TUBA1A-related Tubulinopathy
TUBA1A-related tubulinopathy is a malformation of cortical development caused by heterozygous, almost always de novo, missense mutations in TUBA1A, which encodes the brain-predominant alpha-1A tubulin isotype. Alpha- and beta-tubulin heterodimers polymerize into the microtubules that drive interkinetic nuclear migration of progenitors and the nucleokinesis of migrating neurons during corticogenesis. TUBA1A mutations cluster in functionally critical regions of the protein (including the GTP-binding pocket and the surfaces that contact beta-tubulin and microtubule-associated proteins), and they impair tubulin heterodimer formation, microtubule dynamics, and binding of motor and microtubule-associated proteins. The resulting defect in microtubule-dependent neuronal migration produces a coherent but broad alpha-tubulin cortical malformation spectrum: classical lissencephaly and pachygyria, lissencephaly with cerebellar hypoplasia (LCH), microlissencephaly, simplified gyration, and polymicrogyria-like cortical dysplasia, characteristically accompanied by dysmorphic basal ganglia, corpus callosum dysgenesis, and brainstem and cerebellar hypoplasia or dysplasia. Affected individuals typically have severe developmental and motor delay, intellectual disability, drug-resistant epilepsy, and frequently ataxia and ocular impairment. TUBA1A is the most commonly mutated tubulin gene in this group. It is modeled here as its own alpha-tubulin pathomechanism entry rather than lumped under generic lissencephaly or polymicrogyria, while remaining part of the broader tubulinopathy family that includes the beta-tubulin (TUBB2B, TUBB3, TUBB5) and gamma-tubulin (TUBG1) disorders.
Ask OpenScientist
Ask a research question about TUBA1A-related Tubulinopathy. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Discussions and Knowledge Gaps
1Show evidence (3 references)
Pathophysiology
3Show evidence (3 references)
Show evidence (2 references)
Show evidence (3 references)
Pathograph
Phenotypes
6Nervous System 3
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Other 3
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
1Show evidence (3 references)
Medical Actions
3Source YAML
click to showname: TUBA1A-related Tubulinopathy
creation_date: "2026-06-10T00:00:00Z"
category: Mendelian
disease_term:
preferred_term: lissencephaly due to TUBA1A mutation
term:
id: MONDO:0012703
label: lissencephaly due to TUBA1A mutation
description: >-
TUBA1A-related tubulinopathy is a malformation of cortical development caused
by heterozygous, almost always de novo, missense mutations in TUBA1A, which
encodes the brain-predominant alpha-1A tubulin isotype. Alpha- and beta-tubulin
heterodimers polymerize into the microtubules that drive interkinetic nuclear
migration of progenitors and the nucleokinesis of migrating neurons during
corticogenesis. TUBA1A mutations cluster in functionally critical regions of
the protein (including the GTP-binding pocket and the surfaces that contact
beta-tubulin and microtubule-associated proteins), and they impair tubulin
heterodimer formation, microtubule dynamics, and binding of motor and
microtubule-associated proteins. The resulting defect in microtubule-dependent
neuronal migration produces a coherent but broad alpha-tubulin cortical
malformation spectrum: classical lissencephaly and pachygyria, lissencephaly
with cerebellar hypoplasia (LCH), microlissencephaly, simplified gyration,
and polymicrogyria-like cortical dysplasia, characteristically accompanied by
dysmorphic basal ganglia, corpus callosum dysgenesis, and brainstem and
cerebellar hypoplasia or dysplasia. Affected individuals typically have severe
developmental and motor delay, intellectual disability, drug-resistant
epilepsy, and frequently ataxia and ocular impairment. TUBA1A is the most
commonly mutated tubulin gene in this group. It is modeled here as its own
alpha-tubulin pathomechanism entry rather than lumped under generic
lissencephaly or polymicrogyria, while remaining part of the broader
tubulinopathy family that includes the beta-tubulin (TUBB2B, TUBB3, TUBB5)
and gamma-tubulin (TUBG1) disorders.
parents:
- congenital nervous system disorder
- disorder of development or morphogenesis
- hereditary neurological disease
- neuronal migration disorder
references:
- reference: PMID:17218254
title: "Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans."
- reference: PMID:17584854
title: "Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A)."
- reference: PMID:20466733
title: "TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins."
- reference: PMID:24860126
title: "The wide spectrum of tubulinopathies: what are the key features for the diagnosis?"
- reference: PMID:28111201
title: Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia.
pathophysiology:
- name: Altered Alpha-Tubulin (TUBA1A) Function
description: >-
Heterozygous de novo missense mutations in TUBA1A alter the brain-predominant
alpha-1A tubulin subunit. Disease-associated substitutions map to functionally
critical regions of the protein, including the GTP-binding pocket, and impair
tubulin heterodimer formation and microtubule assembly. Additional
mutations disrupt the binding surfaces for microtubule-associated proteins,
so that the alpha-tubulin lesion compromises microtubule structure and the
docking of the regulatory and motor proteins that act on microtubules. This
establishes altered alpha-tubulin function as the initiating molecular lesion
of the malformation.
conforms_to: microtubule_dependent_neuronal_migration_failure#Microtubule Apparatus Perturbation
cell_types:
- preferred_term: cortical progenitor and migrating neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: tubulin heterodimer assembly
term:
id: GO:0007021
label: tubulin complex assembly
modifier: DECREASED
- preferred_term: microtubule cytoskeleton organization
term:
id: GO:0000226
label: microtubule cytoskeleton organization
modifier: DYSREGULATED
- preferred_term: microtubule-based process
term:
id: GO:0007017
label: microtubule-based process
modifier: DYSREGULATED
evidence:
- reference: PMID:17218254
reference_title: "Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
the causative mutation lies in the guanosine triphosphate (GTP) binding
pocket of alpha-1 tubulin (Tuba1) and affects tubulin heterodimer
formation
explanation: >-
Identifies the GTP-binding pocket of alpha-1 tubulin as the site of the
causative mutation and shows that it impairs tubulin heterodimer
formation, the molecular lesion underlying TUBA1A disease.
- reference: PMID:20466733
reference_title: "TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Tubulin alpha1A (TUBA1A), encoding a critical structural subunit of
microtubules, has recently been implicated in LIS
explanation: >-
Establishes TUBA1A as encoding a critical structural subunit of
microtubules implicated in lissencephaly.
- reference: PMID:20466733
reference_title: "TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
LIS-associated mutations of TUBA1A operate via diverse mechanisms that
include disruption of binding sites for microtubule-associated proteins
(MAPs)
explanation: >-
Cellular and structural data show that TUBA1A mutations act through
diverse mechanisms including disruption of microtubule-associated protein
binding sites.
downstream:
- target: Impaired Microtubule-Dependent Neuronal Migration
description: >-
Defective alpha-tubulin and microtubule function impairs the
microtubule-dependent nucleokinesis and translocation required for
cortical neuronal migration.
- name: Impaired Microtubule-Dependent Neuronal Migration
description: >-
Microtubules generated from TUBA1A-containing heterodimers drive the
interkinetic nuclear migration of progenitors and the nucleokinesis of
migrating neurons. Because TUBA1A is expressed at high levels throughout
central nervous system development, the alpha-tubulin defect impairs
microtubule-dependent neuronal migration. The phenotype recapitulates that
of Doublecortin (DCX) and LIS1 deficiency, reflecting the functional
importance of the microtubule/DCX migration machinery on which the
alpha-tubulin lesion converges.
conforms_to: microtubule_dependent_neuronal_migration_failure#Microtubule-Based Neuronal Motility Failure
cell_types:
- preferred_term: migrating cortical neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: neuron migration
term:
id: GO:0001764
label: neuron migration
modifier: DECREASED
- preferred_term: microtubule-based movement
term:
id: GO:0007018
label: microtubule-based movement
modifier: DYSREGULATED
evidence:
- reference: PMID:17218254
reference_title: "Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
abnormalities in the laminar architecture of the hippocampus and cortex,
accompanied by impaired neuronal migration
explanation: >-
The ENU-induced alpha-tubulin mouse mutant shows impaired neuronal
migration with abnormal hippocampal and cortical lamination, the model
phenotype that prompted screening of human migration disorders.
- reference: PMID:17584854
reference_title: "Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
highlight the importance of the MTs/DCX complex in the neuronal migration
process
explanation: >-
Frames TUBA1A disease within the microtubule/DCX migration machinery,
consistent with impaired microtubule-dependent neuronal migration as the
core mechanism.
downstream:
- target: Cortical Dyslamination and Lissencephaly Spectrum
description: >-
Failed neuronal migration leaves neurons mispositioned, producing the
smooth or abnormally folded, abnormally laminated cortex of the
malformation spectrum.
- name: Cortical Dyslamination and Lissencephaly Spectrum
description: >-
Impaired migration of cortical neurons disrupts the normal six-layered
architecture of the neocortex, producing a coherent but broad alpha-tubulin
malformation spectrum that ranges from microlissencephaly and classical
lissencephaly through pachygyria and simplified gyration to
polymicrogyria-like cortical dysplasia. The malformation is characteristically
accompanied by dysmorphic basal ganglia, corpus callosum dysgenesis, and
cerebellar and brainstem hypoplasia or dysplasia, reflecting the shared
requirement for microtubule function across these developing structures.
conforms_to: microtubule_dependent_neuronal_migration_failure#Cortical Dyslamination and Neuronal Ectopia
cell_types:
- preferred_term: cortical neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: cerebral cortex development
term:
id: GO:0021987
label: cerebral cortex development
modifier: DYSREGULATED
- preferred_term: neuron migration
term:
id: GO:0001764
label: neuron migration
modifier: DECREASED
evidence:
- reference: PMID:24860126
reference_title: "The wide spectrum of tubulinopathies: what are the key features for the diagnosis?"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
commonly referred to as tubulinopathies, are a heterogeneous group of
conditions with a wide spectrum of clinical severity
explanation: >-
Establishes the tubulinopathies, including TUBA1A, as a heterogeneous
group of cortical malformation conditions with a wide spectrum of
severity.
- reference: PMID:24860126
reference_title: "The wide spectrum of tubulinopathies: what are the key features for the diagnosis?"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The core phenotype of TUBA1A and TUBG1 tubulinopathies are
lissencephalies and microlissencephalies
explanation: >-
Identifies lissencephaly and microlissencephaly as the core cortical
malformation phenotype of TUBA1A tubulinopathy.
- reference: PMID:20466733
reference_title: "TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We identified novel and recurrent TUBA1A mutations in approximately 1% of
children with classic LIS and in approximately 30% of children with LCH,
making this the first major gene associated with the rare LCH phenotype
explanation: >-
Quantifies the contribution of TUBA1A mutations to classic lissencephaly
and to lissencephaly with cerebellar hypoplasia, defining the breadth of
the cortical dyslamination spectrum.
phenotypes:
- name: Lissencephaly
description: >-
Smooth brain (agyria/pachygyria) from arrested neuronal migration is the
core cortical malformation of TUBA1A tubulinopathy, ranging to
microlissencephaly at the severe end of the spectrum.
phenotype_term:
preferred_term: Lissencephaly
term:
id: HP:0001339
label: Lissencephaly
evidence:
- reference: PMID:24860126
reference_title: "The wide spectrum of tubulinopathies: what are the key features for the diagnosis?"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The core phenotype of TUBA1A and TUBG1 tubulinopathies are
lissencephalies and microlissencephalies
explanation: >-
Identifies lissencephaly/microlissencephaly as the core phenotype of
TUBA1A tubulinopathy.
- reference: PMID:20466733
reference_title: "TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We identified novel and recurrent TUBA1A mutations in approximately 1% of
children with classic LIS and in approximately 30% of children with LCH
explanation: >-
Documents TUBA1A mutations in classic lissencephaly and in lissencephaly
with cerebellar hypoplasia cohorts.
- name: Pachygyria
description: >-
Abnormally broad, thick gyri with shallow sulci are part of the TUBA1A
cortical dysgenesis spectrum, intermediate between agyria and normal
gyration.
phenotype_term:
preferred_term: Pachygyria
term:
id: HP:0001302
label: Pachygyria
evidence:
- reference: PMID:17584854
reference_title: "Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
the identification of TUBA1A mutations in two patients with lissencephaly
and pachygyria, respectively
explanation: >-
Documents pachygyria as part of the TUBA1A cortical malformation
spectrum.
