Woodhouse-Sakati Syndrome
Woodhouse-Sakati syndrome (WSS) is a rare autosomal recessive multisystem neuroendocrine disorder caused by biallelic pathogenic variants in DCAF17, which encodes a substrate receptor of the CRL4 (cullin-4 RING) ubiquitin ligase complex. Virtually all affected individuals have hypogonadism and progressive alopecia; more than half develop a progressive extrapyramidal movement disorder, sensorineural hearing loss, and mild intellectual disability. Diabetes mellitus, hypothyroidism, dysarthria, and dysphagia are also recognized features. WSS is classified within the neurodegeneration with brain iron accumulation (NBIA) spectrum.
Ask OpenScientist
Ask a research question about Woodhouse-Sakati Syndrome. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (2 references)
Subtypes
2Show evidence (1 reference)
Show evidence (1 reference)
Pathophysiology
3Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Pathograph
Phenotypes
12Digestive 1
Show evidence (1 reference)
Ear 1
Show evidence (2 references)
Endocrine 3
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Genitourinary 1
Show evidence (2 references)
Integument 1
Show evidence (1 reference)
Nervous System 4
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (2 references)
Other 1
Show evidence (2 references)
Genetic Associations
1Show evidence (3 references)
Medical Actions
5Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Differential Diagnoses
1Conditions with similar clinical presentations that must be differentiated from Woodhouse-Sakati Syndrome:
- WSS combines a movement disorder with hypogonadism and progressive alopecia.
- WSS is caused by biallelic DCAF17 variants rather than other NBIA genes.
Show evidence (1 reference)
Clinical Trials
1Show evidence (1 reference)
Source YAML
click to showname: Woodhouse-Sakati Syndrome
creation_date: "2026-06-03T00:00:00Z"
description: >-
Woodhouse-Sakati syndrome (WSS) is a rare autosomal recessive multisystem
neuroendocrine disorder caused by biallelic pathogenic variants in DCAF17,
which encodes a substrate receptor of the CRL4 (cullin-4 RING) ubiquitin
ligase complex. Virtually all affected individuals have hypogonadism and
progressive alopecia; more than half develop a progressive extrapyramidal
movement disorder, sensorineural hearing loss, and mild intellectual
disability. Diabetes mellitus, hypothyroidism, dysarthria, and dysphagia are
also recognized features. WSS is classified within the neurodegeneration with
brain iron accumulation (NBIA) spectrum.
category: Mendelian
parents:
- hereditary disease
- neurodegeneration with brain iron accumulation
disease_term:
preferred_term: Woodhouse-Sakati syndrome
term:
id: MONDO:0009419
label: Woodhouse-Sakati syndrome
references:
- reference: PMID:27489925
title: "Woodhouse-Sakati Syndrome."
tags:
- GeneReviews
- reference: PMID:36721231
title: "Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: a systematic review."
- reference: PMID:31726291
title: "Patterns of neurological manifestations in Woodhouse-Sakati Syndrome."
- reference: PMID:35002959
title: "Case Report: A Chinese Family of Woodhouse-Sakati Syndrome With Diabetes Mellitus, With a Novel Biallelic Deletion Mutation of the DCAF17 Gene."
- reference: PMID:37895404
title: "The Successful Management of Primary Amenorrhea in Woodhouse-Sakati Syndrome: A Case Report and a Literature Review."
- reference: PMID:39342163
title: "Woodhouse-sakati syndrome with no reportable MRI findings: a case report."
- reference: PMID:38320940
title: "Woodhouse-Sakati syndrome: A review."
- reference: PMID:39239833
title: "Single-cell RNA sequencing reveals the important role of Dcaf17 in spermatogenesis of golden hamsters."
- reference: PMID:40235137
title: "Clinical and Genetic Characterization of Woodhouse-Sakati Syndrome in Iranian Patients: A Case Series."
inheritance:
- name: Autosomal recessive inheritance
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
description: >-
WSS is caused by biallelic pathogenic variants in DCAF17 and is inherited in
an autosomal recessive manner. The disease is especially prevalent in the
Greater Middle East, where consanguinity is common.
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
WSS is inherited in an autosomal recessive manner.
explanation: >-
The GeneReviews chapter directly states the autosomal recessive
inheritance pattern.
- reference: PMID:36721231
reference_title: "Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: a systematic review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Woodhouse-Sakati syndrome (WSS) is a rare, autosomal recessive genetic disorder with variable clinical manifestations mainly affecting the endocrine and nervous systems.
explanation: >-
This systematic review confirms autosomal recessive inheritance with
variable expressivity.
has_subtypes:
- name: Type 1
display_name: Type 1 (Rapidly Progressive Neurologic)
description: >-
Disabling, rapidly progressive neurological pattern (Neurological Impairment
Scale [NIS] 3-4) seen in roughly 47% of patients, with severe disability
within a mean of ~7.4 years and earlier onset of neurological manifestations
(mean 12.6 years). Type 1 has a significantly higher rate of intellectual
disability.
evidence:
- reference: PMID:31726291
reference_title: "Patterns of neurological manifestations in Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
A disabling, rapidly progressive pattern (NIS of 3-4; Type 1) was noted in eighteen patients (12 males, 6 females; 47.4%) with severe disability that occurs within a mean duration of 7.4 ± 3.6 years.
explanation: >-
Bohlega 2019 defines Type 1 as the rapidly progressive, disabling
neurological pattern.
- name: Type 2
display_name: Type 2 (Mild Neurologic)
description: >-
Absent or mild neurological involvement (Neurological Impairment Scale [NIS]
0-1) seen in roughly 53% of patients, with preserved activities of daily
living and later onset of neurological manifestations (mean 18.1 years).
evidence:
- reference: PMID:31726291
reference_title: "Patterns of neurological manifestations in Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Type 2 WSS was identified in twenty patients (8 males, 12 females; 52.6%), and showed either absent or mild neurological involvement with preserved activities of daily living (NIS of 0-1).
explanation: >-
Bohlega 2019 defines Type 2 as the mild or absent neurological
involvement pattern.
pathophysiology:
- name: DCAF17 substrate-receptor deficiency
description: >-
DCAF17 (DDB1- and CUL4-associated factor 17) is a substrate receptor of the
CRL4 (cullin-4 RING) ubiquitin E3 ligase complex. Biallelic loss-of-function
variants impair recognition and ubiquitination of target substrates,
disrupting ubiquitin-mediated proteostasis in endocrine, neural, and
ectodermal tissues.
genes:
- preferred_term: DCAF17
term:
id: hgnc:25784
label: DCAF17
biological_processes:
- preferred_term: protein ubiquitination
term:
id: GO:0016567
label: protein ubiquitination
modifier: DECREASED
- preferred_term: ubiquitin-dependent protein catabolic process
term:
id: GO:0006511
label: ubiquitin-dependent protein catabolic process
modifier: DECREASED
molecular_functions:
- preferred_term: ubiquitin protein ligase binding
term:
id: GO:0031625
label: ubiquitin protein ligase binding
modifier: ABNORMAL
evidence:
- reference: PMID:39239833
reference_title: "Single-cell RNA sequencing reveals the important role of Dcaf17 in spermatogenesis of golden hamsters."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Dcaf17, also known as DDB1- and CUL4-associated factor 17, is a member of the DCAF family and acts as the receptor for the CRL4 ubiquitin E3 ligase complex.
explanation: >-
This directly establishes DCAF17 as the substrate receptor of the CRL4
ubiquitin E3 ligase complex, the molecular basis of WSS.
downstream:
- target: Impaired tissue proteostasis
description: >-
Loss of DCAF17 substrate-receptor function impairs CRL4-mediated
ubiquitination, disrupting protein degradation in affected tissues.
causal_link_type: DIRECT
evidence:
- reference: PMID:39239833
reference_title: "Single-cell RNA sequencing reveals the important role of Dcaf17 in spermatogenesis of golden hamsters."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
These data emphasize the significant regulatory role played by Dcaf17 in early spermatogenic cells, with many biological processes being affected, including spermatogenesis and protein degradation.
explanation: >-
Dcaf17 deficiency in a hamster model disrupts protein degradation,
supporting impaired ubiquitin-mediated proteostasis as a downstream
consequence.
- name: Impaired tissue proteostasis
description: >-
Disrupted CRL4-DCAF17 ubiquitin-mediated protein degradation leads to
pleiotropic dysfunction across endocrine (hypogonadal axis, pancreatic,
thyroid), neural (extrapyramidal, auditory, cognitive), and ectodermal
(hair follicle) tissues, producing the multisystem WSS phenotype.
biological_processes:
- preferred_term: proteasome-mediated ubiquitin-dependent protein catabolic process
term:
id: GO:0043161
label: proteasome-mediated ubiquitin-dependent protein catabolic process
modifier: ABNORMAL
cell_types:
- preferred_term: pancreatic beta cell
term:
id: CL:0000169
label: type B pancreatic cell
evidence:
- reference: PMID:36721231
reference_title: "Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: a systematic review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Woodhouse-Sakati syndrome (WSS) is a rare, autosomal recessive genetic disorder with variable clinical manifestations mainly affecting the endocrine and nervous systems.
explanation: >-
The multisystem endocrine and neurological involvement is the clinical
manifestation of impaired DCAF17-dependent proteostasis.
- reference: PMID:35002959
reference_title: "Case Report: A Chinese Family of Woodhouse-Sakati Syndrome With Diabetes Mellitus, With a Novel Biallelic Deletion Mutation of the DCAF17 Gene."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The main features of WSS include diabetes, hypogonadism, alopecia, deafness, intellectual disability and progressive extrapyramidal syndrome.
explanation: >-
The multisystem phenotype reflects DCAF17 dysfunction across endocrine,
ectodermal, auditory, and neural tissues.
downstream:
- target: Hypogonadism
description: Endocrine dysfunction of the hypothalamic-pituitary-gonadal axis.
causal_link_type: DIRECT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Virtually all individuals with Woodhouse-Sakati syndrome (WSS) have the endocrine findings of hypogonadism (evident at puberty)
explanation: >-
Hypogonadism is a near-universal endocrine manifestation of the
DCAF17-driven multisystem disease.
