjiro Kosaki, and Hideyuki Okano. Charge syndrome modeling using patient-ipscs reveals defective migration of neural crest cells harboring chd7 mutations. eLife, Nov 2017. URL: https://doi.org/10.7554/elife.21114, doi:10.7554/elife.21114. This article has 74 citations and is from a domain leading peer-reviewed journal.
-
(sanosaka2022chromatinremodelerchd7 pages 2-3): Tsukasa Sanosaka, Hironobu Okuno, Noriko Mizota, Tomoko Andoh-Noda, Miki Sato, Ryo Tomooka, Satoe Banno, Jun Kohyama, and Hideyuki Okano. Chromatin remodeler chd7 targets active enhancer region to regulate cell type-specific gene expression in human neural crest cells. Scientific Reports, Dec 2022. URL: https://doi.org/10.1038/s41598-022-27293-6, doi:10.1038/s41598-022-27293-6. This article has 16 citations and is from a peer-reviewed journal.
-
(sanosaka2022chromatinremodelerchd7 pages 1-2): Tsukasa Sanosaka, Hironobu Okuno, Noriko Mizota, Tomoko Andoh-Noda, Miki Sato, Ryo Tomooka, Satoe Banno, Jun Kohyama, and Hideyuki Okano. Chromatin remodeler chd7 targets active enhancer region to regulate cell type-specific gene expression in human neural crest cells. Scientific Reports, Dec 2022. URL: https://doi.org/10.1038/s41598-022-27293-6, doi:10.1038/s41598-022-27293-6. This article has 16 citations and is from a peer-reviewed journal.
-
(nie2022chd7regulatesotic pages 1-2): Jing Nie, Yoshitomo Ueda, Alexander J. Solivais, and Eri Hashino. Chd7 regulates otic lineage specification and hair cell differentiation in human inner ear organoids. Nature Communications, Nov 2022. URL: https://doi.org/10.1038/s41467-022-34759-8, doi:10.1038/s41467-022-34759-8. This article has 43 citations and is from a highest quality peer-reviewed journal.
-
(nostrand2014inappropriatep53activation pages 1-2): Jeanine L. Van Nostrand, Colleen A. Brady, Heiyoun Jung, Daniel R. Fuentes, Margaret M. Kozak, Thomas M. Johnson, Chieh-Yu Lin, Chien-Jung Lin, Donald L. Swiderski, Hannes Vogel, Jonathan A. Bernstein, Tania Attié-Bitach, Ching-Pin Chang, Joanna Wysocka, Donna M. Martin, and Laura D. Attardi. Inappropriate p53 activation during development induces features of charge syndrome. Aug 2014. URL: https://doi.org/10.1038/nature13585, doi:10.1038/nature13585. This article has 171 citations and is from a highest quality peer-reviewed journal.
-
(whittaker2017distinctcerebellarfoliation pages 3-6): Danielle E. Whittaker, Sahrunizam Kasah, Alex P. A. Donovan, Jacob Ellegood, Kimberley L. H. Riegman, Holger A. Volk, Imelda McGonnell, Jason P. Lerch, and M. Albert Basson. Distinct cerebellar foliation anomalies in a chd7 haploinsufficient mouse model of charge syndrome. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 175:n/a-n/a, Nov 2017. URL: https://doi.org/10.1002/ajmg.c.31595, doi:10.1002/ajmg.c.31595. This article has 34 citations.
-
(hancock2026multiomicanalysesidentify pages 1-3): Melody B. Hancock, Dana R. Ruby, Rachael A. Bieler, D. Chris Cole, and Kurt C. Marsden. Multi-omic analyses identify molecular targets of chd7 that contribute to charge syndrome model phenotypes. Disease Models & Mechanisms, Mar 2026. URL: https://doi.org/10.1242/dmm.052592, doi:10.1242/dmm.052592. This article has 1 citations and is from a domain leading peer-reviewed journal.
-
(hancock2026multiomicanalysesidentify pages 11-13): Melody B. Hancock, Dana R. Ruby, Rachael A. Bieler, D. Chris Cole, and Kurt C. Marsden. Multi-omic analyses identify molecular targets of chd7 that contribute to charge syndrome model phenotypes. Disease Models & Mechanisms, Mar 2026. URL: https://doi.org/10.1242/dmm.052592, doi:10.1242/dmm.052592. This article has 1 citations and is from a domain leading peer-reviewed journal.
-
(whittaker2017distinctcerebellarfoliation pages 1-2): Danielle E. Whittaker, Sahrunizam Kasah, Alex P. A. Donovan, Jacob Ellegood, Kimberley L. H. Riegman, Holger A. Volk, Imelda McGonnell, Jason P. Lerch, and M. Albert Basson. Distinct cerebellar foliation anomalies in a chd7 haploinsufficient mouse model of charge syndrome. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 175:n/a-n/a, Nov 2017. URL: https://doi.org/10.1002/ajmg.c.31595, doi:10.1002/ajmg.c.31595. This article has 34 citations.
-
(webb2021aframeworkfor pages 8-10): Bryn D. Webb, Irini Manoli, Elizabeth C. Engle, and Ethylin W. Jabs. A framework for the evaluation of patients with congenital facial weakness. Orphanet Journal of Rare Diseases, Apr 2021. URL: https://doi.org/10.1186/s13023-021-01736-1, doi:10.1186/s13023-021-01736-1. This article has 14 citations and is from a peer-reviewed journal.