- name: Cerebellar Hypoplasia
description: >-
A disproportionately small cerebellum, frequently with cerebellar dysplasia,
is a characteristic accompaniment of TUBA1A lissencephaly and defines the
lissencephaly-with-cerebellar-hypoplasia (LCH) subgroup.
phenotype_term:
preferred_term: Cerebellar hypoplasia
term:
id: HP:0001321
label: Cerebellar hypoplasia
evidence:
- reference: PMID:24860126
reference_title: "The wide spectrum of tubulinopathies: what are the key features for the diagnosis?"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
mild to severe cerebellar hypoplasia and dysplasia (63/80; 78.7%)
explanation: >-
Documents cerebellar hypoplasia and dysplasia in the great majority of
tubulinopathy patients.
- reference: PMID:20466733
reference_title: "TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
in approximately 30% of children with LCH, making this the first major
gene associated with the rare LCH phenotype
explanation: >-
Establishes TUBA1A as the first major gene of lissencephaly with
cerebellar hypoplasia, linking the gene to the cerebellar phenotype.
- name: Agenesis of the Corpus Callosum
description: >-
Partial or complete corpus callosum dysgenesis is a frequent commissural
abnormality in TUBA1A tubulinopathy and can occur even without overt
lissencephaly.
phenotype_term:
preferred_term: Agenesis of corpus callosum
term:
id: HP:0001274
label: Agenesis of corpus callosum
evidence:
- reference: PMID:24860126
reference_title: "The wide spectrum of tubulinopathies: what are the key features for the diagnosis?"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
high prevalence of corpus callosum agenesis (32/80; 40%)
explanation: >-
Documents corpus callosum agenesis in a high proportion of tubulinopathy
patients.
- reference: PMID:20466733
reference_title: "TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
a TUBA1A mutation in one child with agenesis of the corpus callosum and
cerebellar hypoplasia without LIS
explanation: >-
Documents corpus callosum agenesis with cerebellar hypoplasia as a TUBA1A
phenotype that can occur in the absence of lissencephaly.
- name: Dysmorphic Basal Ganglia
description: >-
Dysmorphic basal ganglia, reflecting impaired migration and morphogenesis of
the deep grey nuclei, are a characteristic and highly prevalent imaging
hallmark of the tubulinopathies including TUBA1A.
phenotype_term:
preferred_term: Abnormal basal ganglia morphology
term:
id: HP:0002134
label: Abnormal basal ganglia morphology
evidence:
- reference: PMID:24860126
reference_title: "The wide spectrum of tubulinopathies: what are the key features for the diagnosis?"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Dysmorphic basal ganglia are the hallmark of tubulinopathies (found in
75% of cases)
explanation: >-
Establishes dysmorphic basal ganglia as the imaging hallmark of
tubulinopathies, present in the majority of cases.
- name: Brainstem Abnormalities
description: >-
Brainstem hypoplasia or dysplasia accompanies the cortical malformation,
part of the infratentorial involvement characteristic of TUBA1A
tubulinopathy.
phenotype_term:
preferred_term: Abnormal brainstem morphology
term:
id: HP:0002363
label: Abnormal brainstem morphology
evidence:
- reference: PMID:17584854
reference_title: "Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
patients with TUBA1A mutations share not only cortical dysgenesis, but
also cerebellar, hippocampal, corpus callosum, and brainstem
abnormalities
explanation: >-
Documents brainstem (and cerebellar, hippocampal, callosal) abnormalities
as shared features of patients with TUBA1A mutations.
genetic:
- name: TUBA1A
association: Causative
gene_term:
preferred_term: TUBA1A (alpha-1A tubulin)
term:
id: hgnc:20766
label: TUBA1A
evidence:
- reference: PMID:17218254
reference_title: "Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We identified two patients with de novo mutations in TUBA3, the human
homolog of Tuba1
explanation: >-
Founding report identifying de novo alpha-tubulin (TUBA1A, then named
TUBA3) mutations as a cause of human lissencephaly.
- reference: PMID:17584854
reference_title: "Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The de novo occurrence was shown for all mutations
explanation: >-
Confirms the de novo occurrence of TUBA1A mutations, consistent with a
dominant, typically sporadic mechanism.
- reference: PMID:24860126
reference_title: "The wide spectrum of tubulinopathies: what are the key features for the diagnosis?"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
45 were found to carry mutations in TUBA1A (42.5%)
explanation: >-
Establishes TUBA1A as the most commonly mutated tubulin gene among
patients with complex cortical malformations.
treatments:
- name: Anti-Seizure Medication
description: >-
Symptomatic management of the frequently drug-resistant epilepsy associated
with TUBA1A tubulinopathy using standard anti-seizure medications selected by
seizure type. No disease-modifying therapy exists; management is supportive.
treatment_term:
preferred_term: pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
- name: Supportive and Rehabilitative Care
description: >-
Multidisciplinary supportive care including physical, occupational and
developmental therapies for the severe motor and intellectual impairment.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
- name: Genetic Counseling
description: >-
Genetic counseling for families, noting that TUBA1A mutations are almost
always de novo with low recurrence risk, while germline mosaicism can
occasionally cause recurrence.
treatment_term:
preferred_term: Genetic Counseling
term:
id: NCIT:C15240
label: Genetic Counseling
discussions:
- discussion_id: gap_tuba1a_human_organoid_translatability
prompt: >-
Which TUBA1A variant effects on microtubule heterodimer formation, neuronal
migration, and progenitor behavior are conserved across biochemical or mouse
systems, and which require human iPSC-derived cortical organoids or fetal
tissue benchmarks to resolve?
kind: HUMAN_MODEL_MISMATCH
status: OPEN
attaches_to:
- pathophysiology#Altered Alpha-Tubulin (TUBA1A) Function
- pathophysiology#Impaired Microtubule-Dependent Neuronal Migration
- pathophysiology#Cortical Dyslamination and Lissencephaly Spectrum
rationale: >-
The TUBA1A pathograph is supported by mouse neuronal-migration phenotypes,
human clinical genetics, and cellular/structural assays, but human cortical
expansion depends on outer radial glia and fetal cortical organization that
are not fully represented in lissencephalic rodents. A TUBA1A-specific
new-approach-model experiment is needed to test whether the alpha-tubulin
mechanism is purely post-mitotic neuronal motility failure or also includes
human progenitor and outer-radial-glia vulnerability.
evidence:
- reference: PMID:17218254
reference_title: "Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Phenotypic similarity with existing mouse models of lissencephaly led us
to screen a cohort of patients with developmental brain anomalies.
explanation: >-
The founding TUBA1A paper explicitly moves from a mouse migration model to
human patient screening, making model-to-human translatability part of the
evidentiary bridge.
- reference: PMID:28111201
reference_title: Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia.
supports: SUPPORT
evidence_source: OTHER
snippet: >-
However, the mouse brain is naturally lissencephalic, suggesting that
certain aspects of cortical development may not be adequately assessed in
mice.
explanation: >-
Supports treating rodent-to-human translation as an explicit knowledge gap
for lissencephaly mechanisms.
- reference: PMID:28111201
reference_title: Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
We saw a cell migration defect that was rescued when we corrected the MDS
causative chromosomal deletion
explanation: >-
Provides precedent that human iPSC-derived cerebral organoids can measure
and rescue a lissencephaly-relevant migration defect.
proposed_experiments:
- experiment_id: exp_tuba1a_isogenic_cortical_organoid_migration
name: TUBA1A isogenic cortical-organoid migration experiment
description: >-
Engineer recurrent TUBA1A missense variants into human iPSCs, correct
patient-derived variants isogenically where available, and compare
cortical organoid neuronal migration, radial-glial organization,
microtubule dynamics, and outer-radial-glia mitosis against mouse and
biochemical expectations.
experiment_type:
preferred_term: patient-derived cortical organoid perturbation experiment
model_systems:
- name: TUBA1A human iPSC-derived cortical organoid
description: >-
Three-dimensional human cortical organoid carrying a pathogenic TUBA1A
variant, with isogenic corrected and knock-in controls.
experimental_model_type: ORGANOID
namo_type: namo:Organoid
organism:
preferred_term: human
term:
id: NCBITaxon:9606
label: Homo sapiens
tissue_term:
preferred_term: cerebral cortex
term:
id: UBERON:0000956
label: cerebral cortex
cell_types:
- preferred_term: radial glial cell
term:
id: CL:0000681
label: radial glial cell
- preferred_term: migrating cortical neuron
term:
id: CL:0000540
label: neuron
conditions:
- TUBA1A-related tubulinopathy
- lissencephaly
- microtubule-dependent neuronal migration failure
cell_source: Patient-derived or CRISPR-engineered human induced pluripotent stem cells
culture_system: Three-dimensional cortical organoid with live-imaging migration assays
perturbations:
- name: TUBA1A variant correction or knock-in
target: pathophysiology#Altered Alpha-Tubulin (TUBA1A) Function
genes:
- preferred_term: TUBA1A
term:
id: hgnc:20766
label: TUBA1A
description: >-
Correct a patient TUBA1A variant or introduce a recurrent missense
variant into an isogenic human iPSC background.
readouts:
- name: Microtubule apparatus and heterodimer function
target: pathophysiology#Altered Alpha-Tubulin (TUBA1A) Function
biological_processes:
- preferred_term: tubulin complex assembly
term:
id: GO:0007021
label: tubulin complex assembly
modifier: DECREASED
- preferred_term: microtubule cytoskeleton organization
term:
id: GO:0000226
label: microtubule cytoskeleton organization
modifier: DYSREGULATED
assays:
- preferred_term: tubulin heterodimer assembly assay
- preferred_term: microtubule polymerization assay
direction: NEGATIVE
- name: Live-imaging cortical neuron migration
target: pathophysiology#Impaired Microtubule-Dependent Neuronal Migration
biological_processes:
- preferred_term: neuron migration
term:
id: GO:0001764
label: neuron migration
modifier: DECREASED
assays:
- preferred_term: live-cell imaging assay
direction: NEGATIVE
- name: Outer radial glial mitotic behavior
target: pathophysiology#Altered Alpha-Tubulin (TUBA1A) Function
biological_processes:
- preferred_term: neurogenesis
term:
id: GO:0022008
label: neurogenesis
modifier: DYSREGULATED
assays:
- preferred_term: time-lapse microscopy
- preferred_term: single-cell transcriptomic profiling
direction: POSITIVE
controls:
- name: Isogenic corrected organoids
description: Variant-corrected patient-derived organoids differentiated in parallel.
- name: Isogenic knock-in organoids
description: Wild-type-background organoids carrying the introduced TUBA1A variant.
decision_criterion: >-
A conserved TUBA1A microtubule-migration skeleton is strengthened if
mutant organoids reproduce reduced migration and microtubule defects that
are rescued by correction and reproduced by knock-in. A subtype-specific
human branch is supported if organoids reveal outer-radial-glia mitotic or
progenitor-output defects not predicted by mouse migration models alone.
would_support:
- pathophysiology#Altered Alpha-Tubulin (TUBA1A) Function
- pathophysiology#Impaired Microtubule-Dependent Neuronal Migration
- pathophysiology#Cortical Dyslamination and Lissencephaly Spectrum
notes: >-
Entry created from cortical-malformation epic 4098 (issue 4083), seeded from
Romero, Bahi-Buisson & Francis 2018 (Sem Cell Dev Biol 76:33-75). Modeled as a
coherent alpha-tubulin (TUBA1A) microtubule-dependent neuronal migration
pathomechanism rather than lumped under generic lissencephaly or
polymicrogyria, per the epic's mechanism-skeleton entry-boundary rule. TUBA1A
is deliberately split from the beta-tubulin (TUBB2B/TUBB3/TUBB5) and
gamma-tubulin (TUBG1) tubulinopathies because the alpha-tubulin
genotype-phenotype pattern (lissencephaly/microlissencephaly core) is distinct,
while all share the tubulin/microtubule skeleton. Clinical features that are
well established but not given a quotable abstract snippet in the cited
cohort/mechanistic papers — severe intellectual disability, motor delay,
drug-resistant epilepsy, ataxia, and ocular impairment — are summarized in the
description rather than asserted as evidenced phenotypes, pending sources with
exact quotable text. The three core nodes now conform to the
microtubule-dependent neuronal migration module while retaining this entry's
TUBA1A-specific alpha-tubulin molecular trigger and lissencephaly with
cerebellar hypoplasia phenotype emphasis.