- target: Alopecia
description: Ectodermal (hair follicle) involvement causing progressive hair loss.
causal_link_type: DIRECT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
progressive childhood-onset hair thinning that often progresses to alopecia totalis in adulthood
explanation: >-
Progressive alopecia is the near-universal ectodermal manifestation of
the DCAF17-driven multisystem disease.
- target: Progressive extrapyramidal movement disorder
description: Neural involvement causing progressive dystonia and movement abnormalities.
causal_link_type: DIRECT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
More than half of individuals have the neurologic findings of progressive extrapyramidal movements (dystonic spasms with dystonic posturing with dysarthria and dysphagia)
explanation: >-
Neural involvement produces the progressive extrapyramidal movement
disorder seen in more than half of affected individuals.
- target: Dystonia
description: Dystonic spasms and posturing are the main movement-disorder manifestation.
causal_link_type: DIRECT
- target: Intellectual disability
description: Neural involvement in WSS commonly includes mild intellectual disability.
causal_link_type: DIRECT
- target: Sensorineural hearing impairment
description: Auditory involvement causing postlingual sensorineural hearing loss.
causal_link_type: DIRECT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
moderate bilateral postlingual sensorineural hearing loss
explanation: >-
Auditory involvement produces moderate bilateral postlingual
sensorineural hearing loss.
- target: Hypothyroidism
description: Endocrine tissue involvement can manifest as hypothyroidism.
causal_link_type: DIRECT
- target: Dysarthria
description: Extrapyramidal neurologic involvement can impair speech articulation.
causal_link_type: DIRECT
- target: Dysphagia
description: Extrapyramidal neurologic involvement can impair swallowing.
causal_link_type: DIRECT
- target: Abnormal cerebral white matter morphology
description: Neurologic involvement in WSS includes variable neuroradiologic white-matter abnormalities.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Male infertility
description: Gonadal-axis and spermatogenic involvement can produce male infertility.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Pancreatic beta cell dysfunction
description: >-
Endocrine pancreatic involvement with impaired insulin secretion driving
diabetes mellitus.
causal_link_type: DIRECT
evidence:
- reference: PMID:35002959
reference_title: "Case Report: A Chinese Family of Woodhouse-Sakati Syndrome With Diabetes Mellitus, With a Novel Biallelic Deletion Mutation of the DCAF17 Gene."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
These findings indicate that the DCAF17 gene mutation may cause pancreatic β cell functional impairment and contribute to the development of diabetes.
explanation: >-
DCAF17 dysfunction impairs pancreatic beta-cell function, linking the
proteostasis defect to diabetes mellitus.
- name: Pancreatic beta cell dysfunction
description: >-
DCAF17 deficiency is associated with impaired pancreatic beta-cell function.
Affected individuals show reduced insulin and C-peptide release after glucose
stimulation, implicating beta-cell secretory failure in the diabetes mellitus
of WSS.
cell_types:
- preferred_term: pancreatic beta cell
term:
id: CL:0000169
label: type B pancreatic cell
biological_processes:
- preferred_term: insulin secretion
term:
id: GO:0030073
label: insulin secretion
modifier: DECREASED
evidence:
- reference: PMID:35002959
reference_title: "Case Report: A Chinese Family of Woodhouse-Sakati Syndrome With Diabetes Mellitus, With a Novel Biallelic Deletion Mutation of the DCAF17 Gene."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We found that the two patients exhibited low insulin and C-peptide release after glucose stimulation by insulin and C-peptide release tests.
explanation: >-
Reduced insulin and C-peptide responses to glucose demonstrate impaired
beta-cell secretory function in WSS patients.
downstream:
- target: Diabetes mellitus
description: >-
Beta-cell secretory failure produces insulin deficiency and diabetes
mellitus.
causal_link_type: DIRECT
evidence:
- reference: PMID:35002959
reference_title: "Case Report: A Chinese Family of Woodhouse-Sakati Syndrome With Diabetes Mellitus, With a Novel Biallelic Deletion Mutation of the DCAF17 Gene."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
These findings indicate that the DCAF17 gene mutation may cause pancreatic β cell functional impairment and contribute to the development of diabetes.
explanation: >-
The authors directly link DCAF17-related beta-cell impairment to the
development of diabetes mellitus.
phenotypes:
- name: Hypogonadism
category: Endocrine
description: >-
Hypogonadism, evident at puberty, is a near-universal feature of WSS and
affects both sexes. It often presents as hypogonadotropic hypogonadism with
delayed puberty, primary amenorrhea, and infertility.
phenotype_term:
preferred_term: Hypogonadotropic hypogonadism
term:
id: HP:0000044
label: Hypogonadotropic hypogonadism
frequency: VERY_FREQUENT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Virtually all individuals with Woodhouse-Sakati syndrome (WSS) have the endocrine findings of hypogonadism (evident at puberty)
explanation: >-
GeneReviews documents hypogonadism in virtually all individuals,
supporting VERY_FREQUENT.
- name: Alopecia
category: Integumentary
description: >-
Progressive childhood-onset hair thinning that often progresses to alopecia
totalis in adulthood is a near-universal ectodermal feature of WSS.
phenotype_term:
preferred_term: Alopecia
term:
id: HP:0001596
label: Alopecia
clinical_course: PROGRESSIVE
frequency: VERY_FREQUENT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Virtually all individuals with Woodhouse-Sakati syndrome (WSS) have the endocrine findings of hypogonadism (evident at puberty) and progressive childhood-onset hair thinning that often progresses to alopecia totalis in adulthood
explanation: >-
GeneReviews documents progressive alopecia as a near-universal feature,
affecting virtually all individuals, supporting VERY_FREQUENT.
- name: Progressive extrapyramidal movement disorder
category: Neurologic
description: >-
More than half of individuals develop progressive extrapyramidal movements,
including dystonic spasms with dystonic posturing, dysarthria, and
dysphagia.
phenotype_term:
preferred_term: Progressive extrapyramidal movement disorder
term:
id: HP:0007153
label: Progressive extrapyramidal movement disorder
frequency: FREQUENT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
More than half of individuals have the neurologic findings of progressive extrapyramidal movements (dystonic spasms with dystonic posturing with dysarthria and dysphagia)
explanation: >-
GeneReviews documents progressive extrapyramidal movements in more than
half of affected individuals, supporting FREQUENT.
- reference: PMID:31726291
reference_title: "Patterns of neurological manifestations in Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Neurological involvement was noted in 31 patients (81.5%).
explanation: >-
In a cohort of 38 genetically confirmed patients, neurological involvement
was present in 81.5%, supporting FREQUENT.
- name: Dystonia
category: Neurologic
description: >-
Dystonic spasms with dystonic posturing are the predominant movement-disorder
manifestation of the extrapyramidal syndrome in WSS.
phenotype_term:
preferred_term: Dystonia
term:
id: HP:0001332
label: Dystonia
frequency: FREQUENT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
progressive extrapyramidal movements (dystonic spasms with dystonic posturing with dysarthria and dysphagia)
explanation: >-
GeneReviews documents dystonic spasms and dystonic posturing as the
movement-disorder feature.
- reference: PMID:31726291
reference_title: "Patterns of neurological manifestations in Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Dystonia was the most common neurological manifestation (67%), followed by intellectual disability (45%) and sensorineural hearing loss (30%).
explanation: >-
In a cohort of 38 genetically confirmed patients, dystonia was the most
common neurological manifestation at 67%, supporting FREQUENT.
- name: Sensorineural hearing impairment
category: Neurologic
description: >-
Moderate bilateral postlingual sensorineural hearing loss develops in more
than half of affected individuals.
phenotype_term:
preferred_term: Sensorineural hearing impairment
term:
id: HP:0000407
label: Sensorineural hearing impairment
frequency: FREQUENT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
moderate bilateral postlingual sensorineural hearing loss
explanation: >-
GeneReviews documents moderate bilateral postlingual sensorineural hearing
loss as part of the neurologic findings present in more than half of
individuals.
- reference: PMID:31726291
reference_title: "Patterns of neurological manifestations in Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Dystonia was the most common neurological manifestation (67%), followed by intellectual disability (45%) and sensorineural hearing loss (30%).
explanation: >-
In a cohort of 38 genetically confirmed patients, sensorineural hearing
loss occurred in 30%, supporting FREQUENT.
- name: Intellectual disability
category: Neurologic
description: >-
Mild intellectual disability is present in more than half of affected
individuals.
phenotype_term:
preferred_term: Intellectual disability
term:
id: HP:0001249
label: Intellectual disability
severity: MILD
frequency: FREQUENT
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
moderate bilateral postlingual sensorineural hearing loss, and mild intellectual disability
explanation: >-
GeneReviews documents mild intellectual disability among the neurologic
findings present in more than half of individuals.
- reference: PMID:31726291
reference_title: "Patterns of neurological manifestations in Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Dystonia was the most common neurological manifestation (67%), followed by intellectual disability (45%) and sensorineural hearing loss (30%).
explanation: >-
In a cohort of 38 genetically confirmed patients, intellectual disability
occurred in 45%, supporting FREQUENT.
- name: Diabetes mellitus
category: Endocrine
description: >-
Diabetes mellitus is a recognized endocrine manifestation of WSS, with
surveillance recommended beginning at age 20 years.
phenotype_term:
preferred_term: Diabetes mellitus
term:
id: HP:0000819
label: Diabetes mellitus
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Standard treatment for diabetes mellitus, hypothyroidism, hearing loss, and intellectual disability.
explanation: >-
GeneReviews lists diabetes mellitus among the recognized manifestations
requiring standard treatment and surveillance.