-
(driesen2024chd7disorder—notcharge pages 8-9): Jef Driesen, Helen Van Hoecke, Leen Maes, Sandra Janssens, Frederic Acke, and Els De Leenheer. Chd7 disorder—not charge syndrome—presenting as isolated cochleovestibular dysfunction. Genes, 15:643, May 2024. URL: https://doi.org/10.3390/genes15050643, doi:10.3390/genes15050643. This article has 3 citations.
-
(polito2024chargesyndromeand pages 7-8): Maria Vincenza Polito, Mario Ferraioli, Alessandra Nocilla, Guido Coppola, Federica D'Auria, Antonio Marzano, Luca Barnabei, Marisa Malinconico, Eduardo Bossone, and Francesco Ferrara. Charge syndrome and congenital heart diseases: systematic review of literature. Monaldi archives for chest disease = Archivio Monaldi per le malattie del torace, Sep 2024. URL: https://doi.org/10.4081/monaldi.2023.2661, doi:10.4081/monaldi.2023.2661. This article has 10 citations.
-
(meisner2020congenitalheartdefects pages 5-6): Joshua K. Meisner and Donna M. Martin. Congenital heart defects in charge: the molecular role of chd7 and effects on cardiac phenotype and clinical outcomes. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 184:81-89, Dec 2020. URL: https://doi.org/10.1002/ajmg.c.31761, doi:10.1002/ajmg.c.31761. This article has 47 citations.
-
(driesen2024chd7disorder—notcharge media 9654fd32): Jef Driesen, Helen Van Hoecke, Leen Maes, Sandra Janssens, Frederic Acke, and Els De Leenheer. Chd7 disorder—not charge syndrome—presenting as isolated cochleovestibular dysfunction. Genes, 15:643, May 2024. URL: https://doi.org/10.3390/genes15050643, doi:10.3390/genes15050643. This article has 3 citations.
-
(bergman2011chd7mutationsand pages 15-19): J. E. H. Bergman, N. Janssen, L. H. Hoefsloot, M. C. J. Jongmans, R. M. W. Hofstra, and C. M. A. van Ravenswaaij-Arts. Chd7 mutations and charge syndrome: the clinical implications of an expanding phenotype. Journal of Medical Genetics, 48:334-342, Mar 2011. URL: https://doi.org/10.1136/jmg.2010.087106, doi:10.1136/jmg.2010.087106. This article has 379 citations and is from a domain leading peer-reviewed journal.
-
(bergman2011chd7mutationsand pages 44-44): J. E. H. Bergman, N. Janssen, L. H. Hoefsloot, M. C. J. Jongmans, R. M. W. Hofstra, and C. M. A. van Ravenswaaij-Arts. Chd7 mutations and charge syndrome: the clinical implications of an expanding phenotype. Journal of Medical Genetics, 48:334-342, Mar 2011. URL: https://doi.org/10.1136/jmg.2010.087106, doi:10.1136/jmg.2010.087106. This article has 379 citations and is from a domain leading peer-reviewed journal.
-
(ouassifi2025chargesyndromein pages 1-4): Kawtar El Ouassifi, Anas Douami, Hind Ouair, Ibtihal Benhsaien, Aziz Bousfiha, Jalila El Bakkouri, Siham Salam, and Fatima Ailal. Charge syndrome in a six-month-old male infant: a case report. Cureus, May 2025. URL: https://doi.org/10.7759/cureus.84181, doi:10.7759/cureus.84181. This article has 2 citations.
-
(rossi2023casereportfunctional pages 5-6): Cesare Rossi, Sherin Ramadan, Cecilia Evangelisti, Simona Ferrari, Maria Accadia, Reha M. Toydemir, and Emanuele Panza. Case report: functional characterization of a novel chd7 intronic variant in patients with charge syndrome. Frontiers in Genetics, Feb 2023. URL: https://doi.org/10.3389/fgene.2023.1082100, doi:10.3389/fgene.2023.1082100. This article has 5 citations and is from a peer-reviewed journal.
-
(wieland2020chargesyndrome pages 7-8): J Wieland, D Wieland, T Estiphan, and B Weakley. Charge syndrome. Encyclopedia of Autism Spectrum Disorders, Feb 2020. URL: https://doi.org/10.1007/978-1-4614-6430-3_38-2, doi:10.1007/978-1-4614-6430-3_38-2. This article has 140 citations.
-
(wieland2020chargesyndrome pages 6-7): J Wieland, D Wieland, T Estiphan, and B Weakley. Charge syndrome. Encyclopedia of Autism Spectrum Disorders, Feb 2020. URL: https://doi.org/10.1007/978-1-4614-6430-3_38-2, doi:10.1007/978-1-4614-6430-3_38-2. This article has 140 citations.
-
(bergman2011chd7mutationsand pages 19-21): J. E. H. Bergman, N. Janssen, L. H. Hoefsloot, M. C. J. Jongmans, R. M. W. Hofstra, and C. M. A. van Ravenswaaij-Arts. Chd7 mutations and charge syndrome: the clinical implications of an expanding phenotype. Journal of Medical Genetics, 48:334-342, Mar 2011. URL: https://doi.org/10.1136/jmg.2010.087106, doi:10.1136/jmg.2010.087106. This article has 379 citations and is from a domain leading peer-reviewed journal.
-
(wieland2020chargesyndrome pages 8-10): J Wieland, D Wieland, T Estiphan, and B Weakley. Charge syndrome. Encyclopedia of Autism Spectrum Disorders, Feb 2020. URL: https://doi.org/10.1007/978-1-4614-6430-3_38-2, doi:10.1007/978-1-4614-6430-3_38-2. This article has 140 citations.
Artifacts