References & Deep Research
References
5Deep Research
1Question: You are an expert researcher providing comprehensive, well-cited information.
Provide detailed information focusing on: 1. Key concepts and definitions with current understanding 2. Recent developments and latest research (prioritize 2023-2024 sources) 3. Current applications and real-world implementations 4. Expert opinions and analysis from authoritative sources 5. Relevant statistics and data from recent studies
Format as a comprehensive research report with proper citations. Include URLs and publication dates where available. Always prioritize recent, authoritative sources and provide specific citations for all major claims.
Disease Characteristics Research Template
Target Disease
- Disease Name: TUBA1A-related Tubulinopathy
- MONDO ID: (if available)
- Category: Mendelian
Research Objectives
Please provide a comprehensive research report on TUBA1A-related Tubulinopathy covering all of the disease characteristics listed below. This report will be used to populate a disease knowledge base entry. Be thorough and cite primary literature (PMID preferred) for all claims.
For each section, suggested databases/resources are listed. These are the first places you should search for information on each topic.
1. Disease Information
Search first: OMIM, Orphanet, ICD-10/ICD-11, MeSH, PubMed
- What is the disease? Provide a concise overview.
- What are the key identifiers? (OMIM, Orphanet, ICD-10/ICD-11, MeSH, Mondo)
- What are the common synonyms and alternative names?
- Is the information derived from individual patients (e.g., EHR) or aggregated disease-level resources?
2. Etiology
- Disease Causal Factors: What are the primary causes? (genetic, environmental, infectious, mechanistic)
- Risk Factors:
Search first: PubMed, Cochrane Library, UpToDate, clinical guidelines, ClinVar, ClinGen, GWAS Catalog, PheGenI, CTD, CDC, WHO, epidemiological databases
- Genetic risk factors (causal variants, susceptibility loci, modifier genes)
- Environmental risk factors (toxins, lifestyle, occupational exposures, age, sex, family history)
- Protective Factors:
Search first: PubMed, Cochrane Library, clinical trial databases, GWAS Catalog, gnomAD, WHO, CDC, nutrition databases
- Genetic protective factors (protective variants, modifier alleles)
- Environmental protective factors (diet, lifestyle, exposures that reduce risk)
- Gene-Environment Interactions: How do genetic and environmental factors interact to influence disease?
Search first: CTD, PubMed, PheGenI, GxE databases
3. Phenotypes
Search first: HPO (Human Phenotype Ontology), OMIM, Orphanet, PubMed, clinicaltrials.gov, MedDRA, SNOMED CT, DECIPHER, LOINC
For each phenotype, provide: - Phenotype type: symptoms, clinical signs, physical manifestations, behavioral changes, or laboratory abnormalities
For symptoms/signs: HPO, OMIM, Orphanet, PubMed For behavioral changes: HPO, DSM, RDoC (Research Domain Criteria), PubMed For laboratory abnormalities: LOINC, SNOMED CT, LabTests Online, PubMed - Phenotype characteristics: Search first: OMIM, Orphanet, HPO, PubMed - Age of symptom onset (neonatal, childhood, adult-onset, late-onset) - Symptom severity (mild, moderate, severe, variable) - Symptom progression (stable, progressive, episodic, fluctuating) - Frequency among affected individuals (percentage or qualitative) - Quality of life impact: Effects on daily functioning and well-being (per-phenotype when possible) Search first: EQ-5D database, SF-36, WHO QOL databases, PubMed - Suggest HPO (Human Phenotype Ontology) terms for each phenotype
4. Genetic/Molecular Information
- Causal Genes: Gene mutations or chromosomal abnormalities responsible for disease (gene symbols, OMIM IDs)
Search first: OMIM, ClinVar, HGMD, Ensembl, NCBI Gene
- Pathogenic Variants:
- Affected genes (gene symbols, HGNC IDs) > Search first: OMIM, NCBI Gene, Ensembl, HGNC, UniProt, GeneCards
- Variant classification (pathogenic, likely pathogenic, VUS per ACMG/AMP guidelines) > Search first: ClinVar, ClinGen, ACMG/AMP guidelines, VarSome
- Variant type/class (missense, frameshift, nonsense, splice-site, structural)
- Allele frequency in population databases > Search first: gnomAD, 1000 Genomes, ExAC, TOPMed, dbSNP
- Somatic vs germline origin > Search first: COSMIC (somatic), ClinVar, ICGC, TCGA
- Functional consequences (loss of function, gain of function, dominant negative)
- Modifier Genes: Genes that modify disease severity or expression
- Epigenetic Information: DNA methylation, histone modifications, chromatin changes affecting disease
Search first: ENCODE, Roadmap Epigenomics, MethBase, DiseaseMeth
- Chromosomal Abnormalities: Large-scale genetic changes (aneuploidy, translocations, inversions)
Search first: DECIPHER, ClinVar, ECARUCA, UCSC Genome Browser
5. Environmental Information
- Environmental Factors: Non-genetic contributing factors (toxins, radiation, pollution, occupational exposure)
Search first: CTD (Comparative Toxicogenomics Database), TOXNET, PubMed, EPA databases
- Lifestyle Factors: Behavioral factors (smoking, diet, exercise, alcohol consumption)
Search first: CDC databases, WHO, PubMed, NHANES
- Infectious Agents: If applicable, pathogens causing or triggering disease (bacteria, viruses, fungi, parasites)
Search first: NCBI Taxonomy, ViPR, BV-BRC, MicrobeDB, GIDEON
6. Mechanism / Pathophysiology
- Molecular Pathways: Specific signaling cascades or biochemical pathways involved (Wnt, MAPK, mTOR, PI3K-AKT, etc.)
Search first: KEGG, Reactome, WikiPathways, PathBank, BioCyc
- Cellular Processes: Cell-level mechanisms (apoptosis, autophagy, cell cycle dysregulation, inflammation, etc.)
Search first: Gene Ontology (GO), Reactome, KEGG, PubMed
- Protein Dysfunction: How protein structure or function is altered (misfolding, aggregation, loss of function, gain of function)
Search first: UniProt, PDB (Protein Data Bank), InterPro, Pfam, AlphaFold
- Metabolic Changes: Alterations in metabolic processes (energy metabolism, lipid metabolism, amino acid metabolism)
Search first: KEGG, BioCyc, HMDB (Human Metabolome Database), BRENDA
- Immune System Involvement: Role of immune response (autoimmunity, immunodeficiency, chronic inflammation)
Search first: ImmPort, Immunome Database, IEDB, Gene Ontology
- Tissue Damage Mechanisms: How tissues/ are injured (oxidative stress, ischemia, fibrosis, necrosis)
Search first: PubMed, Gene Ontology, Reactome
- Biochemical Abnormalities: Specific molecular defects (enzyme deficiencies, receptor dysfunction, ion channel defects)
Search first: BRENDA, UniProt, KEGG, OMIM, PubMed
- Epigenetic Changes: DNA methylation, histone modifications affecting gene expression in disease
Search first: ENCODE, Roadmap Epigenomics, MethBase, DiseaseMeth
- Molecular Profiling (if available):
- Transcriptomics/gene expression changes > Search first: GEO (Gene Expression Omnibus), ArrayExpress, GTEx, Human Cell Atlas, SRA
- Proteomics findings > Search first: PRIDE, ProteomeXchange, Human Protein Atlas, STRING, BioGRID
- Metabolomics signatures > Search first: MetaboLights, Metabolomics Workbench, HMDB, METLIN
- Lipidomics alterations > Search first: LIPID MAPS, SwissLipids, LipidHome, Metabolomics Workbench
- Genomic structural features > Search first: UCSC Genome Browser, Ensembl, NCBI, dbVar, DGV
- Advanced Technologies (if applicable):
- Single-cell analysis findings (cell-type specific mechanisms, cellular heterogeneity) > Search first: Human Cell Atlas, Single Cell Portal, GEO, CELLxGENE
- Spatial transcriptomics findings > Search first: GEO, Spatial Research, Vizgen, 10x Genomics data
- Multi-omics integration results > Search first: TCGA, ICGC, cBioPortal, LinkedOmics, PubMed
- Functional genomics screens (CRISPR, RNAi) > Search first: DepMap, GenomeRNAi, PubMed, BioGRID ORCS
For each mechanism, describe: - The causal chain from initial trigger to clinical manifestation - Which mechanisms are upstream vs downstream - What cell types and biological processes are involved - Suggest GO terms for biological processes and CL terms for cell types
7. Anatomical Structures Affected
- Organ Level:
- Primary organs directly affected
- Secondary organ involvement (complications, secondary effects)
- Body systems involved (cardiovascular, nervous, digestive, respiratory, endocrine, etc.)