- reference: PMID:35002959
reference_title: "Case Report: A Chinese Family of Woodhouse-Sakati Syndrome With Diabetes Mellitus, With a Novel Biallelic Deletion Mutation of the DCAF17 Gene."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
These findings indicate that the DCAF17 gene mutation may cause pancreatic β cell functional impairment and contribute to the development of diabetes.
explanation: >-
This case report links DCAF17-related beta-cell impairment to diabetes
mellitus in WSS patients.
- name: Hypothyroidism
category: Endocrine
description: >-
Hypothyroidism is a recognized endocrine manifestation of WSS, with
surveillance recommended beginning at age 20 years.
phenotype_term:
preferred_term: Hypothyroidism
term:
id: HP:0000821
label: Hypothyroidism
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Standard treatment for diabetes mellitus, hypothyroidism, hearing loss, and intellectual disability.
explanation: >-
GeneReviews lists hypothyroidism among the recognized endocrine
manifestations requiring standard treatment and surveillance.
- name: Dysarthria
category: Neurologic
description: >-
Dysarthria accompanies the extrapyramidal movement disorder and often
benefits from speech therapy.
phenotype_term:
preferred_term: Dysarthria
term:
id: HP:0001260
label: Dysarthria
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Dysarthria often benefits from consultation with a speech therapist.
explanation: >-
GeneReviews documents dysarthria as part of the WSS neurologic phenotype.
- name: Dysphagia
category: Neurologic
description: >-
Dysphagia accompanies the extrapyramidal movement disorder and may
eventually require a gastrostomy to maintain caloric intake.
phenotype_term:
preferred_term: Dysphagia
term:
id: HP:0002015
label: Dysphagia
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Those with dysphagia often require measures to reduce oral secretions, use of thickened liquids and pureed foods to avoid aspiration, and eventually a gastrostomy to help maintain caloric intake.
explanation: >-
GeneReviews documents dysphagia as a WSS neurologic manifestation
requiring dietary and surgical management.
- name: Abnormal cerebral white matter morphology
category: Neurologic
description: >-
Neuroimaging in WSS frequently shows white matter abnormalities, and brain
iron accumulation places WSS within the NBIA spectrum. Imaging is variable,
however: at least one genetically confirmed case had no reportable MRI
abnormalities.
phenotype_term:
preferred_term: Abnormal cerebral white matter morphology
term:
id: HP:0002500
label: Abnormal cerebral white matter morphology
evidence:
- reference: PMID:38320940
reference_title: "Woodhouse-Sakati syndrome: A review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Patients with WSS are characterized by endocrinological and neurological manifestations and neuroradiological findings.
explanation: >-
This review documents characteristic neuroradiological findings in WSS;
WSS is described as part of the NBIA spectrum with brain iron and white
matter changes.
- reference: PMID:39342163
reference_title: "Woodhouse-sakati syndrome with no reportable MRI findings: a case report."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
all previously reported cases having positive MRI findings, unlike our case.
explanation: >-
While most WSS cases show positive MRI findings such as white-matter
changes, this report documents a genetically confirmed case with no
reportable MRI abnormalities, indicating the imaging phenotype is variable.
- name: Male infertility
category: Reproductive
description: >-
Hypogonadism in affected males is associated with
oligoasthenoteratozoospermia and infertility.
phenotype_term:
preferred_term: Male infertility
term:
id: HP:0003251
label: Male infertility
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Virtually all individuals with Woodhouse-Sakati syndrome (WSS) have the endocrine findings of hypogonadism (evident at puberty)
explanation: >-
Near-universal hypogonadism in WSS underlies reproductive dysfunction,
including infertility in affected males.
- reference: PMID:39239833
reference_title: "Single-cell RNA sequencing reveals the important role of Dcaf17 in spermatogenesis of golden hamsters."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Several previous studies have reported that mutations in Dcaf17 cause Woodhouse-Sakati syndrome, which results in oligoasthenoteratozoospermia and male infertility.
explanation: >-
This study summarizes that DCAF17 mutations in WSS result in
oligoasthenoteratozoospermia and male infertility. The supporting model
organism (golden hamster) recapitulates the infertility phenotype.
genetic:
- name: DCAF17
association: Loss of function mutation
gene_term:
preferred_term: DCAF17
term:
id: hgnc:25784
label: DCAF17
notes: >-
WSS is caused by biallelic pathogenic variants in DCAF17. A recurrent
frameshift deletion (c.436delC, p.Ala147Hisfs*9) is a founder variant in
Arab populations.
evidence:
- reference: PMID:36721231
reference_title: "Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: a systematic review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Thirteen different DCAF17 variants were associated with WSS development (including 8 identified in the GME region).
explanation: >-
This systematic review identifies DCAF17 as the disease gene with multiple
pathogenic variants reported worldwide.
- reference: PMID:36721231
reference_title: "Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: a systematic review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The most frequent variant was a frameshift deletion variant (c.436delC, p.Ala147Hisfs*9) unique to Arabs that was reported in 11 cases from Tunisia, Kuwait, Qatar, Bahrain, and Saudi Arabia.
explanation: >-
Supports the recurrent founder c.436delC frameshift deletion in Arab
populations.
- reference: PMID:35002959
reference_title: "Case Report: A Chinese Family of Woodhouse-Sakati Syndrome With Diabetes Mellitus, With a Novel Biallelic Deletion Mutation of the DCAF17 Gene."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Whole-exome sequencing revealed a homozygous deletion NM_025000.4:c.1488_1489delAG in the DCAF17 gene, which resulted in a frameshift mutation that led to stop codon formation.
explanation: >-
Documents a distinct loss-of-function frameshift DCAF17 variant
(c.1488_1489delAG) outside the Arab founder, identified in a Chinese
family, illustrating the worldwide allelic heterogeneity of WSS.
diagnosis:
- name: DCAF17 molecular genetic testing
description: >-
The diagnosis of WSS is established in a proband with suggestive clinical,
neuroimaging, and neurophysiologic findings by identification of biallelic
pathogenic variants in DCAF17 on molecular genetic testing.
diagnosis_term:
preferred_term: molecular genetic testing
term:
id: MAXO:0000533
label: molecular genetic testing
qualifiers:
- predicate:
preferred_term: has participant
term:
id: RO:0000057
label: has participant
value:
preferred_term: DCAF17
term:
id: hgnc:25784
label: DCAF17
results: Biallelic pathogenic DCAF17 variant.
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The diagnosis of WSS is established in a proband with suggestive clinical, neuroimaging, and neurophysiologic findings by identification of biallelic pathogenic variants in DCAF17 on molecular genetic testing.
explanation: >-
GeneReviews specifies molecular genetic confirmation of biallelic DCAF17
variants as the diagnostic standard.
treatments:
- name: Hormone replacement therapy
description: >-
Hypogonadism requires hormone replacement therapy to induce secondary sex
characteristics and promote bone health at the usual age of puberty.
treatment_term:
preferred_term: hormone replacement therapy
term:
id: NCIT:C15599
label: Hormone Replacement Therapy
target_mechanisms:
- target: Hypogonadism
treatment_effect: MODULATES
description: >-
Hormone replacement compensates for the hypogonadal endocrine deficiency.
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hypogonadism requires hormone replacement therapy to induce secondary sex characteristics and promote bone health at the usual age of puberty.
explanation: >-
GeneReviews recommends hormone replacement therapy for the hypogonadism
of WSS.
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hypogonadism requires hormone replacement therapy to induce secondary sex characteristics and promote bone health at the usual age of puberty.
explanation: >-
GeneReviews recommends hormone replacement therapy as core management of
WSS hypogonadism.
- reference: PMID:37895404
reference_title: "The Successful Management of Primary Amenorrhea in Woodhouse-Sakati Syndrome: A Case Report and a Literature Review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Treatment includes hormonal replacement therapy for the induction of puberty.
explanation: >-
This case report describes successful hormone replacement therapy for
pubertal induction in a WSS patient with primary amenorrhea.
- name: Pharmacotherapy for dystonia
description: >-
Treatment for dystonia is routine; oral medications are tried first and
followed in some instances by botulinum toxin injection and/or deep-brain
stimulation.
treatment_term:
preferred_term: pharmacotherapy
term:
id: MAXO:0000058
label: pharmacotherapy
target_mechanisms:
- target: Dystonia
treatment_effect: MODULATES
description: >-
Oral antidystonia medications are first-line symptomatic treatment.
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Treatment for dystonia is routine; oral medications are tried first and followed in some instances by botulinum toxin injection and/or deep-brain stimulation.
explanation: >-
GeneReviews describes oral pharmacotherapy as first-line dystonia
management.
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Treatment for dystonia is routine; oral medications are tried first and followed in some instances by botulinum toxin injection and/or deep-brain stimulation.
explanation: >-
GeneReviews documents oral pharmacotherapy as the first-line approach for
WSS-associated dystonia.
- name: Botulinum toxin injection
description: >-
Botulinum toxin injection is used for dystonia in some individuals when oral
medications are insufficient.
treatment_term:
preferred_term: botulinum toxin therapy
term:
id: MAXO:0000058
label: pharmacotherapy
therapeutic_agent:
- preferred_term: botulinum toxin
term:
id: CHEBI:3160
label: Botulinum toxin type A
target_mechanisms:
- target: Dystonia
treatment_effect: MODULATES
description: >-
Botulinum toxin reduces focal dystonic muscle activity.
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
oral medications are tried first and followed in some instances by botulinum toxin injection and/or deep-brain stimulation
explanation: >-
GeneReviews lists botulinum toxin injection as second-line dystonia
treatment.