Search first: Uberon, FMA (Foundational Model of Anatomy), OMIM, HPO, ICD-11, MeSH, SNOMED CT
- Tissue and Cell Level:
- Specific tissue types affected (epithelial, connective, muscle, nervous)
- Specific cell populations targeted (with Cell Ontology terms)
Search first: Uberon, Human Protein Atlas, Cell Ontology, Human Cell Atlas, CellMarker, PanglaoDB
- Subcellular Level:
- Cellular compartments involved (mitochondria, nucleus, ER, lysosomes) (with GO Cellular Component terms)
Search first: Gene Ontology (Cellular Component), UniProt, Human Protein Atlas
- Localization:
- Specific anatomical sites (with UBERON terms) > Search first: FMA, Uberon, NeuroNames (for brain), SNOMED CT
- Lateralization (unilateral, bilateral, asymmetric) > Search first: HPO, clinical literature, imaging databases
8. Temporal Development
- Onset:
- Typical age of onset (congenital, pediatric, adult, geriatric)
- Onset pattern (acute, subacute, chronic, insidious)
Search first: OMIM, Orphanet, HPO, PubMed
- Progression:
- Disease stages (early, intermediate, advanced, end-stage) > Search first: Cancer Staging Manual (AJCC), WHO classifications, PubMed
- Progression rate (rapid, slow, variable)
- Disease course pattern (episodic, relapsing-remitting, progressive, stable)
- Disease duration (self-limited, chronic lifelong)
Search first: Disease registries, longitudinal cohort databases, natural history studies, PubMed, Orphanet, OMIM
- Patterns:
- Remission patterns (spontaneous, treatment-induced) > Search first: Clinical trial databases, disease registries, PubMed
- Critical periods (time windows of vulnerability or opportunity for intervention) > Search first: PubMed, developmental biology databases, clinical guidelines
9. Inheritance and Population
- Epidemiology:
- Prevalence (cases per 100,000 at given time)
- Incidence (new cases per 100,000 per year)
Search first: Orphanet, CDC, WHO, GBD (Global Burden of Disease), national registries, SEER, disease registries
- For Genetic Etiology:
- Inheritance pattern (AD, AR, X-linked, mitochondrial, multifactorial, polygenic) > Search first: OMIM, Orphanet, ClinVar, GTR (Genetic Testing Registry)
- Penetrance (complete, incomplete, age-dependent) > Search first: ClinVar, OMIM, PubMed, ClinGen
- Expressivity (variable, consistent) > Search first: OMIM, ClinVar, PubMed
- Genetic anticipation (increasing severity in successive generations) > Search first: OMIM, PubMed (especially for repeat expansion disorders)
- Germline mosaicism > Search first: ClinVar, OMIM, genetic counseling literature, PubMed
- Founder effects (population-specific mutations) > Search first: gnomAD, population genetics databases, PubMed
- Consanguinity role > Search first: OMIM, population studies, genetic counseling resources
- Carrier frequency > Search first: gnomAD, carrier screening databases, GeneReviews, GTR
- Population Demographics:
- Affected populations (ethnic or demographic groups with higher prevalence) > Search first: gnomAD, 1000 Genomes, PAGE Study, PubMed, population registries
- Geographic distribution (endemic areas, regional variation) > Search first: WHO, CDC, GBD, Orphanet, geographic epidemiology databases
- Geographic distribution of specific variants
- Sex ratio (male:female) > Search first: Disease registries, OMIM, PubMed, epidemiological databases
- Age distribution of affected individuals > Search first: CDC, disease registries, SEER, Orphanet
10. Diagnostics
- Clinical Tests:
- Laboratory tests (blood, urine, tissue chemistry, specific enzyme assays) > Search first: LOINC, LabTests Online, PubMed
- Biomarkers (proteins, metabolites, genetic markers, circulating biomarkers) > Search first: FDA Biomarker List, BEST (Biomarkers, EndpointS, and other Tools), PubMed
- Imaging studies (X-ray, CT, MRI, PET, ultrasound) > Search first: RadLex, DICOM, Radiopaedia, imaging databases
- Functional tests (pulmonary function, cardiac stress tests) > Search first: LOINC, clinical guidelines, PubMed
- Electrophysiology (EEG, EMG, ECG, nerve conduction studies) > Search first: LOINC, clinical neurophysiology databases, PubMed
- Biopsy findings (histopathology, immunohistochemistry) > Search first: SNOMED CT, College of American Pathologists resources, PubMed
- Pathology findings (microscopic examination) > Search first: SNOMED CT, Digital Pathology databases, PubMed
- Genetic Testing:
Search first: GTR (Genetic Testing Registry), GeneReviews, ClinGen
- Overview of recommended genetic testing approach
- Whole genome sequencing (WGS) utility > Search first: GTR, ClinVar, GEL (Genomics England), gnomAD
- Whole exome sequencing (WES) utility > Search first: GTR, ClinVar, OMIM, GeneMatcher
- Gene panels (which panels, which genes) > Search first: GTR, ClinVar, laboratory-specific databases
- Single gene testing > Search first: GTR, ClinVar, OMIM, GeneReviews
- Chromosomal microarray (CMA) > Search first: DECIPHER, ClinVar, dbVar, ECARUCA
- Karyotyping > Search first: Chromosome Abnormality Database, ClinVar, cytogenetics resources
- FISH > Search first: ClinVar, cytogenetics databases, PubMed
- Mitochondrial DNA testing > Search first: MITOMAP, MSeqDR, ClinVar, GTR
- Repeat expansion testing > Search first: GTR, ClinVar, repeat expansion databases, PubMed
- Omics-Based Diagnostics (if applicable):
- RNA sequencing / transcriptomics > Search first: GEO, ArrayExpress, GTEx, RNA-seq databases
- Proteomics > Search first: PRIDE, ProteomeXchange, FDA Biomarker database
- Metabolomics > Search first: MetaboLights, Metabolomics Workbench, HMDB
- Epigenomics > Search first: GEO, ENCODE, Roadmap Epigenomics, MethBase
- Liquid biopsy > Search first: COSMIC, ClinVar, liquid biopsy databases, PubMed
- Clinical Criteria:
- Standardized diagnostic criteria (DSM, ICD, society guidelines) > Search first: DSM-5, ICD-11, clinical society guidelines, UpToDate
- Differential diagnosis (other conditions to rule out, with distinguishing features) > Search first: DynaMed, UpToDate, clinical decision support systems
- Screening:
- Screening methods for asymptomatic individuals (newborn screening, carrier screening, cascade screening) > Search first: ACMG recommendations, CDC newborn screening, GTR
11. Outcome/Prognosis
- Survival and Mortality:
- Survival rate (5-year, 10-year, overall) > Search first: SEER, cancer registries, disease-specific registries, PubMed
- Life expectancy (with and without treatment if applicable) > Search first: Orphanet, disease registries, actuarial databases, PubMed
- Mortality rate > Search first: CDC, WHO, GBD, national mortality databases
- Disease-specific mortality (deaths directly attributable to disease) > Search first: Disease registries, CDC Wonder, GBD, PubMed
- Morbidity and Function:
- Morbidity (disease-related disability and health impacts) > Search first: GBD, WHO, disability databases, PubMed
- Disability outcomes (long-term functional impairments) > Search first: ICF (International Classification of Functioning), disability registries
- Quality of life measures (EQ-5D, SF-36, PROMIS, disease-specific tools) > Search first: EQ-5D database, SF-36, PROMIS, PubMed
- Disease Course:
- Complications (secondary problems: infections, organ failure, etc.) > Search first: ICD codes, disease registries, clinical databases, PubMed
- Recovery potential (likelihood and extent of recovery, with vs without treatment) > Search first: Natural history studies, rehabilitation databases, PubMed
- Prediction:
- Prognostic factors (age, disease severity, biomarkers, treatment response) > Search first: Prognostic models databases, clinical calculators, PubMed
- Prognostic biomarkers (molecular markers predicting disease course) > Search first: FDA Biomarker database, PubMed, cancer prognostic databases
12. Treatment
- Pharmacotherapy:
- Pharmacological treatments (drug names, drug classes, mechanisms of action) > Search first: DrugBank, RxNorm, ATC classification, DailyMed, FDA databases
- Pharmacogenomics (how genetic variants affect drug metabolism, efficacy, toxicity) > Search first: PharmGKB, CPIC (Clinical Pharmacogenetics), FDA Table of PGx Biomarkers
- Advanced Therapeutics:
- Gene therapy (viral vectors, CRISPR, gene replacement, gene editing) > Search first: ClinicalTrials.gov, FDA gene therapy database, ASGCT resources
- Cell therapy (stem cell transplant, CAR-T, cellular therapeutics) > Search first: ClinicalTrials.gov, FDA cell therapy database, FACT standards
- RNA-based therapies (ASOs, siRNA, mRNA therapies) > Search first: ClinicalTrials.gov, FDA approvals, PubMed
- Targeted therapies (treatments directed at specific molecular targets) > Search first: My Cancer Genome, OncoKB, ClinicalTrials.gov, FDA approvals
- Immunotherapies (checkpoint inhibitors, monoclonal antibodies) > Search first: Cancer Immunotherapy Database, FDA approvals, ClinicalTrials.gov
- Surgical and Interventional:
- Surgical interventions (types of surgery, timing, outcomes) > Search first: CPT codes, surgical registries, clinical guidelines, PubMed
- Supportive and Rehabilitative:
- Supportive care (symptom management, pain control, nutrition) > Search first: Clinical guidelines, Cochrane Library, PubMed
- Rehabilitation (physical therapy, occupational therapy, speech therapy) > Search first: Rehabilitation medicine databases, clinical guidelines, PubMed
- Experimental:
- Experimental treatments in clinical trials (with NCT identifiers if available) > Search first: ClinicalTrials.gov, EU Clinical Trials Register, WHO ICTRP
- Treatment Outcomes:
- Treatment response rates > Search first: Clinical trial databases, FDA reviews, systematic reviews, PubMed
- Side effects and adverse events > Search first: FDA Adverse Event Reporting System (FAERS), MedWatch, PubMed
- Treatment Strategy:
- Treatment algorithms (clinical pathways, decision trees) > Search first: Clinical practice guidelines, NCCN Guidelines, UpToDate
- Combination therapies > Search first: ClinicalTrials.gov, treatment guidelines, PubMed
- Personalized medicine approaches (genotype-guided treatment) > Search first: My Cancer Genome, CIViC, PharmGKB, precision medicine databases
For each treatment, suggest MAXO (Medical Action Ontology) terms where applicable.
13. Prevention
- Prevention Levels:
- Primary prevention (preventing disease occurrence: vaccination, risk factor modification) > Search first: CDC, WHO, USPSTF recommendations, Cochrane Library
- Secondary prevention (early detection and treatment: screening programs, early intervention) > Search first: USPSTF, CDC screening guidelines, WHO
- Tertiary prevention (preventing complications in those with disease) > Search first: Clinical guidelines, disease management protocols, PubMed
- Immunization: Vaccine strategies (if applicable)
Search first: CDC vaccine schedules, WHO immunization, FDA vaccine database
- Screening and Early Detection:
- Screening programs (population-based: newborn screening, cancer screening) > Search first: CDC screening programs, USPSTF, cancer screening databases
- Genetic screening (carrier screening, preimplantation genetic diagnosis, prenatal testing) > Search first: ACMG recommendations, ACOG guidelines, GTR
- Risk stratification (identifying high-risk individuals for targeted prevention) > Search first: Risk prediction models, clinical calculators, PubMed
- Behavioral Interventions: Lifestyle modifications to reduce risk
Search first: CDC, WHO, behavioral intervention databases, Cochrane Library
- Counseling: Genetic counseling (risk assessment, family planning guidance)
Search first: NSGC resources, ACMG guidelines, GeneReviews
- Public Health:
- Public health interventions (sanitation, vector control, health education) > Search first: CDC, WHO, public health databases, PubMed
- Environmental interventions (reducing environmental risk factors) > Search first: EPA databases, WHO environmental health, PubMed
- Prophylaxis: Preventive medications or procedures
Search first: Clinical guidelines, FDA approvals, PubMed
14. Other Species / Natural Disease
- Taxonomy: Species affected (with NCBI Taxon identifiers)
Search first: NCBI Taxonomy
- Breed: Specific breeds affected (with VBO identifiers if applicable)
Search first: VBO (Vertebrate Breed Ontology)
- Gene: Orthologous genes in other species (with NCBI Gene IDs)
Search first: NCBI Gene
- Natural Disease:
- Naturally occurring disease in other species (companion animals, wildlife) > Search first: OMIA (Online Mendelian Inheritance in Animals), VetCompass, PubMed
- Veterinary relevance and importance in animal health > Search first: OMIA, veterinary databases, PubMed
- Comparative Biology:
- Comparative pathology (similarities and differences across species) > Search first: OMIA, comparative pathology databases, PubMed
- Evolutionary conservation of disease mechanisms > Search first: HomoloGene, OrthoMCL, Alliance of Genome Resources
- Transmission (if applicable):
- Zoonotic potential > Search first: CDC zoonotic diseases, WHO zoonoses, GIDEON
- Cross-species susceptibility > Search first: NCBI Taxonomy, veterinary databases, PubMed
15. Model Organisms
- Model Types:
- Model organism type (mammalian, invertebrate, cellular, in vitro) > Search first: Alliance of Genome Resources, model organism databases
- Specific model systems (mouse, rat, zebrafish, Drosophila, C. elegans, yeast, cell lines, organoids, iPSCs) > Search first: MGI, RGD, ZFIN, FlyBase, WormBase, SGD, ATCC, Cellosaurus
- Induced models (drug treatment, surgical intervention, environmental manipulation) > Search first: MGI, model organism databases, PubMed
- Genetic Models:
- Types available (knockout, knock-in, transgenic, conditional, humanized) > Search first: MGI, IMPC, KOMP, EuMMCR, IMSR
- Model Characteristics:
- Phenotype recapitulation (how well model reproduces human disease features) > Search first: Model organism databases, comparative studies, PubMed
- Model limitations (aspects of human disease not captured) > Search first: Model organism databases, PubMed, review articles
- Applications:
- Research applications (what aspects of disease can be studied) > Search first: Model organism databases, PubMed
- Resources:
- Model databases > Search first: MGI, RGD, ZFIN, FlyBase, WormBase, IMSR, EMMA, MMRRC
Citation Requirements
- Cite primary literature (PMID preferred) for all mechanistic and clinical claims
- Prioritize recent reviews and landmark papers
- Include direct quotes from abstracts where possible to support key statements
- Distinguish evidence source types: human clinical, model organism, in vitro, computational
Output Format
Structure your response as a comprehensive narrative organized by the sections above. For each section, provide: - Factual content with specific details (numbers, percentages, gene names, variant nomenclature) - Ontology term suggestions (HPO, GO, CL, UBERON, CHEBI, MAXO, MONDO) where applicable - Evidence citations with PMIDs - Direct quotes from abstracts to support key claims - Clear indication when information is not available or not applicable for this disease
This report will be used to populate a disease knowledge base entry with: - Pathophysiology descriptions with causal chains - Gene/protein annotations (HGNC, GO terms) - Phenotype associations (HP terms) with frequencies - Cell type involvement (CL terms) - Anatomical locations (UBERON terms) - Chemical entities (CHEBI terms) - Treatment annotations (MAXO terms) - Evidence items with PMIDs and exact abstract quotes - Epidemiology, prognosis, diagnostic, and prevention information - Animal model descriptions with phenotype recapitulation details
TUBA1A-related Tubulinopathy (TUBA1A-associated tubulinopathy / LIS3)
Executive summary
TUBA1A-related tubulinopathy is an autosomal-dominant neurodevelopmental disorder caused predominantly by heterozygous (usually de novo) TUBA1A missense variants that disrupt microtubule function during cortical development, leading to malformations of cortical development (MCD) such as lissencephaly/pachygyria, polymicrogyria-like dysgenesis, and associated abnormalities of the corpus callosum, basal ganglia, cerebellum/brainstem, and ventricles, with frequent global developmental delay and epilepsy. Large aggregated analyses (166 individuals) and more recent phenotype-deep cohorts (10 individuals with detailed epilepsy phenotyping) provide quantitative phenotype frequencies and expanding genotype–phenotype correlations, while real-world genomic testing cohorts show high diagnostic yield for “tubulinopathy” imaging patterns and prominent contribution from TUBA1A. (hebebrand2019themutationaland pages 1-2, hebebrand2019themutationaland pages 2-3, kooshavar2024diagnosticutilityof pages 4-5, schroter2022complementingthephenotypical pages 1-2)
| Study (year, journal) | Cohort | Key phenotype stats | Variant/genetic stats | Diagnostic/testing stats | URL/DOI |
|---|---|---|---|---|---|
| Hebebrand et al. 2019, Orphanet Journal of Rare Diseases | 166 affected individuals total (146 born, 20 fetuses); HPO-standardized clinical data available for 107 cases | Developmental delay 98.1% (52/53); corpus callosum anomalies 96.2% (102/106); microcephaly 76.0% (57/75); lissencephaly/agyria-pachygyria 70.0% (67/96) (hebebrand2019themutationaland pages 1-2, hebebrand2019themutationaland pages 2-3, hebebrand2019themutationaland media ec7b3e06) | 121 distinct TUBA1A variants identified, including 15 recurrent variants; missense variants clustered in the C-terminal region; Arg402 was the most commonly affected residue (13.3% of cases/variants reviewed) (hebebrand2019themutationaland pages 1-2, hebebrand2019themutationaland pages 5-6) | Exome sequencing identified heterozygous de novo missense variants in new cases; study also curated ClinVar/DECIPHER/denovo-db and applied ACMG-style interpretation workflows (hebebrand2019themutationaland pages 1-2, hebebrand2019themutationaland pages 2-3) | https://doi.org/10.1186/s13023-019-1020-x |
| Schröter et al. 2022, European Journal of Human Genetics | 10 unrelated individuals (8 living; 2 terminated pregnancies) | Epilepsy 75% (6/8); infantile onset among epilepsies 83%; refractory epilepsy 50%; global developmental delay 63%; severe motor impairment/tetraparesis 50% (schroter2022complementingthephenotypical pages 1-2, schroter2022complementingthephenotypical pages 2-3) | 9 missense variants reported (4 novel, 5 previously published); hotspot residues Arg264/Arg402/Arg422 together accounted for 55% of reported cases in the broader literature summarized by the authors (N=57) (schroter2022complementingthephenotypical pages 6-7, schroter2022complementingthephenotypical pages 5-6) | Systematic MRI re-evaluation plus protein-structure/prediction modeling; all reported MRIs abnormal; study emphasizes TUBA1A as a cause of congenital brain malformation with early-onset epilepsy (schroter2022complementingthephenotypical pages 2-3, schroter2022complementingthephenotypical pages 1-2, schroter2022complementingthephenotypical pages 7-8) | https://doi.org/10.1038/s41431-021-01027-0 |
| Kooshavar et al. 2024, Brain Communications | 102 children with brain malformations in the Australian Genomics Brain Malformation Flagship; tubulinopathy subgroup n=10 | Tubulinopathy represented ~10% of the imaged/sequenced cohort; mean age at ES 5.4 years (kooshavar2024diagnosticutilityof pages 1-3, kooshavar2024diagnosticutilityof pages 3-4) | TUBA1A was the most frequent genetic diagnosis; 8/37 diagnoses from clinical singleton ES were TUBA1A (22% of solved clinical ES cases) (kooshavar2024diagnosticutilityof pages 1-3, kooshavar2024diagnosticutilityof pages 4-5) | Clinical singleton exome sequencing yield 36% (37/102), rising to 43% (44/102) after research reanalysis; tubulinopathy subgroup yield 90% (9/10); workflow included mandatory CMA first and phenotype-guided ES/reanalysis (kooshavar2024diagnosticutilityof pages 1-3, kooshavar2024diagnosticutilityof pages 5-6, kooshavar2024diagnosticutilityof pages 4-5, kooshavar2024diagnosticutilityof pages 3-4) | https://doi.org/10.1093/braincomms/fcae056 |
Table: This table compiles the most clinically actionable quantitative findings from key TUBA1A-related tubulinopathy studies, including phenotype frequencies, variant hotspots, and real-world exome sequencing performance. It is useful for rapid knowledge-base curation and evidence-backed clinical summary.
1. Disease information
1.1 Definition and overview
“TUBA1A-associated tubulinopathy” is described as clinically heterogeneous, with principal manifestations including “brain malformations, microcephaly, developmental delay and epilepsy.” (hebebrand2019themutationaland pages 1-2)
Recent clinical reviews emphasize that tubulinopathies are disorders of microtubule proteins critical for mitosis and neuronal migration; TUBA1A is commonly highlighted as the most frequently implicated tubulin gene with severe neurological manifestations and complex cortical/subcortical malformations. (pavone2023casereportstructural pages 1-2)
1.2 Key identifiers (available from retrieved sources)
- Causal gene: TUBA1A (α-tubulin isotype) (hebebrand2019themutationaland pages 1-2)
- TUBA1A gene identifiers:
- OMIM (gene): TUBA1A, MIM#602529 (hebebrand2019themutationaland pages 1-2, pavone2023casereportstructural pages 1-2)
- Disease OMIM identifier used in disease literature:
- “TUBA1A (OMIM #611603)” is explicitly referenced in a 2024 familial mosaicism report discussing tubulinopathies and recurrence risk. (tang2024parentalmosaicismrather pages 1-2)
- Note: In the retrieved evidence, OMIM #611603 is used in the context of the disorder (commonly referred to in the broader literature as Lissencephaly 3 / LIS3), but a full OMIM disease entry text was not retrieved in this run.
Not available in retrieved texts: MONDO ID, Orphanet ID, ICD-10/ICD-11 codes, and MeSH term(s). These typically require direct querying of curated ontology/clinical coding databases (e.g., Orphanet/MONDO/ICD browsers), which were not retrievable from the current document set.
1.3 Common synonyms / alternative names (from retrieved texts and standard usage in papers)
- TUBA1A-associated tubulinopathy (hebebrand2019themutationaland pages 1-2)
- TUBA1A tubulinopathy (schroter2022complementingthephenotypical pages 1-2)
- Often presented clinically under MCD labels such as lissencephaly (agyria–pachygyria), polymicrogyria-like cortical dysgenesis, and other complex MCDs. (hebebrand2019themutationaland pages 2-3, schroter2022complementingthephenotypical pages 1-2)
1.4 Evidence type
The current synthesis is derived primarily from: - Aggregated disease-level evidence compiled from published cases (systematic HPO standardization across studies). (hebebrand2019themutationaland pages 1-2, hebebrand2019themutationaland pages 2-3) - More recent multicenter case series with systematic MRI re-evaluation and seizure phenotyping. (schroter2022complementingthephenotypical pages 1-2) - Real-world diagnostic cohort data from clinical exome sequencing pipelines with research reanalysis. (kooshavar2024diagnosticutilityof pages 4-5)
2. Etiology
2.1 Disease causal factors
Primary cause: heterozygous pathogenic variants in TUBA1A leading to impaired microtubule function during brain development. TUBA1A-associated tubulinopathy is described as “an autosomal dominant disorder mostly caused by de novo variants in TUBA1A.” (hebebrand2019themutationaland pages 1-2)
2.2 Risk factors
Genetic risk factors
- De novo heterozygous missense variation is the dominant causal pattern in most reported cases. A recent multicenter series states: “Except for three familial cases, TUBA1A-tubulinopathy is exclusively caused by de novo missense variants scattered throughout the gene.” (schroter2022complementingthephenotypical pages 1-2)
- Variant hotspots / recurrent residues:
- Large standardized synopsis identified 121 specific variants, including 15 recurrent variants, with clustering around Arg402 (reported as the most commonly affected position; 13.3%). (hebebrand2019themutationaland pages 1-2)
- The 2022 multicenter series highlights enrichment at residues including Arg264, Arg402, Arg422 and reports that Arg264/Arg402/Arg422 account for 55% (N=57) of reported cases summarized by the authors. (schroter2022complementingthephenotypical pages 6-7)
Environmental risk factors
No specific environmental risk factors were identified in the retrieved disease-focused literature. TUBA1A-related tubulinopathy is primarily a monogenic developmental disorder; any gene–environment contributors to severity (e.g., prenatal exposures) are not established in the cited evidence.
2.3 Protective factors
No protective genetic or environmental factors were identified in the retrieved evidence set.
2.4 Gene–environment interactions
No gene–environment interaction evidence specific to TUBA1A-related tubulinopathy was identified in the retrieved texts.