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
oral medications are tried first and followed in some instances by botulinum toxin injection and/or deep-brain stimulation
explanation: >-
GeneReviews documents botulinum toxin injection as a treatment option for
WSS dystonia.
- name: Deep brain stimulation
description: >-
Deep brain stimulation is used in some individuals with refractory dystonia.
A reported WSS patient achieved significant improvement following DBS.
therapeutic_modality: DEVICE
treatment_term:
preferred_term: deep brain stimulation
term:
id: NCIT:C21024
label: Deep Brain Stimulation
target_mechanisms:
- target: Dystonia
treatment_effect: MODULATES
description: >-
DBS modulates basal-ganglia output to reduce refractory dystonia.
evidence:
- reference: PMID:40235137
reference_title: "Clinical and Genetic Characterization of Woodhouse-Sakati Syndrome in Iranian Patients: A Case Series."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Patients had variable responses to common therapies, and one patient achieved significant improvement following DBS.
explanation: >-
This case series reports significant improvement with DBS in a WSS
patient with refractory dystonia.
evidence:
- reference: PMID:40235137
reference_title: "Clinical and Genetic Characterization of Woodhouse-Sakati Syndrome in Iranian Patients: A Case Series."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We highlight the importance of considering WSS in patients with both neurological and endocrine symptoms and suggest DBS as a potential treatment option.
explanation: >-
This case series proposes DBS as a treatment option for WSS-associated
movement disorder.
- reference: PMID:39342163
reference_title: "Woodhouse-sakati syndrome with no reportable MRI findings: a case report."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Treatment provided includes botulinum toxin injections and deep brain stimulation, providing better dystonia control, with progress in walking and strength exercises, and overall remarkable improvement.
explanation: >-
This case report documents that deep brain stimulation (with botulinum
toxin and neurorehabilitation) produced remarkable improvement in dystonia
control and ambulation in a WSS patient.
- name: Supportive care
description: >-
Treatment is symptomatic and should be managed by a multidisciplinary team,
including speech therapy for dysarthria, dysphagia measures, and standard
treatment of diabetes mellitus, hypothyroidism, hearing loss, and
intellectual disability.
treatment_term:
preferred_term: Supportive Care
term:
id: NCIT:C15747
label: Supportive Care
evidence:
- reference: PMID:27489925
reference_title: "Woodhouse-Sakati Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Treatment of manifestations: Treatment is symptomatic and should be managed by a multidisciplinary team.
explanation: >-
GeneReviews describes WSS management as symptomatic multidisciplinary
supportive care.
differential_diagnoses:
- name: Neurodegeneration with brain iron accumulation
description: >-
WSS is classified within the NBIA spectrum and shares progressive
extrapyramidal movement disorder and brain iron accumulation with other
NBIA disorders, but is distinguished by its prominent endocrine and
ectodermal features (hypogonadism, alopecia, diabetes).
distinguishing_features:
- WSS combines a movement disorder with hypogonadism and progressive alopecia.
- WSS is caused by biallelic DCAF17 variants rather than other NBIA genes.
disease_term:
preferred_term: neurodegeneration with brain iron accumulation
term:
id: MONDO:0018307
label: neurodegeneration with brain iron accumulation
evidence:
- reference: PMID:38320940
reference_title: "Woodhouse-Sakati syndrome: A review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Woodhouse-Sakati Syndrome (WSS) is one of the rarest NBIAs.
explanation: >-
This review places WSS within the NBIA spectrum, supporting NBIA disorders
as the relevant differential category.
clinical_trials:
- name: NCT05522374
status: RECRUITING
description: >-
TIRCON International NBIA Patient Registry and Natural History Study, a
prospective observational registry collecting longitudinal clinical and
natural-history data on neurodegeneration with brain iron accumulation
(NBIA) disorders, the spectrum within which Woodhouse-Sakati syndrome is
classified.
target_phenotypes:
- preferred_term: Progressive extrapyramidal movement disorder
term:
id: HP:0007153
label: Progressive extrapyramidal movement disorder
evidence:
- reference: clinicaltrials:NCT05522374
reference_title: "TIRCON International NBIA (Neurodegeneration Associated With Brain Iron Accumulation) Patient Registry and Natural History Study"
supports: SUPPORT
snippet: >-
continue the provision of a global registry and natural history study for NBIA disorders
explanation: >-
This registry provides a global natural-history study for NBIA disorders,
the disease spectrum that includes Woodhouse-Sakati syndrome.
datasets: []
References & Deep Research
References
9Deep Research
11. Disease Information
1.1 Definition and current understanding
Woodhouse–Sakati syndrome is a rare multisystem neuroendocrine disorder with core endocrine involvement (hypogonadism, diabetes, thyroid abnormalities) and progressive neurologic manifestations, caused by biallelic pathogenic variants in DCAF17 and inherited in an autosomal recessive pattern. (bakhsh2023thesuccessfulmanagement pages 1-2, louro2019woodhouse–sakatisyndromefirst pages 1-2, wakim2024woodhousesakatisyndromegenotype–phenotype pages 1-2)
Abstract-supported definition (direct quotes): - A 2023 systematic review states: “Woodhouse-Sakati syndrome (WSS) is a rare, autosomal recessive genetic disorder with variable clinical manifestations mainly affecting the endocrine and nervous systems.” (Kohil et al., Orphanet J Rare Dis, published Jan 2023; DOI: https://doi.org/10.1186/s13023-023-02614-8) (kohil2023geneticepidemiologyof pages 1-2) - A 2024 case report states WSS is “a rare autosomal recessive condition caused by biallelic pathogenic variants in the DCAF17 gene” with “fewer than 200 cases reported” and symptoms that “first emerge in middle-late adolescence.” (Irvine & Ahmad, BMC Neurology, published Sep 2024; DOI: https://doi.org/10.1186/s12883-024-03865-z) (irvine2024woodhousesakatisyndromewith pages 1-3)
1.2 Synonyms / alternative names
- Woodhouse–Sakati syndrome (WSS) (bakhsh2023thesuccessfulmanagement pages 1-2, louro2019woodhouse–sakatisyndromefirst pages 1-2)
- Sometimes described as a DCAF17-linked NBIA (neurodegeneration with brain iron accumulation) phenotype due to basal ganglia iron deposition on MRI in many patients. (louro2019woodhouse–sakatisyndromefirst pages 1-2)
1.3 Evidence source types
Evidence in this report is primarily from: - Aggregated disease-level resources: systematic reviews and narrative reviews (2023–2024 prioritized). (kohil2023geneticepidemiologyof pages 1-2, wakim2024woodhousesakatisyndromegenotype–phenotype pages 2-4) - Human clinical evidence: patient cohorts and case reports (including quantified cohort neurologic data). (bohlega2019patternsofneurological pages 2-3, louro2019woodhouse–sakatisyndromefirst pages 1-2, irvine2024woodhousesakatisyndromewith pages 3-5)
2. Etiology
2.1 Disease causal factors
Primary cause: Germline loss-of-function variants in DCAF17 (biallelic) with autosomal recessive inheritance. (wakim2024woodhousesakatisyndromegenotype–phenotype pages 1-2, kohil2023geneticepidemiologyof pages 1-2, amalnath2024woodhouse–sakatisyndromein pages 1-3)
DCAF17 is described as encoding nucleolar proteins (two main transcripts) and is implicated (in reviews) in nucleolar functions and possibly ubiquitin-ligase associated biology; truncating variants likely impair function through truncated protein and/or nonsense-mediated decay. (wakim2024woodhousesakatisyndromegenotype–phenotype pages 4-5, kohil2023geneticepidemiologyof pages 6-7)
2.2 Risk factors
Because WSS is Mendelian, “risk factors” are primarily genetic and population-structure related.
Genetic risk factors - Biallelic pathogenic variants in DCAF17 are necessary and sufficient for the disorder in reported families. (wakim2024woodhousesakatisyndromegenotype–phenotype pages 1-2, kohil2023geneticepidemiologyof pages 1-2)
Population risk factors / epidemiologic correlates - Consanguinity is common among reported families in high-prevalence regions: the 2023 systematic review found that in the Greater Middle East (GME) region, “consanguineous marriages were common (67%).” (Kohil et al., 2023) (kohil2023geneticepidemiologyof pages 1-2)
2.3 Protective factors / gene–environment interactions
No protective genetic variants or environmental protective factors have been established in the retrieved literature for WSS. The intrafamilial phenotypic variability noted in cohorts suggests possible modifier factors, but specific genes or environmental interactions are not clearly defined. (bohlega2019patternsofneurological pages 1-2)
3. Phenotypes
3.1 Key phenotypic domains
WSS typically involves: - Endocrine/reproductive: hypogonadism with delayed/absent puberty and primary amenorrhea; diabetes mellitus; hypothyroidism; low IGF-1. (bakhsh2023thesuccessfulmanagement pages 1-2, louro2019woodhouse–sakatisyndromefirst pages 1-2, zhou2021casereporta pages 1-2) - Dermatologic: alopecia (often temporal/frontotemporal), sparse eyebrows; progeroid skin changes. (wakim2024woodhousesakatisyndromegenotype–phenotype pages 2-4, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7) - Neurologic: progressive extrapyramidal movement disorder (dystonia prominent), dysarthria/dysphagia; intellectual disability; seizures in a subset; hearing loss. (bohlega2019patternsofneurological pages 2-3, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7) - Imaging: many cases show basal ganglia iron deposition and leukodystrophy/white matter changes; exceptions occur. (louro2019woodhouse–sakatisyndromefirst pages 1-2, irvine2024woodhousesakatisyndromewith pages 1-3)
3.2 Quantitative phenotype frequencies (human cohort data)
A detailed neurologic cohort (n=38, genetically confirmed; founder DCAF17 c.436delC) reported:
- Neurologic involvement: 31/38 (81.5%)
- Dystonia: 25/38 (65.7%)
- Intellectual disability: 36.8% (also described as 45% in another excerpt of same study)
- Sensorineural hearing loss: 31.5%
- Seizures: 10.5%
- Rigidity: 5.2%
- Tremor/ataxia/choreoathetosis: 2.6%
Additionally, patients clustered into a severe phenotype (47.4%) with earlier onset and progressive disability: mean age of first neurologic symptoms 12.6 ± 4.5 years; loss of ambulation over 7.4 ± 3.6 years. (Bohlega et al., Parkinsonism Relat Disord, published Dec 2019; DOI: https://doi.org/10.1016/j.parkreldis.2019.10.007) (bohlega2019patternsofneurological pages 2-3, bohlega2019patternsofneurological pages 1-2)
3.3 Typical age of onset and progression
- Endocrine/alopecia manifestations often become apparent around puberty/adolescence, with neurologic symptoms frequently emerging later and progressing variably. (irvine2024woodhousesakatisyndromewith pages 1-3, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7)
3.4 Suggested HPO terms (non-exhaustive)
(These are ontology suggestions based on described clinical features; HPO IDs should be verified against the HPO database.) - Hypogonadism; primary amenorrhea; delayed puberty (bakhsh2023thesuccessfulmanagement pages 1-2, louro2019woodhouse–sakatisyndromefirst pages 1-2) - Alopecia; sparse eyebrows (wakim2024woodhousesakatisyndromegenotype–phenotype pages 2-4, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7) - Diabetes mellitus (often adolescent/young adult onset) (louro2019woodhouse–sakatisyndromefirst pages 1-2) - Hypothyroidism (louro2019woodhouse–sakatisyndromefirst pages 1-2) - Dystonia; dysarthria; dysphagia (bohlega2019patternsofneurological pages 2-3, louro2019woodhouse–sakatisyndromefirst pages 1-2) - Intellectual disability (bohlega2019patternsofneurological pages 2-3) - Sensorineural hearing impairment (bohlega2019patternsofneurological pages 2-3, louro2019woodhouse–sakatisyndromefirst pages 1-2) - Abnormal brain iron accumulation; leukodystrophy/white matter abnormalities (louro2019woodhouse–sakatisyndromefirst pages 1-2)
3.5 Quality-of-life impacts
Direct standardized QoL instruments specific to WSS were not identified in the retrieved papers; however, severe dystonia and progressive disability including wheelchair dependence are reported, implying major functional burden. (irvine2024woodhousesakatisyndromewith pages 3-5, bohlega2019patternsofneurological pages 2-3)
4. Genetic / Molecular Information
4.1 Causal gene
- DCAF17 (DDB1 and CUL4-associated factor 17; formerly C2orf37). (kohil2023geneticepidemiologyof pages 1-2, louro2019woodhouse–sakatisyndromefirst pages 1-2)
4.2 Pathogenic variant spectrum
A 2023 systematic review identified 185 patients in 97 families from 12 countries and reported 13 distinct DCAF17 variants linked to WSS. (Kohil et al., 2023) (kohil2023geneticepidemiologyof pages 1-2)
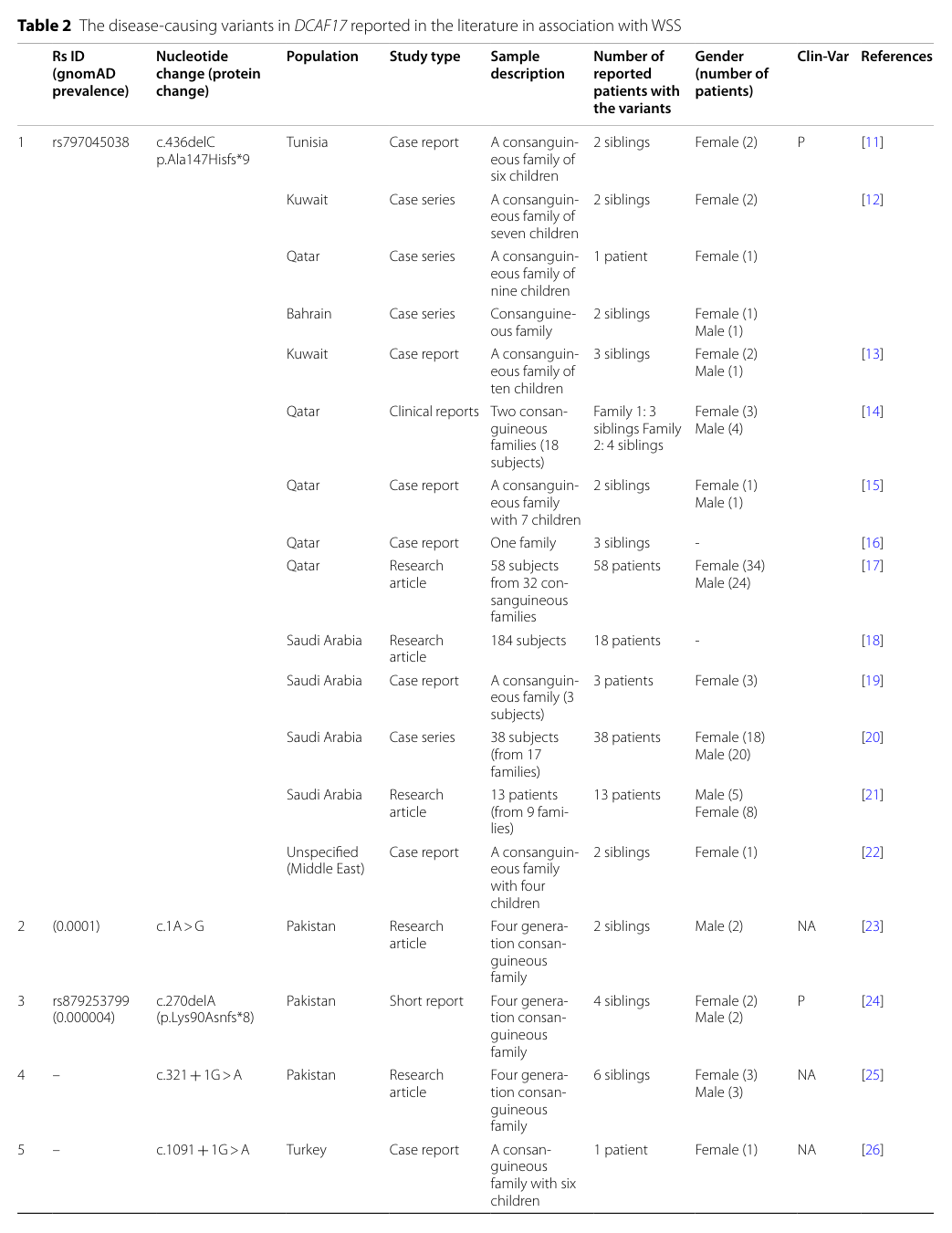
Key recurrent/founder and representative variants include: - c.436delC (p.Ala147Hisfs*9) (frameshift; recurrent/founder in Arab populations) (kohil2023geneticepidemiologyof pages 4-6, kohil2023geneticepidemiologyof pages 1-2, kohil2023geneticepidemiologyof pages 6-7) - Splice-site variants such as c.321+1G>A and c.1091+1G>A (kohil2023geneticepidemiologyof pages 4-6) - c.1488_1489delAG (frameshift; reported in China; gnomAD prevalence noted as 0.000011 in one excerpt) (kohil2023geneticepidemiologyof pages 6-7) - c.1091+2T>C (Portuguese case; splice-site) (louro2019woodhouse–sakatisyndromefirst pages 1-2) - Novel truncating c.153G>A (p.Trp51*) in an Indian patient; absent from population databases cited (gnomAD/IndiGenomes) (Amalnath et al., Am J Med Genet A, published Sep 2024; DOI: https://doi.org/10.1002/ajmg.a.63405) (amalnath2024woodhouse–sakatisyndromein pages 1-3)
Visual evidence: A table of reported DCAF17 variants and countries is available from the 2023 systematic review (Table 2). (kohil2023geneticepidemiologyof media b5c57154, kohil2023geneticepidemiologyof media 150533d1)
4.3 Founder effects and geographic distribution
- The 2023 systematic review describes c.436delC (p.Ala147Hisfs*9) as “unique to Arabs,” reported across Tunisia, Kuwait, Qatar, Bahrain, and Saudi Arabia. (kohil2023geneticepidemiologyof pages 1-2)
- The same review reports high representation of families from the Greater Middle East and an association with consanguinity. (kohil2023geneticepidemiologyof pages 1-2)
- A 2024 case report reiterates that “most of the patients have been reported from Greater Middle Eastern countries.” (Amalnath et al., 2024) (amalnath2024woodhouse–sakatisyndromein pages 1-3)
4.4 Modifier genes / epigenetics / chromosomal abnormalities
No validated modifier genes, disease-specific epigenetic signatures, or recurrent chromosomal abnormalities were identified in the retrieved evidence.
5. Environmental Information
WSS is primarily genetic; no environmental triggers, toxins, lifestyle exposures, or infectious agents have been established as causal or modifying factors in the retrieved sources.
6. Mechanism / Pathophysiology
6.1 Current mechanistic understanding
Mechanistic understanding remains incomplete. However, reviews and systematic summaries converge on a nucleolar DCAF17 biology with downstream multisystem effects.