3. Phenotypes
3.1 Core clinical phenotype (with frequencies)
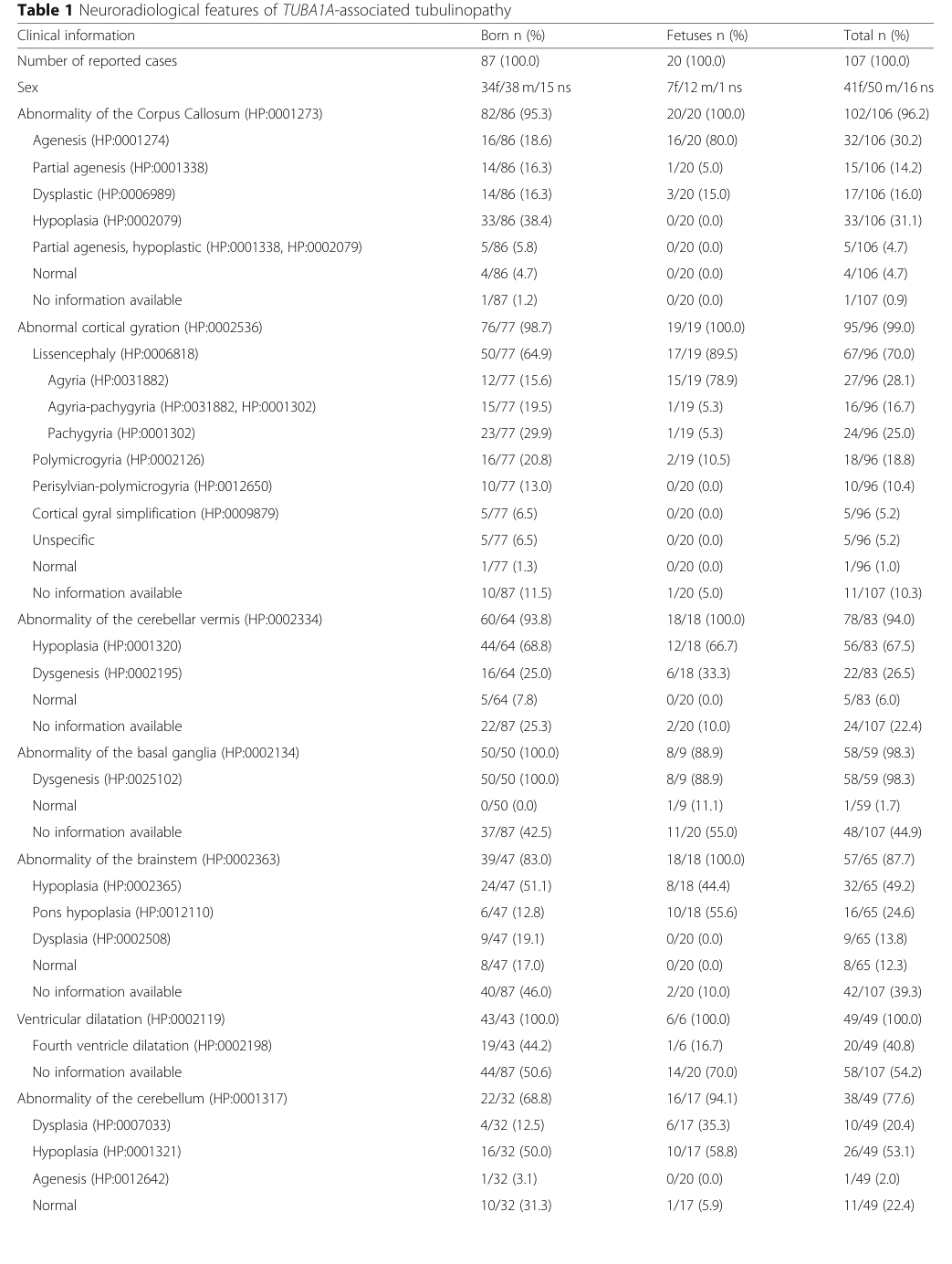
Largest standardized review (166 individuals; HPO standardized): - Developmental delay: 98% (hebebrand2019themutationaland pages 1-2) - Corpus callosum anomalies: 96% (hebebrand2019themutationaland pages 1-2) - Microcephaly: 76% (hebebrand2019themutationaland pages 1-2) - Lissencephaly (agyria–pachygyria): 70% (hebebrand2019themutationaland pages 1-2)
These frequencies are also captured in the paper’s summary tables (image-extracted table regions). (hebebrand2019themutationaland media ec7b3e06, hebebrand2019themutationaland media f96aba65, hebebrand2019themutationaland media 2322f156)
Detailed epilepsy-focused cohort (10 individuals; 8 living): - “Epilepsy was observed in 75% of the cases, which showed infantile onset in 83% and a refractory course in 50%.” (schroter2022complementingthephenotypical pages 1-2) - “Global developmental delay and severe motor impairment with tetraparesis was present in 63% and 50% of the subjects, respectively.” (schroter2022complementingthephenotypical pages 1-2)
3.2 Neuroimaging / structural brain abnormalities
High-frequency MRI abnormalities in the large synopsis include corpus callosum abnormality, abnormal cortical gyration/lissencephaly, cerebellar vermis abnormality, basal ganglia dysgenesis, brainstem abnormalities, and ventricular dilatation. (hebebrand2019themutationaland pages 2-3)
The 2022 cohort further emphasizes heterogeneity including “cobblestone lissencephaly and subcortical band heterotopia” and reports hydrocephalus with posterior infarction in two cases. (schroter2022complementingthephenotypical pages 1-2)
3.3 Epilepsy and electrophysiology
A tubulinopathy epilepsy review states epilepsy can be variable but suggests a generally less aggressive treatment stance in some cohorts: “epilepsy in tubulinopathies when present has a favorable evolution over time suggesting a not particularly aggressive therapeutic approach.” (romaniello2019epilepsyintubulinopathy pages 1-3)
In contrast, the 2022 TUBA1A-focused series notes a substantial refractory burden: “Their anti-epileptic treatment is challenging as epilepsy predominantly shows an infantile onset and treatment-resistant course...” (schroter2022complementingthephenotypical pages 1-2)
3.4 Example phenotype-to-HPO mapping (suggested)
(These are ontology suggestions for knowledge-base structuring; frequencies vary by cohort.) - Global developmental delay — HP:0001263 - Intellectual disability — HP:0001249 - Microcephaly — HP:0000252 - Seizures — HP:0001250; Infantile-onset seizures — HP:0003593 - Lissencephaly — HP:0001339 - Pachygyria — HP:0001302 - Polymicrogyria — HP:0002126 - Agenesis/dysgenesis of corpus callosum — HP:0001274 - Cerebellar hypoplasia — HP:0001321 - Ventriculomegaly/hydrocephalus — HP:0002119 / HP:0000238 - Spasticity — HP:0001257 - Hypotonia — HP:0001252 - Nystagmus — HP:0000639; Strabismus — HP:0000486
4. Genetic / molecular information
4.1 Causal gene
- TUBA1A (α-tubulin). (hebebrand2019themutationaland pages 1-2)
4.2 Variant spectrum and classes
- Predominantly heterozygous missense variants reported across cohorts and reviews. (hebebrand2019themutationaland pages 1-2, schroter2022complementingthephenotypical pages 1-2)
- Large synopsis: 121 specific variants (15 recurrent), with clustering in the C-terminal region and around Arg402. (hebebrand2019themutationaland pages 1-2)
4.3 Inheritance
- Autosomal dominant; “mostly caused by de novo variants.” (hebebrand2019themutationaland pages 1-2)
- Familial recurrence can occur through parental mosaicism, which is clinically important for counseling and recurrence-risk estimation. (tang2024parentalmosaicismrather pages 1-2, tang2024parentalmosaicismrather pages 4-5)
4.4 Mosaicism and recurrence risk (2024 development)
A 2024 report highlights that parental mosaicism can explain sibling recurrence even when parental leukocyte testing is negative and summarizes recurrence-risk estimates tied to variant allele fraction (VAF) in parental blood (≥1% associated with ~24% recurrence risk; >6% up to ~50%). (tang2024parentalmosaicismrather pages 1-2, tang2024parentalmosaicismrather pages 5-7)
4.5 Population frequency
Disease-causing variants are typically ultra-rare/absent in population databases in reported cases (e.g., a de novo variant absent in gnomAD; and a 2024 case report noting absence from multiple population datasets). (hebebrand2019themutationaland pages 5-6, saidin2024anovelpathogenic pages 6-8)
5. Environmental information
No robust environmental or lifestyle contributors are established in the retrieved evidence for TUBA1A-related tubulinopathy.
6. Mechanism / pathophysiology
6.1 Current mechanistic understanding (causal chain)
Upstream event: pathogenic TUBA1A variants alter α/β-tubulin heterodimer behavior and/or microtubule lattice properties. (hoff2022themolecularbiology pages 10-11, hoff2022themolecularbiology pages 11-12)
Cellular consequence: disrupted microtubule dynamics and/or impaired binding/function of microtubule-associated proteins (MAPs) and motors (notably dynein), affecting neuronal migration, neurite outgrowth, and cortical organization. (cushion2023mappingtubulinmutations pages 6-7, zocchi2023decipheringthetubulin pages 19-20)
Tissue-level outcome: malformations of cortical development (lissencephaly/pachygyria, polymicrogyria-like dysgenesis, heterotopia) and associated deep gray matter, commissural, cerebellar/brainstem and ventricular abnormalities. (hebebrand2019themutationaland pages 2-3, schroter2022complementingthephenotypical pages 1-2)
6.2 MAP/motor protein interaction mechanism (R402 hotspot)
A mechanistic review summarizes that α-tubulin residue R402 is a pathogenic hotspot whose substitutions commonly cause lissencephaly through defective neuronal migration. R402 (with E415) stabilizes a C-terminal hairpin important for MAP binding and also interacts with dynein; R402 substitutions incorporate into microtubules yet impair dynein processivity (yeast models) and cause severe neuronal migration defects with altered microtubule-associated proteome (mouse conditional R402H). (cushion2023mappingtubulinmutations pages 6-7)
6.3 Variant-specific mechanistic diversity (developmental vs degenerative)
Mechanisms appear variant-specific and include: - Dominant “poisoning” / dominant-negative or neomorphic effects after incorporation into microtubules (e.g., R402 mutants impair dynein activity and neuronal migration). (zocchi2023decipheringthetubulin pages 19-20, hoff2022themolecularbiology pages 11-12) - Heterodimer destabilization / reduced incorporation (e.g., N102D prevents incorporation and reduces total α-tubulin; associated with neonatal lethality in model evidence summarized in review). (hoff2022themolecularbiology pages 10-11) - Proteostasis and aggregation phenotypes (2023): a novel p.I384N variant reduced TUBA1A stability and microtubule incorporation and increased aggregation; proteasome inhibition increased mutant tubulin levels and promoted insoluble aggregates, suggesting a mechanistic bridge to neurodegeneration (spastic paraplegia/ataxia phenotype). (zocchi2023novellossof pages 1-2, zocchi2023novellossof pages 8-12)
6.4 Suggested ontology annotations
GO (Biological Process): - Microtubule-based process — GO:0007017 - Microtubule cytoskeleton organization — GO:0000226 - Neuron migration — GO:0001764 - Axon guidance — GO:0007411 - Intracellular transport — GO:0046907
CL (cell types; major implicated populations): - Radial glial cell — CL:0000675 (neuronal migration scaffold; common in MCD mechanism models) - Cortical excitatory neuron — CL:0002600 (or broader cortical neuron terms)
UBERON (anatomy): - Cerebral cortex — UBERON:0000956 - Corpus callosum — UBERON:0002020 - Basal ganglion — UBERON:0002420 - Cerebellum — UBERON:0002037 - Brainstem — UBERON:0002298 - Lateral ventricle — UBERON:0002083
7. Anatomical structures affected
Predominantly central nervous system structures, consistent with TUBA1A’s role in neuronal microtubules: - Cerebral cortex (MCD including lissencephaly/polymicrogyria-like patterns) (hebebrand2019themutationaland pages 2-3, schroter2022complementingthephenotypical pages 1-2) - Corpus callosum anomalies (high frequency in aggregated series) (hebebrand2019themutationaland pages 1-2, hebebrand2019themutationaland media ec7b3e06) - Basal ganglia/internal capsule abnormalities and thalamic abnormalities (hebebrand2019themutationaland pages 2-3, schroter2022complementingthephenotypical pages 1-2) - Cerebellum/vermis and brainstem abnormalities (hebebrand2019themutationaland pages 2-3, schroter2022complementingthephenotypical pages 1-2) - Ventricular dilatation/hydrocephalus (hebebrand2019themutationaland pages 2-3, schroter2022complementingthephenotypical pages 1-2)
8. Temporal development
- Typically congenital/neurodevelopmental onset, with brain malformations detectable prenatally or in infancy in severe cases; pregnancy termination due to brain malformations is reported in TUBA1A cohorts. (schroter2022complementingthephenotypical pages 1-2)
- Epilepsy often starts in infancy/neonatal period in detailed cohorts (infantile onset 83% among epilepsy cases). (schroter2022complementingthephenotypical pages 1-2)
9. Inheritance and population
9.1 Inheritance
- Autosomal dominant, predominantly de novo. (hebebrand2019themutationaland pages 1-2, schroter2022complementingthephenotypical pages 1-2)
9.2 Epidemiology
Population prevalence/incidence is not established in the retrieved evidence set.
However, multiple sources state that TUBA1A accounts for a measurable fraction of lissencephaly: - “TUBA1A accounts for 4–5% of all lissencephaly cases.” (hebebrand2019themutationaland pages 1-2)
10. Diagnostics
10.1 Clinical diagnostic approach (current practice)
TUBA1A-related disease is typically suspected based on MRI patterns of malformations of cortical development and associated midline/deep gray matter anomalies, followed by genomic testing (often exome sequencing) to identify pathogenic variants. (hebebrand2019themutationaland pages 2-3, schroter2022complementingthephenotypical pages 1-2)
A narrative review recommends screening: individuals with cortical and subcortical anomalies “should be screened also for pathogenic variants in TUBA1A.” (pavone2023casereportstructural pages 1-2)
10.2 Genetic testing utility and real-world yields (2024)
A 2024 national cohort study of children with brain malformations (Australian Genomics Brain Malformation Flagship; n=102) provides real-world performance data: - Overall diagnostic yield: 36% (37/102) from clinical singleton exome sequencing, rising to 43% (44/102) after research reanalysis. (kooshavar2024diagnosticutilityof pages 1-3, kooshavar2024diagnosticutilityof pages 5-6) - TUBA1A contribution: 8/37 (22%) of clinical singleton-exome diagnoses were due to TUBA1A. (kooshavar2024diagnosticutilityof pages 4-5) - Tubulinopathy subgroup: 9/10 (90%) diagnostic rate via clinical singleton exome sequencing. (kooshavar2024diagnosticutilityof pages 5-6)
The same study documents common implementation steps: mandatory chromosomal microarray prior to exome testing and exclusion of congenital CMV in polymicrogyria cases, underscoring multidisciplinary diagnostic workflows. (kooshavar2024diagnosticutilityof pages 1-3)
10.3 Differential diagnosis
In practice, differential diagnosis overlaps with other malformations of cortical development and genetic lissencephalies/tubulinopathies (other tubulin genes), as well as non-genetic causes of polymicrogyria (e.g., congenital CMV, per Kooshavar cohort protocol). (kooshavar2024diagnosticutilityof pages 1-3, pavone2023casereportstructural pages 1-2)
11. Outcome / prognosis
Prognosis is variable and driven by severity of brain malformations and epilepsy burden: - In the epilepsy-focused TUBA1A cohort, severe motor impairment (tetraparesis) occurred in 50%, and epilepsy was refractory in 50% of epilepsy cases, reflecting substantial neurodisability in a sizable subset. (schroter2022complementingthephenotypical pages 1-2) - Some tubulinopathy epilepsy series suggest seizures may improve over time in subsets (“favorable evolution over time”), but this may not generalize to all TUBA1A phenotypes given the treatment-resistant course described in more recent TUBA1A-focused cohorts. (romaniello2019epilepsyintubulinopathy pages 1-3, schroter2022complementingthephenotypical pages 1-2)
12. Treatment
12.1 Disease-modifying therapies
No disease-modifying therapies specific to TUBA1A-related tubulinopathy were identified in the retrieved evidence.