- The 2023 systematic review notes that DCAF17 encodes nucleolar proteins and that mutant DCAF17 has been suggested to cause “defective ribosome biogenesis,” “reduced splicing efficiency,” and loss-of-function effects. (kohil2023geneticepidemiologyof pages 1-2)
- A 2019 Portuguese case report describes DCAF17 as a nucleolar protein that “may act as a substrate receptor for the CUL4-DDB1 E3 ubiquitin ligase complex,” providing a plausible link to proteostasis/regulatory pathways. (louro2019woodhouse–sakatisyndromefirst pages 1-2)
6.2 Causal chain (evidence-based, with uncertainty)
1) Biallelic DCAF17 LOF → 2) nucleolar dysfunction (proposed ribosome/splicing deficits; uncertain) → 3) selective vulnerability in endocrine tissues and nervous system → 4) clinical syndrome with hypogonadism/diabetes/thyroid dysfunction and progressive dystonia/intellectual disability/hearing loss. (kohil2023geneticepidemiologyof pages 1-2, louro2019woodhouse–sakatisyndromefirst pages 1-2, bohlega2019patternsofneurological pages 2-3)
6.3 Tissue-level pathology proxies (imaging)
MRI findings in many patients include basal ganglia iron deposition and leukodystrophy/white matter changes, supporting classification alongside NBIA phenotypes for some individuals; however, normal MRI is possible. (louro2019woodhouse–sakatisyndromefirst pages 1-2, irvine2024woodhousesakatisyndromewith pages 1-3)
6.4 Suggested GO / CL terms (high-level suggestions)
(These are ontology suggestions inferred from described biology and are not directly asserted as experimentally demonstrated in WSS-specific studies in the retrieved evidence.) - GO biological process candidates: ribosome biogenesis; RNA splicing; protein ubiquitination; DNA repair; cell cycle regulation; apoptosis (wakim2024woodhousesakatisyndromegenotype–phenotype pages 4-5, kohil2023geneticepidemiologyof pages 1-2) - CL cell types likely involved clinically: pancreatic beta cell; gonadal cells (ovarian/testicular); neurons of basal ganglia; oligodendrocytes/myelin-related systems (clinical proxy via leukodystrophy) (zhou2021casereporta pages 1-2, louro2019woodhouse–sakatisyndromefirst pages 1-2)
6.5 Molecular profiling / multi-omics / single-cell / spatial
No WSS-specific transcriptomic, proteomic, metabolomic, or single-cell/spatial multi-omics datasets were identified in the retrieved evidence.
7. Anatomical Structures Affected
7.1 Organ- and system-level
- Endocrine system: gonads/HPG axis, pancreas (beta-cell dysfunction suggested), thyroid. (zhou2021casereporta pages 1-2, louro2019woodhouse–sakatisyndromefirst pages 1-2)
- Nervous system: basal ganglia/extrapyramidal circuitry; white matter involvement in many cases. (louro2019woodhouse–sakatisyndromefirst pages 1-2, bohlega2019patternsofneurological pages 2-3)
- Integumentary system: scalp hair follicles (alopecia). (wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7)
- Auditory system: sensorineural hearing loss. (bohlega2019patternsofneurological pages 2-3, louro2019woodhouse–sakatisyndromefirst pages 1-2)
7.2 Suggested UBERON terms (conceptual)
- Ovary/uterus (absent ovaries on ultrasound; uterine/adnexal hypoplasia in some cases) (wakim2024woodhousesakatisyndromegenotype–phenotype pages 2-4, baigh2026woodhousesakatisyndromedue pages 2-4)
- Basal ganglia; globus pallidus; substantia nigra; white matter (louro2019woodhouse–sakatisyndromefirst pages 1-2)
- Pancreas (pancreatic atrophy in one family; functional impairment evidence) (zhou2021casereporta pages 1-2)
8. Temporal Development
8.1 Onset
Symptoms commonly emerge in middle-late adolescence with endocrine features such as delayed puberty/amenorrhea and metabolic abnormalities, and later neurologic deterioration in many patients. (irvine2024woodhousesakatisyndromewith pages 1-3, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7)
8.2 Progression
Progression is variable. In the 38-patient cohort, a severe phenotype included loss of ambulation over ~7 years after neurologic onset, whereas a milder/absent neurologic phenotype occurred in roughly half. (bohlega2019patternsofneurological pages 2-3)
9. Inheritance and Population
9.1 Inheritance
- Autosomal recessive with biallelic DCAF17 pathogenic variants. (wakim2024woodhousesakatisyndromegenotype–phenotype pages 1-2, kohil2023geneticepidemiologyof pages 1-2)
9.2 Epidemiology (counts and geography)
Robust prevalence/incidence estimates were not identified.
Best available summary from literature aggregation: - Across 25 studies, 185 patients in 97 families from 12 countries were identified (systematic review to June 2022). (Kohil et al., 2023) (kohil2023geneticepidemiologyof pages 1-2) - Strong geographic clustering in the Greater Middle East, with consanguinity common. (kohil2023geneticepidemiologyof pages 1-2, amalnath2024woodhouse–sakatisyndromein pages 1-3)
10. Diagnostics
10.1 Clinical clues
Alopecia + hypogonadism (often primary amenorrhea) + diabetes mellitus + progressive dystonia/extrapyramidal signs are recurrent diagnostic clues. (bakhsh2023thesuccessfulmanagement pages 1-2, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7)
10.2 Laboratory testing
- Endocrine: gonadotropins/sex steroids consistent with hypergonadotropic hypogonadism; glucose/HbA1c; thyroid tests; IGF-1 may be low. (louro2019woodhouse–sakatisyndromefirst pages 1-2, zhou2021casereporta pages 1-2)
- Diabetes physiology: OGTT-derived measures, insulin and C-peptide testing can show impaired secretion and low HOMA-b in some cases. (zhou2021casereporta pages 1-2)
10.3 Imaging
- Brain MRI: typical findings include progressive periventricular leukodystrophy/white-matter changes and iron deposition in globus pallidus/substantia nigra/red nucleus. (louro2019woodhouse–sakatisyndromefirst pages 1-2)
- Important recent development: a 2024 report described a genetically confirmed WSS patient with no reportable abnormalities on T2/ADC/SWI MRI sequences. (irvine2024woodhousesakatisyndromewith pages 1-3)
10.4 Genetic testing (definitive)
Definitive diagnosis relies on identifying biallelic pathogenic variants in DCAF17, commonly via targeted sequencing, multigene panels, or exome sequencing. (irvine2024woodhousesakatisyndromewith pages 1-3, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7)
Example diagnostic implementations: - Whole-exome sequencing identified DCAF17 c.1488_1489delAG in a Chinese family with WSS and diabetes phenotype. (Frontiers Endocrinology, Dec 2021; DOI: https://doi.org/10.3389/fendo.2021.770871) (zhou2021casereporta pages 1-2)
10.5 Differential diagnosis
Differential diagnosis includes other leukodystrophies and other NBIA disorders (PKAN, PLA2G6-associated disease), where genetic testing is decisive. (louro2019woodhouse–sakatisyndromefirst pages 1-2, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7)
11. Outcome / Prognosis
Systematic survival statistics are not available in the retrieved evidence.
- A 2023 management-focused case report/literature review notes: “it is generally believed that individuals with this syndrome have a normal lifespan,” while progressive comorbidities can affect long-term quality of life. (Bakhsh et al., Life, Oct 2023; DOI: https://doi.org/10.3390/life13102022) (bakhsh2023thesuccessfulmanagement pages 1-2)
- Severe neurologic phenotypes can lead to substantial disability and loss of ambulation. (bohlega2019patternsofneurological pages 2-3)
12. Treatment
12.1 Current applications / real-world implementations
There is no disease-specific curative therapy; management is symptomatic and multidisciplinary. (irvine2024woodhousesakatisyndromewith pages 1-3, bakhsh2023thesuccessfulmanagement pages 6-8)
Endocrine / reproductive - Hormone replacement therapy (HRT) is used to induce puberty and menstruation in affected females. (bakhsh2023thesuccessfulmanagement pages 1-2) - A 2023 case report describes incremental estrogen/progesterone therapy over four years with pubertal development and reversal of primary amenorrhea. (bakhsh2023thesuccessfulmanagement pages 6-8)
Diabetes management Standard diabetes care is applied (lifestyle, oral agents, insulin as needed). (bakhsh2023thesuccessfulmanagement pages 6-8)
Neurologic management (dystonia/extrapyramidal) - Botulinum toxin for focal dystonia and deep brain stimulation (DBS) for refractory dystonia have been applied. - A 2024 BMC Neurology case reported DBS with immediate and subsequent improvement after activation and “remarkable improvement,” including regained assisted ambulation (~20 m with support versus previously unable). (Irvine & Ahmad, Sep 2024) (irvine2024woodhousesakatisyndromewith pages 3-5) - A 2025 Iranian case series notes one patient achieved “significant improvement” after GPi DBS, suggesting DBS as a treatment option. (Khosravi et al., J Mov Disord, Jul 2025; DOI: https://doi.org/10.14802/jmd.25043) (khosravi2025clinicalandgenetic pages 1-3)
Supportive rehabilitation Intensive physiotherapy/occupational therapy/speech-language therapy is reported with functional gains, especially when paired with dystonia control (e.g., DBS). (irvine2024woodhousesakatisyndromewith pages 3-5)
12.2 Suggested MAXO terms (conceptual)
- Hormone replacement therapy; pubertal induction therapy (bakhsh2023thesuccessfulmanagement pages 1-2)
- Diabetes pharmacotherapy; insulin therapy (bakhsh2023thesuccessfulmanagement pages 6-8)
- Botulinum toxin injection therapy (irvine2024woodhousesakatisyndromewith pages 3-5)
- Deep brain stimulation (pallidal DBS) (irvine2024woodhousesakatisyndromewith pages 3-5, khosravi2025clinicalandgenetic pages 1-3)
- Rehabilitation therapy (PT/OT/SLT) (irvine2024woodhousesakatisyndromewith pages 3-5)
13. Prevention
No primary prevention exists beyond genetic counseling and carrier/family testing in at-risk families.