12.2 Symptomatic management
Epilepsy: anti-seizure medications are standard, but treatment can be challenging due to early-onset and refractory seizures in many patients. (schroter2022complementingthephenotypical pages 1-2)
Supportive care: disease reviews emphasize broad supportive management (developmental/rehabilitative care) as central, but detailed standardized protocols were not retrievable in the current text set.
12.3 MAXO suggestions (supportive actions)
- Anti-seizure pharmacotherapy — MAXO:0000746 (anti-epileptic drug therapy; term may vary by version)
- Physical therapy — MAXO:0000011
- Occupational therapy — MAXO:0000012
- Speech and language therapy — MAXO:0000013
- Nutritional support / enteral feeding when needed — MAXO:0000112 (term may vary)
12.4 Clinical trials
A ClinicalTrials.gov search using broad terms (TUBA1A/tubulinopathy/lissencephaly) did not return clearly relevant interventional trials in the retrieved tool output. (kooshavar2024diagnosticutilityof pages 3-4)
13. Prevention
Prevention is primarily genetic (reproductive risk management) rather than environmental.
13.1 Genetic counseling and reproductive options
Parental mosaicism is increasingly recognized as a cause of sibling recurrence and changes recurrence-risk counseling. The 2024 report emphasizes offering genetic counseling and prioritizing prenatal diagnosis and/or preimplantation genetic testing (PGT-M) for subsequent pregnancies when mosaicism is suspected. (tang2024parentalmosaicismrather pages 4-5, tang2024parentalmosaicismrather pages 5-7)
14. Other species / natural disease
No naturally occurring veterinary analogs were identified in the retrieved evidence.
15. Model organisms and experimental systems
Evidence for models relevant to mechanism and translation includes: - Yeast models of the R402-equivalent mutation demonstrate mutant tubulin incorporation with specific impairment of dynein processivity despite normal recruitment, supporting a dominant mechanism. (cushion2023mappingtubulinmutations pages 6-7) - Mouse models (conditional Tuba1a R402H) show severe neuronal migration defects and altered microtubule-associated proteome composition; neuron culture tracking shows impaired dynein-mediated lysosomal transport. (cushion2023mappingtubulinmutations pages 6-7) - Cellular overexpression systems (HEK-293, COS-1; neural progenitors) were used to demonstrate variant-specific effects on tubulin stability, microtubule incorporation, and aggregation/proteostasis (e.g., p.I384N). (zocchi2023novellossof pages 6-8, zocchi2023novellossof pages 8-12)
Figures/tables supporting phenotype frequencies
Phenotype frequency tables for the large 2019 TUBA1A synopsis (including developmental delay, corpus callosum anomalies, microcephaly, and lissencephaly) were retrieved as cropped table images from the source article. (hebebrand2019themutationaland media ec7b3e06, hebebrand2019themutationaland media f96aba65, hebebrand2019themutationaland media 2322f156)
Notes on evidence gaps
- MONDO/Orphanet/ICD/MeSH identifiers were not present in the retrieved full-text excerpts; adding these would require direct database retrieval.
- Standardized treatment algorithms and rehabilitation protocols specific to TUBA1A-related tubulinopathy were not available in the retrieved texts; current evidence supports symptomatic and supportive management with challenging epilepsy control in many cases.
References
-
(hebebrand2019themutationaland pages 1-2): Moritz Hebebrand, Ulrike Hüffmeier, Regina Trollmann, Ute Hehr, Steffen Uebe, Arif B. Ekici, Cornelia Kraus, Mandy Krumbiegel, André Reis, Christian T. Thiel, and Bernt Popp. The mutational and phenotypic spectrum of tuba1a-associated tubulinopathy. Orphanet Journal of Rare Diseases, Feb 2019. URL: https://doi.org/10.1186/s13023-019-1020-x, doi:10.1186/s13023-019-1020-x. This article has 100 citations and is from a peer-reviewed journal.
-
(hebebrand2019themutationaland pages 2-3): Moritz Hebebrand, Ulrike Hüffmeier, Regina Trollmann, Ute Hehr, Steffen Uebe, Arif B. Ekici, Cornelia Kraus, Mandy Krumbiegel, André Reis, Christian T. Thiel, and Bernt Popp. The mutational and phenotypic spectrum of tuba1a-associated tubulinopathy. Orphanet Journal of Rare Diseases, Feb 2019. URL: https://doi.org/10.1186/s13023-019-1020-x, doi:10.1186/s13023-019-1020-x. This article has 100 citations and is from a peer-reviewed journal.
-
(kooshavar2024diagnosticutilityof pages 4-5): Daniz Kooshavar, David J Amor, Kirsten Boggs, Naomi Baker, Christopher Barnett, Michelle G de Silva, Samantha Edwards, Michael C Fahey, Justine E Marum, Penny Snell, Kiymet Bozaoglu, Kate Pope, Shekeeb S Mohammad, Kate Riney, Rani Sachdev, Ingrid E Scheffer, Sarah Schenscher, John Silberstein, Nicholas Smith, Melanie Tom, Tyson L Ware, Paul J Lockhart, and Richard J Leventer. Diagnostic utility of exome sequencing followed by research reanalysis in human brain malformations. Brain Communications, Feb 2024. URL: https://doi.org/10.1093/braincomms/fcae056, doi:10.1093/braincomms/fcae056. This article has 6 citations and is from a peer-reviewed journal.
-
(schroter2022complementingthephenotypical pages 1-2): Julian Schröter, Bernt Popp, Heiko Brennenstuhl, Jan H. Döring, Stephany H. Donze, Emilia K. Bijlsma, Arie van Haeringen, Dagmar Huhle, Leonie Jestaedt, Andreas Merkenschlager, Maria Arelin, Daniel Gräfe, Sonja Neuser, Stephanie Oates, Deb K. Pal, Michael J. Parker, Johannes R. Lemke, Georg F. Hoffmann, Stefan Kölker, Inga Harting, and Steffen Syrbe. Complementing the phenotypical spectrum of tuba1a tubulinopathy and its role in early-onset epilepsies. European Journal of Human Genetics, 30:298-306, Jan 2022. URL: https://doi.org/10.1038/s41431-021-01027-0, doi:10.1038/s41431-021-01027-0. This article has 26 citations and is from a domain leading peer-reviewed journal.
-
(hebebrand2019themutationaland media ec7b3e06): Moritz Hebebrand, Ulrike Hüffmeier, Regina Trollmann, Ute Hehr, Steffen Uebe, Arif B. Ekici, Cornelia Kraus, Mandy Krumbiegel, André Reis, Christian T. Thiel, and Bernt Popp. The mutational and phenotypic spectrum of tuba1a-associated tubulinopathy. Orphanet Journal of Rare Diseases, Feb 2019. URL: https://doi.org/10.1186/s13023-019-1020-x, doi:10.1186/s13023-019-1020-x. This article has 100 citations and is from a peer-reviewed journal.
-
(hebebrand2019themutationaland pages 5-6): Moritz Hebebrand, Ulrike Hüffmeier, Regina Trollmann, Ute Hehr, Steffen Uebe, Arif B. Ekici, Cornelia Kraus, Mandy Krumbiegel, André Reis, Christian T. Thiel, and Bernt Popp. The mutational and phenotypic spectrum of tuba1a-associated tubulinopathy. Orphanet Journal of Rare Diseases, Feb 2019. URL: https://doi.org/10.1186/s13023-019-1020-x, doi:10.1186/s13023-019-1020-x. This article has 100 citations and is from a peer-reviewed journal.
-
(schroter2022complementingthephenotypical pages 2-3): Julian Schröter, Bernt Popp, Heiko Brennenstuhl, Jan H. Döring, Stephany H. Donze, Emilia K. Bijlsma, Arie van Haeringen, Dagmar Huhle, Leonie Jestaedt, Andreas Merkenschlager, Maria Arelin, Daniel Gräfe, Sonja Neuser, Stephanie Oates, Deb K. Pal, Michael J. Parker, Johannes R. Lemke, Georg F. Hoffmann, Stefan Kölker, Inga Harting, and Steffen Syrbe. Complementing the phenotypical spectrum of tuba1a tubulinopathy and its role in early-onset epilepsies. European Journal of Human Genetics, 30:298-306, Jan 2022. URL: https://doi.org/10.1038/s41431-021-01027-0, doi:10.1038/s41431-021-01027-0. This article has 26 citations and is from a domain leading peer-reviewed journal.
-
(schroter2022complementingthephenotypical pages 6-7): Julian Schröter, Bernt Popp, Heiko Brennenstuhl, Jan H. Döring, Stephany H. Donze, Emilia K. Bijlsma, Arie van Haeringen, Dagmar Huhle, Leonie Jestaedt, Andreas Merkenschlager, Maria Arelin, Daniel Gräfe, Sonja Neuser, Stephanie Oates, Deb K. Pal, Michael J. Parker, Johannes R. Lemke, Georg F. Hoffmann, Stefan Kölker, Inga Harting, and Steffen Syrbe. Complementing the phenotypical spectrum of tuba1a tubulinopathy and its role in early-onset epilepsies. European Journal of Human Genetics, 30:298-306, Jan 2022. URL: https://doi.org/10.1038/s41431-021-01027-0, doi:10.1038/s41431-021-01027-0. This article has 26 citations and is from a domain leading peer-reviewed journal.
-
(schroter2022complementingthephenotypical pages 5-6): Julian Schröter, Bernt Popp, Heiko Brennenstuhl, Jan H. Döring, Stephany H. Donze, Emilia K. Bijlsma, Arie van Haeringen, Dagmar Huhle, Leonie Jestaedt, Andreas Merkenschlager, Maria Arelin, Daniel Gräfe, Sonja Neuser, Stephanie Oates, Deb K. Pal, Michael J. Parker, Johannes R. Lemke, Georg F. Hoffmann, Stefan Kölker, Inga Harting, and Steffen Syrbe. Complementing the phenotypical spectrum of tuba1a tubulinopathy and its role in early-onset epilepsies. European Journal of Human Genetics, 30:298-306, Jan 2022. URL: https://doi.org/10.1038/s41431-021-01027-0, doi:10.1038/s41431-021-01027-0. This article has 26 citations and is from a domain leading peer-reviewed journal.
-
(schroter2022complementingthephenotypical pages 7-8): Julian Schröter, Bernt Popp, Heiko Brennenstuhl, Jan H. Döring, Stephany H. Donze, Emilia K. Bijlsma, Arie van Haeringen, Dagmar Huhle, Leonie Jestaedt, Andreas Merkenschlager, Maria Arelin, Daniel Gräfe, Sonja Neuser, Stephanie Oates, Deb K. Pal, Michael J. Parker, Johannes R. Lemke, Georg F. Hoffmann, Stefan Kölker, Inga Harting, and Steffen Syrbe. Complementing the phenotypical spectrum of tuba1a tubulinopathy and its role in early-onset epilepsies. European Journal of Human Genetics, 30:298-306, Jan 2022. URL: https://doi.org/10.1038/s41431-021-01027-0, doi:10.1038/s41431-021-01027-0. This article has 26 citations and is from a domain leading peer-reviewed journal.