- Family screening/cascade testing is recommended in management literature. (bakhsh2023thesuccessfulmanagement pages 1-2)
14. Other Species / Natural Disease
No naturally occurring veterinary analogs were identified in the retrieved evidence.
15. Model Organisms
No directly retrieved WSS-specific animal model papers were available in the accessible corpus in this run; therefore, model organism phenotypic recapitulation cannot be summarized here.
Recent developments (2023–2024 emphasized)
1) 2023 systematic review (genetic epidemiology): consolidated global case counts (185 patients/97 families/12 countries), variant spectrum (13 variants), and consanguinity burden in GME (67%). (Kohil et al., Jan 2023) (kohil2023geneticepidemiologyof pages 1-2) 2) 2024 phenotype/genotype review: emphasized expanding phenotypic spectrum and structured diagnostic workup recommendations (endocrine panel, ECG, hearing, MRI, genetic confirmation). (Wakim et al., Jul 2024) (wakim2024woodhousesakatisyndromegenotype–phenotype pages 7-8) 3) 2024 MRI-negative case report: reported genetically confirmed WSS with no reportable MRI abnormalities, challenging the assumption that MRI is always positive. (Irvine & Ahmad, Sep 2024) (irvine2024woodhousesakatisyndromewith pages 1-3) 4) 2024 therapeutic report: DBS combined with intensive rehabilitation produced marked functional improvements in severe dystonia. (irvine2024woodhousesakatisyndromewith pages 3-5) 5) 2024 novel pathogenic variants in underrepresented populations: novel truncating DCAF17 variant reported from India with fatal pulmonary hemorrhage complications despite intervention, highlighting phenotypic expansion and medical complexity. (Amalnath et al., Sep 2024) (amalnath2024woodhouse–sakatisyndromein pages 1-3)
Clinical trials / registries
No WSS-specific interventional trials were identified in the retrieved ClinicalTrials.gov search results; however, WSS is included in a major NBIA registry.
- TIRCON International NBIA Registry / Natural History Study
- NCT: NCT05522374
- Type: Observational, prospective patient registry (started 2012; actively recruiting)
- Target enrollment: ~2000; duration: 30 years
- Includes: explicitly lists “Woodhouse Sakati Syndrome” among NBIA conditions
- Data collected: clinical outcomes (e.g., BAD scale, UPDRS, PedsQL) and disease progression encoded as HPO terms, plus biospecimens (DNA/RNA/plasma/urine). (ClinicalTrials.gov record; accessed via trial chunks) (NCT05522374 chunk 1, NCT05522374 chunk 2)
Summary table
| Domain | Key findings/statistics | Best supporting citation IDs |
|---|---|---|
| Identifiers | Woodhouse–Sakati syndrome (WSS); autosomal recessive multisystem neuroendocrine disorder caused by biallelic DCAF17 variants; MONDO:0009419; OMIM:241080 (disease); DCAF17 OMIM:612515 | (OpenTargets Search: Woodhouse-Sakati syndrome, wakim2024woodhousesakatisyndromegenotype–phenotype pages 1-2, kohil2023geneticepidemiologyof pages 1-2) |
| Core phenotype | Hallmark features: hypogonadism and alopecia; additional common findings include diabetes mellitus, hypothyroidism, sensorineural hearing loss, intellectual disability, dysarthria/dysphagia, and progressive extrapyramidal signs; adolescence/puberty is a typical presentation window | (bakhsh2023thesuccessfulmanagement pages 1-2, louro2019woodhouse–sakatisyndromefirst pages 1-2, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7) |
| Neurologic phenotype frequencies | In a genetically confirmed n=38 cohort: neurologic involvement 31/38 (81.5%); dystonia 25/38 (65.7%); intellectual disability 36.8%–45%; sensorineural hearing loss 31.5%–30%; seizures 10.5%; rigidity 5.2%; tremor/ataxia/choreoathetosis 2.6%. Severe phenotype in 18/38 (47.4%) with mean neurologic onset 12.6 ± 4.5 y and loss of ambulation over 7.4 ± 3.6 y; milder/absent neurologic phenotype in 20/38 (52.6%) with later onset 18.1 ± 4.3 y | (bohlega2019patternsofneurological pages 2-3, bohlega2019patternsofneurological pages 1-2) |
| Endocrine phenotype | Diabetes and hypothyroidism are frequent; review estimates about ~50% diabetes and ~30% hypothyroidism. Females often present with delayed/absent puberty and primary amenorrhea; hypergonadotropic hypogonadism, low estradiol, absent/underdeveloped ovaries, and low IGF-1 are reported. In one c.436delC table subset: hypogonadism 100%, diabetes 28%, hypothyroidism 20% | (wakim2024woodhousesakatisyndromegenotype–phenotype pages 2-4, louro2019woodhouse–sakatisyndromefirst pages 1-2, wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7, zhou2021casereporta pages 1-2) |
| Imaging findings | Typical MRI: progressive periventricular/frontoparietal white-matter abnormalities or leukodystrophy and iron deposition in globus pallidus ± substantia nigra/red nucleus; small pituitary also reported. However, a 2024 case showed no reportable T2/ADC/SWI MRI abnormalities, expanding the spectrum | (louro2019woodhouse–sakatisyndromefirst pages 1-2, irvine2024woodhousesakatisyndromewith pages 1-3) |
| Genetics/variants | Systematic review found 185 patients from 97 families in 12 countries and 13 pathogenic DCAF17 variants. Most frequent founder/recurrent Arab variant: c.436delC (p.Ala147Hisfs*9), reported across Tunisia, Kuwait, Qatar, Bahrain, and Saudi Arabia; other variants include c.321+1G>A, c.1091+2T>C, c.1488_1489delAG, c.153G>A (p.Trp51*), c.270dup, c.1111delA, c.1238delA. No clear genotype–phenotype correlation established | (kohil2023geneticepidemiologyof pages 4-6, kohil2023geneticepidemiologyof pages 1-2, wakim2024woodhousesakatisyndromegenotype–phenotype pages 4-5, amalnath2024woodhouse–sakatisyndromein pages 1-3, kohil2023geneticepidemiologyof pages 6-7, kohil2023geneticepidemiologyof media b5c57154) |
| Management/treatment | No disease-specific curative therapy; management is multidisciplinary and symptom-directed. Reported approaches: hormone replacement therapy for puberty induction/amenorrhea (case report showed pubertal development and reversal of amenorrhea over 4 years), diabetes treatment with lifestyle/oral agents/insulin, botulinum toxin for focal dystonia, deep brain stimulation (GPi DBS) for refractory dystonia, plus physiotherapy/OT/SLT. A 2024 case reported remarkable improvement in dystonia control and ambulation after DBS + intensive rehab | (irvine2024woodhousesakatisyndromewith pages 3-5, irvine2024woodhousesakatisyndromewith pages 1-3, bakhsh2023thesuccessfulmanagement pages 1-2, bakhsh2023thesuccessfulmanagement pages 6-8, khosravi2025clinicalandgenetic pages 1-3) |
| Epidemiology/consanguinity | Extremely rare; literature-based review concentrated cases in the Greater Middle East. Among reviewed studies, 67% of GME families had consanguinity. Most genetically confirmed cases/families were from GME populations, consistent with founder effects and autosomal recessive inheritance | (kohil2023geneticepidemiologyof pages 1-2, amalnath2024woodhouse–sakatisyndromein pages 1-3, kohil2023geneticepidemiologyof media b5c57154) |
Table: This table condenses the most actionable identifiers, phenotype statistics, genetics, imaging, treatment, and epidemiology for Woodhouse–Sakati syndrome. It is designed as a quick-reference summary for building or validating a disease knowledge base entry.
Key evidence visualization
A visual table summarizing disease-causing DCAF17 variants and their geographic distribution is available from the 2023 Orphanet Journal of Rare Diseases systematic review (Table 2). (kohil2023geneticepidemiologyof media b5c57154, kohil2023geneticepidemiologyof media 150533d1)
Limitations / gaps
- No robust prevalence/incidence rates were found in the retrieved evidence.
- Limited mechanistic data and limited disease-specific multi-omics resources were identified in accessible texts.
- Animal models were not directly retrievable in this run; further targeted searches (e.g., “Dcaf17 knockout mouse infertility”, “Dcaf17 nucleolar function”) would be required.
References
-
(OpenTargets Search: Woodhouse-Sakati syndrome): Open Targets Query (Woodhouse-Sakati syndrome, 25 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(louro2019woodhouse–sakatisyndromefirst pages 1-2): Pedro Louro, João Durães, Diana Oliveira, Sandra Paiva, Lina Ramos, and Maria Carmo Macário. Woodhouse–sakati syndrome: first report of a portuguese case. American Journal of Medical Genetics Part A, 179:2237-2240, Jul 2019. URL: https://doi.org/10.1002/ajmg.a.61303, doi:10.1002/ajmg.a.61303. This article has 15 citations.
-
(zhou2021casereporta pages 1-2): Min Zhou, Ningjie Shi, Juan Zheng, Yang Chen, Siqi Wang, Kang-li Xiao, Zhen-hai Cui, Kangli Qiu, F. Zhu, and Hui-qing Li. Case report: a chinese family of woodhouse-sakati syndrome with diabetes mellitus, with a novel biallelic deletion mutation of the dcaf17 gene. Frontiers in Endocrinology, Dec 2021. URL: https://doi.org/10.3389/fendo.2021.770871, doi:10.3389/fendo.2021.770871. This article has 7 citations.
-
(wakim2024woodhousesakatisyndromegenotype–phenotype pages 1-2): Victor Wakim, Mohammad El Dassouki, Ahlam Azar, Abeer Hani, Cybel Mehawej, Eliane Chouery, Marie-Jeanne Baroudi, and Gerard Wakim. Woodhouse-sakati syndrome: genotype–phenotype review and case of intra-familial heterogeneity. Journal of Rare Diseases, Jul 2024. URL: https://doi.org/10.1007/s44162-024-00045-y, doi:10.1007/s44162-024-00045-y. This article has 0 citations.
-
(bakhsh2023thesuccessfulmanagement pages 1-2): Hanadi Bakhsh, Norah Alqntash, and Ebtesam Almajed. The successful management of primary amenorrhea in woodhouse–sakati syndrome: a case report and a literature review. Life, 13:2022, Oct 2023. URL: https://doi.org/10.3390/life13102022, doi:10.3390/life13102022. This article has 2 citations.
-
(kohil2023geneticepidemiologyof pages 1-2): Amira Kohil, Atiyeh M. Abdallah, Khalid Hussain, and Mashael Al-Shafai. Genetic epidemiology of woodhouse-sakati syndrome in the greater middle east region and beyond: a systematic review. Orphanet Journal of Rare Diseases, Jan 2023. URL: https://doi.org/10.1186/s13023-023-02614-8, doi:10.1186/s13023-023-02614-8. This article has 12 citations and is from a peer-reviewed journal.
-
(irvine2024woodhousesakatisyndromewith pages 1-3): Rebecca Eilish Irvine and Arshia Ahmad. Woodhouse-sakati syndrome with no reportable mri findings: a case report. BMC Neurology, Sep 2024. URL: https://doi.org/10.1186/s12883-024-03865-z, doi:10.1186/s12883-024-03865-z. This article has 3 citations and is from a peer-reviewed journal.
-
(wakim2024woodhousesakatisyndromegenotype–phenotype pages 2-4): Victor Wakim, Mohammad El Dassouki, Ahlam Azar, Abeer Hani, Cybel Mehawej, Eliane Chouery, Marie-Jeanne Baroudi, and Gerard Wakim. Woodhouse-sakati syndrome: genotype–phenotype review and case of intra-familial heterogeneity. Journal of Rare Diseases, Jul 2024. URL: https://doi.org/10.1007/s44162-024-00045-y, doi:10.1007/s44162-024-00045-y. This article has 0 citations.
-
(bohlega2019patternsofneurological pages 2-3): Saeed Bohlega, Ali H. Abusrair, Fahad S. Al-Ajlan, Norah Alharbi, Abdulaziz Al-Semari, Balsam Bohlega, Dalya Abualsaud, and Fowzan Alkuraya. Patterns of neurological manifestations in woodhouse-sakati syndrome. Dec 2019. URL: https://doi.org/10.1016/j.parkreldis.2019.10.007, doi:10.1016/j.parkreldis.2019.10.007. This article has 26 citations and is from a peer-reviewed journal.
-
(irvine2024woodhousesakatisyndromewith pages 3-5): Rebecca Eilish Irvine and Arshia Ahmad. Woodhouse-sakati syndrome with no reportable mri findings: a case report. BMC Neurology, Sep 2024. URL: https://doi.org/10.1186/s12883-024-03865-z, doi:10.1186/s12883-024-03865-z. This article has 3 citations and is from a peer-reviewed journal.
-
(amalnath2024woodhouse–sakatisyndromein pages 1-3): S. Deepak Amalnath, Jothivanan, Junko Oshima, Jillian G. Buchan, and Sarah Paolucci. Woodhouse–sakati syndrome in an indian patient with a novel pathogenic variant. American Journal of Medical Genetics Part A, 194:100-102, Sep 2024. URL: https://doi.org/10.1002/ajmg.a.63405, doi:10.1002/ajmg.a.63405. This article has 4 citations.
-
(wakim2024woodhousesakatisyndromegenotype–phenotype pages 4-5): Victor Wakim, Mohammad El Dassouki, Ahlam Azar, Abeer Hani, Cybel Mehawej, Eliane Chouery, Marie-Jeanne Baroudi, and Gerard Wakim. Woodhouse-sakati syndrome: genotype–phenotype review and case of intra-familial heterogeneity. Journal of Rare Diseases, Jul 2024. URL: https://doi.org/10.1007/s44162-024-00045-y, doi:10.1007/s44162-024-00045-y. This article has 0 citations.
-
(kohil2023geneticepidemiologyof pages 6-7): Amira Kohil, Atiyeh M. Abdallah, Khalid Hussain, and Mashael Al-Shafai. Genetic epidemiology of woodhouse-sakati syndrome in the greater middle east region and beyond: a systematic review. Orphanet Journal of Rare Diseases, Jan 2023. URL: https://doi.org/10.1186/s13023-023-02614-8, doi:10.1186/s13023-023-02614-8. This article has 12 citations and is from a peer-reviewed journal.
-
(bohlega2019patternsofneurological pages 1-2): Saeed Bohlega, Ali H. Abusrair, Fahad S. Al-Ajlan, Norah Alharbi, Abdulaziz Al-Semari, Balsam Bohlega, Dalya Abualsaud, and Fowzan Alkuraya. Patterns of neurological manifestations in woodhouse-sakati syndrome. Dec 2019. URL: https://doi.org/10.1016/j.parkreldis.2019.10.007, doi:10.1016/j.parkreldis.2019.10.007. This article has 26 citations and is from a peer-reviewed journal.
-
(wakim2024woodhousesakatisyndromegenotype–phenotype pages 5-7): Victor Wakim, Mohammad El Dassouki, Ahlam Azar, Abeer Hani, Cybel Mehawej, Eliane Chouery, Marie-Jeanne Baroudi, and Gerard Wakim. Woodhouse-sakati syndrome: genotype–phenotype review and case of intra-familial heterogeneity. Journal of Rare Diseases, Jul 2024. URL: https://doi.org/10.1007/s44162-024-00045-y, doi:10.1007/s44162-024-00045-y. This article has 0 citations.
-
(kohil2023geneticepidemiologyof pages 4-6): Amira Kohil, Atiyeh M. Abdallah, Khalid Hussain, and Mashael Al-Shafai. Genetic epidemiology of woodhouse-sakati syndrome in the greater middle east region and beyond: a systematic review. Orphanet Journal of Rare Diseases, Jan 2023. URL: https://doi.org/10.1186/s13023-023-02614-8, doi:10.1186/s13023-023-02614-8. This article has 12 citations and is from a peer-reviewed journal.
-
(kohil2023geneticepidemiologyof media b5c57154): Amira Kohil, Atiyeh M. Abdallah, Khalid Hussain, and Mashael Al-Shafai. Genetic epidemiology of woodhouse-sakati syndrome in the greater middle east region and beyond: a systematic review. Orphanet Journal of Rare Diseases, Jan 2023. URL: https://doi.org/10.1186/s13023-023-02614-8, doi:10.1186/s13023-023-02614-8. This article has 12 citations and is from a peer-reviewed journal.
-
(kohil2023geneticepidemiologyof media 150533d1): Amira Kohil, Atiyeh M. Abdallah, Khalid Hussain, and Mashael Al-Shafai. Genetic epidemiology of woodhouse-sakati syndrome in the greater middle east region and beyond: a systematic review. Orphanet Journal of Rare Diseases, Jan 2023. URL: https://doi.org/10.1186/s13023-023-02614-8, doi:10.1186/s13023-023-02614-8. This article has 12 citations and is from a peer-reviewed journal.
-
(baigh2026woodhousesakatisyndromedue pages 2-4): ZH Baigh, JA Sheikh, BMO Dawar, Z Baigh, and BO Dawar. Woodhouse-sakati syndrome due to the rare dcaf17 c. 321+ 1g> a mutation: the second case report worldwide. Unknown journal, 2026.
-
(bakhsh2023thesuccessfulmanagement pages 6-8): Hanadi Bakhsh, Norah Alqntash, and Ebtesam Almajed. The successful management of primary amenorrhea in woodhouse–sakati syndrome: a case report and a literature review. Life, 13:2022, Oct 2023. URL: https://doi.org/10.3390/life13102022, doi:10.3390/life13102022. This article has 2 citations.
-
(khosravi2025clinicalandgenetic pages 1-3): Sepehr Khosravi, Toktam Moosavian, Shadab Salehpour, Seyed Amir Hassan Habibi, Afagh Alavi, and Mohammad Rohani. Clinical and genetic characterization of woodhouse-sakati syndrome in iranian patients: a case series. Journal of Movement Disorders, 18:257-261, Jul 2025. URL: https://doi.org/10.14802/jmd.25043, doi:10.14802/jmd.25043. This article has 0 citations and is from a peer-reviewed journal.
-
(wakim2024woodhousesakatisyndromegenotype–phenotype pages 7-8): Victor Wakim, Mohammad El Dassouki, Ahlam Azar, Abeer Hani, Cybel Mehawej, Eliane Chouery, Marie-Jeanne Baroudi, and Gerard Wakim. Woodhouse-sakati syndrome: genotype–phenotype review and case of intra-familial heterogeneity. Journal of Rare Diseases, Jul 2024. URL: https://doi.org/10.1007/s44162-024-00045-y, doi:10.1007/s44162-024-00045-y. This article has 0 citations.
-
(NCT05522374 chunk 1): Prof. Thomas Klopstock. TIRCON International NBIA Registry. LMU Klinikum. 2012. ClinicalTrials.gov Identifier: NCT05522374
-
(NCT05522374 chunk 2): Prof. Thomas Klopstock. TIRCON International NBIA Registry. LMU Klinikum. 2012. ClinicalTrials.gov Identifier: NCT05522374