-
(kooshavar2024diagnosticutilityof pages 1-3): Daniz Kooshavar, David J Amor, Kirsten Boggs, Naomi Baker, Christopher Barnett, Michelle G de Silva, Samantha Edwards, Michael C Fahey, Justine E Marum, Penny Snell, Kiymet Bozaoglu, Kate Pope, Shekeeb S Mohammad, Kate Riney, Rani Sachdev, Ingrid E Scheffer, Sarah Schenscher, John Silberstein, Nicholas Smith, Melanie Tom, Tyson L Ware, Paul J Lockhart, and Richard J Leventer. Diagnostic utility of exome sequencing followed by research reanalysis in human brain malformations. Brain Communications, Feb 2024. URL: https://doi.org/10.1093/braincomms/fcae056, doi:10.1093/braincomms/fcae056. This article has 6 citations and is from a peer-reviewed journal.
-
(kooshavar2024diagnosticutilityof pages 3-4): Daniz Kooshavar, David J Amor, Kirsten Boggs, Naomi Baker, Christopher Barnett, Michelle G de Silva, Samantha Edwards, Michael C Fahey, Justine E Marum, Penny Snell, Kiymet Bozaoglu, Kate Pope, Shekeeb S Mohammad, Kate Riney, Rani Sachdev, Ingrid E Scheffer, Sarah Schenscher, John Silberstein, Nicholas Smith, Melanie Tom, Tyson L Ware, Paul J Lockhart, and Richard J Leventer. Diagnostic utility of exome sequencing followed by research reanalysis in human brain malformations. Brain Communications, Feb 2024. URL: https://doi.org/10.1093/braincomms/fcae056, doi:10.1093/braincomms/fcae056. This article has 6 citations and is from a peer-reviewed journal.
-
(kooshavar2024diagnosticutilityof pages 5-6): Daniz Kooshavar, David J Amor, Kirsten Boggs, Naomi Baker, Christopher Barnett, Michelle G de Silva, Samantha Edwards, Michael C Fahey, Justine E Marum, Penny Snell, Kiymet Bozaoglu, Kate Pope, Shekeeb S Mohammad, Kate Riney, Rani Sachdev, Ingrid E Scheffer, Sarah Schenscher, John Silberstein, Nicholas Smith, Melanie Tom, Tyson L Ware, Paul J Lockhart, and Richard J Leventer. Diagnostic utility of exome sequencing followed by research reanalysis in human brain malformations. Brain Communications, Feb 2024. URL: https://doi.org/10.1093/braincomms/fcae056, doi:10.1093/braincomms/fcae056. This article has 6 citations and is from a peer-reviewed journal.
-
(pavone2023casereportstructural pages 1-2): Piero Pavone, Pasquale Striano, Giovanni Cacciaguerra, Simona Domenica Marino, Enrico Parano, Xena Giada Pappalardo, Raffaele Falsaperla, and Martino Ruggieri. Case report: structural brain abnormalities in tuba1a-tubulinopathies: a narrative review. Frontiers in Pediatrics, Sep 2023. URL: https://doi.org/10.3389/fped.2023.1210272, doi:10.3389/fped.2023.1210272. This article has 7 citations.
-
(tang2024parentalmosaicismrather pages 1-2): Hai Xuan Tang, Y‐Thanh Lu, Thi Minh Thi Ha, Nhat‐Thang Tran, Doan Minh Dang, Son Xuan Ly, Thu Ha Thi Bui, Son Ta Vo, Minh Doan Thai, Vu Dinh Nguyen, Thong Van Nguyen, Linh Thuy Dinh, Lan‐Anh Thi Luong, Kim‐Phuong Doan, Kim Huong Thi Nguyen, Thanh‐Thuy Thi Do, Dinh‐Kiet Truong, Hoa Giang, Hoai‐Nghia Nguyen, Thu Huong Nhut Trinh, and Hung Sang Tang. Parental mosaicism rather than de novo variants in foxg1 ‐related syndrome and tuba1a ‐associated tubulinopathy: familial case reports. Molecular Genetics & Genomic Medicine, Jun 2024. URL: https://doi.org/10.1002/mgg3.2484, doi:10.1002/mgg3.2484. This article has 0 citations and is from a peer-reviewed journal.
-
(hebebrand2019themutationaland media f96aba65): Moritz Hebebrand, Ulrike Hüffmeier, Regina Trollmann, Ute Hehr, Steffen Uebe, Arif B. Ekici, Cornelia Kraus, Mandy Krumbiegel, André Reis, Christian T. Thiel, and Bernt Popp. The mutational and phenotypic spectrum of tuba1a-associated tubulinopathy. Orphanet Journal of Rare Diseases, Feb 2019. URL: https://doi.org/10.1186/s13023-019-1020-x, doi:10.1186/s13023-019-1020-x. This article has 100 citations and is from a peer-reviewed journal.
-
(hebebrand2019themutationaland media 2322f156): Moritz Hebebrand, Ulrike Hüffmeier, Regina Trollmann, Ute Hehr, Steffen Uebe, Arif B. Ekici, Cornelia Kraus, Mandy Krumbiegel, André Reis, Christian T. Thiel, and Bernt Popp. The mutational and phenotypic spectrum of tuba1a-associated tubulinopathy. Orphanet Journal of Rare Diseases, Feb 2019. URL: https://doi.org/10.1186/s13023-019-1020-x, doi:10.1186/s13023-019-1020-x. This article has 100 citations and is from a peer-reviewed journal.
-
(romaniello2019epilepsyintubulinopathy pages 1-3): Romina Romaniello, Claudio Zucca, Filippo Arrigoni, Paolo Bonanni, Elena Panzeri, Maria T. Bassi, and Renato Borgatti. Epilepsy in tubulinopathy: personal series and literature review. Cells, 8:669, Jul 2019. URL: https://doi.org/10.3390/cells8070669, doi:10.3390/cells8070669. This article has 48 citations.
-
(tang2024parentalmosaicismrather pages 4-5): Hai Xuan Tang, Y‐Thanh Lu, Thi Minh Thi Ha, Nhat‐Thang Tran, Doan Minh Dang, Son Xuan Ly, Thu Ha Thi Bui, Son Ta Vo, Minh Doan Thai, Vu Dinh Nguyen, Thong Van Nguyen, Linh Thuy Dinh, Lan‐Anh Thi Luong, Kim‐Phuong Doan, Kim Huong Thi Nguyen, Thanh‐Thuy Thi Do, Dinh‐Kiet Truong, Hoa Giang, Hoai‐Nghia Nguyen, Thu Huong Nhut Trinh, and Hung Sang Tang. Parental mosaicism rather than de novo variants in foxg1 ‐related syndrome and tuba1a ‐associated tubulinopathy: familial case reports. Molecular Genetics & Genomic Medicine, Jun 2024. URL: https://doi.org/10.1002/mgg3.2484, doi:10.1002/mgg3.2484. This article has 0 citations and is from a peer-reviewed journal.
-
(tang2024parentalmosaicismrather pages 5-7): Hai Xuan Tang, Y‐Thanh Lu, Thi Minh Thi Ha, Nhat‐Thang Tran, Doan Minh Dang, Son Xuan Ly, Thu Ha Thi Bui, Son Ta Vo, Minh Doan Thai, Vu Dinh Nguyen, Thong Van Nguyen, Linh Thuy Dinh, Lan‐Anh Thi Luong, Kim‐Phuong Doan, Kim Huong Thi Nguyen, Thanh‐Thuy Thi Do, Dinh‐Kiet Truong, Hoa Giang, Hoai‐Nghia Nguyen, Thu Huong Nhut Trinh, and Hung Sang Tang. Parental mosaicism rather than de novo variants in foxg1 ‐related syndrome and tuba1a ‐associated tubulinopathy: familial case reports. Molecular Genetics & Genomic Medicine, Jun 2024. URL: https://doi.org/10.1002/mgg3.2484, doi:10.1002/mgg3.2484. This article has 0 citations and is from a peer-reviewed journal.
-
(saidin2024anovelpathogenic pages 6-8): Akzam Saidin, Anet Papazovska Cherepnalkovski, Zeeshan Shaukat, Todor Arsov, Rashid Hussain, Ben J. Roberts, Marija Bucat, Klara Cogelja, Michael G. Ricos, and Leanne M. Dibbens. A novel pathogenic tuba1a variant in a croatian infant is linked to a severe tubulinopathy with walker–warburg-like features. Genes, 15:1031, Aug 2024. URL: https://doi.org/10.3390/genes15081031, doi:10.3390/genes15081031. This article has 0 citations.
-
(hoff2022themolecularbiology pages 10-11): Katelyn J. Hoff, Andrew J. Neumann, and Jeffrey K. Moore. The molecular biology of tubulinopathies: understanding the impact of variants on tubulin structure and microtubule regulation. Frontiers in Cellular Neuroscience, Nov 2022. URL: https://doi.org/10.3389/fncel.2022.1023267, doi:10.3389/fncel.2022.1023267. This article has 41 citations.
-
(hoff2022themolecularbiology pages 11-12): Katelyn J. Hoff, Andrew J. Neumann, and Jeffrey K. Moore. The molecular biology of tubulinopathies: understanding the impact of variants on tubulin structure and microtubule regulation. Frontiers in Cellular Neuroscience, Nov 2022. URL: https://doi.org/10.3389/fncel.2022.1023267, doi:10.3389/fncel.2022.1023267. This article has 41 citations.
-
(cushion2023mappingtubulinmutations pages 6-7): Thomas D. Cushion, Ines Leca, and David A. Keays. Mapping tubulin mutations. Frontiers in Cell and Developmental Biology, Feb 2023. URL: https://doi.org/10.3389/fcell.2023.1136699, doi:10.3389/fcell.2023.1136699. This article has 27 citations.
-
(zocchi2023decipheringthetubulin pages 19-20): Riccardo Zocchi, Claudia Compagnucci, Enrico Bertini, and Antonella Sferra. Deciphering the tubulin language: molecular determinants and readout mechanisms of the tubulin code in neurons. International Journal of Molecular Sciences, 24:2781, Feb 2023. URL: https://doi.org/10.3390/ijms24032781, doi:10.3390/ijms24032781. This article has 15 citations.
-
(zocchi2023novellossof pages 1-2): Riccardo Zocchi, Emanuele Bellacchio, Michela Piccione, Raffaella Scardigli, Valentina D’Oria, Stefania Petrini, Kristin Baranano, Enrico Bertini, and Antonella Sferra. Novel loss of function mutation in tuba1a gene compromises tubulin stability and proteostasis causing spastic paraplegia and ataxia. Frontiers in Cellular Neuroscience, Jun 2023. URL: https://doi.org/10.3389/fncel.2023.1162363, doi:10.3389/fncel.2023.1162363. This article has 11 citations.
-
(zocchi2023novellossof pages 8-12): Riccardo Zocchi, Emanuele Bellacchio, Michela Piccione, Raffaella Scardigli, Valentina D’Oria, Stefania Petrini, Kristin Baranano, Enrico Bertini, and Antonella Sferra. Novel loss of function mutation in tuba1a gene compromises tubulin stability and proteostasis causing spastic paraplegia and ataxia. Frontiers in Cellular Neuroscience, Jun 2023. URL: https://doi.org/10.3389/fncel.2023.1162363, doi:10.3389/fncel.2023.1162363. This article has 11 citations.
-
(zocchi2023novellossof pages 6-8): Riccardo Zocchi, Emanuele Bellacchio, Michela Piccione, Raffaella Scardigli, Valentina D’Oria, Stefania Petrini, Kristin Baranano, Enrico Bertini, and Antonella Sferra. Novel loss of function mutation in tuba1a gene compromises tubulin stability and proteostasis causing spastic paraplegia and ataxia. Frontiers in Cellular Neuroscience, Jun 2023. URL: https://doi.org/10.3389/fncel.2023.1162363, doi:10.3389/fncel.2023.1162363. This article has 11 citations.