Sifrim-Hitz-Weiss syndrome
Sifrim-Hitz-Weiss syndrome (SIHIWES; also called CHD4 neurodevelopmental disorder or CHD4-related syndrome) is a rare autosomal dominant multisystemic neurodevelopmental disorder caused by de novo, mostly nontruncating (missense) variants in CHD4. CHD4 encodes the catalytic ATPase subunit of the nucleosome remodeling and deacetylase (NuRD) chromatin-remodeling complex, which couples ATP-dependent chromatin remodeling with histone deacetylase activity to repress developmental gene-expression programs. SIHIWES variants alter CHD4 ATP-hydrolysis and nucleosome-remodeling activity, dysregulating transcriptional programs required for neurodevelopment and organogenesis. Core manifestations include global developmental delay, speech delay, mild-to-moderate intellectual disability, hypotonia, brain anomalies (including ventriculomegaly), congenital heart defects, dysmorphic facial features, and frequently macrocephaly. Additional features include hearing impairment, ophthalmic abnormalities, skeletal and limb anomalies, and hypogonadism (cryptorchidism) in males.
Ask OpenScientist
Ask a research question about Sifrim-Hitz-Weiss syndrome. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (1 reference)
Discussions and Knowledge Gaps
1Show evidence (1 reference)
Pathophysiology
3Show evidence (4 references)
Show evidence (2 references)
Show evidence (2 references)
Pathograph
- Target 'Dysregulated developmental gene expression' (from 'CHD4/NuRD chromatin remodeling dysfunction') not found in named elements
- Target 'Brain structural anomalies' (from 'Impaired NuRD-dependent cortical neurogenesis') not found in named elements
- Target 'Congenital heart defects' (from 'NuRD-dependent cardiac transcriptional repression defect') not found in named elements
Phenotypes
15Cardiovascular 1
Show evidence (1 reference)
Ear 1
Show evidence (2 references)
Endocrine 1
Show evidence (2 references)
Genitourinary 1
Show evidence (1 reference)
Head and Neck 2
Show evidence (2 references)
Show evidence (2 references)
Musculoskeletal 3
Show evidence (1 reference)
Show evidence (1 reference)
Nervous System 4
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (2 references)
Other 2
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
1Show evidence (3 references)
Medical Actions
3Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Source YAML
click to showname: Sifrim-Hitz-Weiss syndrome
creation_date: "2026-06-04T00:00:00Z"
category: Mendelian
description: >-
Sifrim-Hitz-Weiss syndrome (SIHIWES; also called CHD4 neurodevelopmental
disorder or CHD4-related syndrome) is a rare autosomal dominant multisystemic
neurodevelopmental disorder caused by de novo, mostly nontruncating
(missense) variants in CHD4. CHD4 encodes the catalytic ATPase subunit of the
nucleosome remodeling and deacetylase (NuRD) chromatin-remodeling complex,
which couples ATP-dependent chromatin remodeling with histone deacetylase
activity to repress developmental gene-expression programs. SIHIWES variants
alter CHD4 ATP-hydrolysis and nucleosome-remodeling activity, dysregulating
transcriptional programs required for neurodevelopment and organogenesis.
Core manifestations include global developmental delay, speech delay,
mild-to-moderate intellectual disability, hypotonia, brain anomalies
(including ventriculomegaly), congenital heart defects, dysmorphic facial
features, and frequently macrocephaly. Additional features include hearing
impairment, ophthalmic abnormalities, skeletal and limb anomalies, and
hypogonadism (cryptorchidism) in males.
disease_term:
preferred_term: Sifrim-Hitz-Weiss syndrome
term:
id: MONDO:0014946
label: Sifrim-Hitz-Weiss syndrome
parents:
- chromatin remodeling disorder
- neurodevelopmental disorder
references:
- reference: PMID:32881470
title: "CHD4 Neurodevelopmental Disorder."
tags:
- GeneReviews
prevalence:
- population: Published literature cohorts
prevalence_class: ULTRA_RARE
percentage: ultra-rare
notes: >-
No robust population prevalence estimate is available. The disorder was
first delineated in 2016 from exome sequencing of congenital heart disease
probands, and the largest clinical series to date described 32 individuals
with mostly de novo CHD4 variants.
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We collected clinical and molecular data from 32 individuals with mostly de novo variants in CHD4, identified through next-generation sequencing."
explanation: The largest published cohort comprised 32 individuals, underscoring the rarity of the disorder.
inheritance:
- name: Autosomal dominant inheritance
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
description: >-
Sifrim-Hitz-Weiss syndrome is an autosomal dominant disorder typically
caused by a de novo pathogenic variant in CHD4.
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Sifrim-Hitz-Weiss syndrome (SIHIWES) is a recently described multisystemic neurodevelopmental disorder caused by de novo variants inCHD4."
explanation: Establishes the disorder as caused by de novo CHD4 variants, consistent with autosomal dominant inheritance.
pathophysiology:
- name: CHD4/NuRD chromatin remodeling dysfunction
description: >-
CHD4 is the catalytic ATPase core of the nucleosome remodeling and
deacetylase (NuRD) complex, which couples ATP-dependent chromatin
remodeling with histone deacetylase activity to repress gene transcription.
SIHIWES-causing variants are predominantly nontruncating substitutions
clustered in the SNF2-like ATPase region; they alter CHD4 ATP hydrolysis
and nucleosome-remodeling activity in a variant-specific manner, perturbing
the NuRD-dependent transcriptional programs required for normal
neurodevelopment and organogenesis.
genes:
- preferred_term: CHD4
term:
id: hgnc:1919
label: CHD4
protein_complexes:
- preferred_term: NuRD complex
term:
id: GO:0016581
label: NuRD complex
biological_processes:
- preferred_term: ATP-dependent chromatin remodeling
term:
id: GO:0006338
label: chromatin remodeling
- preferred_term: nucleosome remodeling/disassembly
term:
id: GO:0006337
label: nucleosome disassembly
- preferred_term: histone/protein deacetylation
term:
id: GO:0006476
label: protein deacetylation
- preferred_term: transcriptional repression
term:
id: GO:0000122
label: negative regulation of transcription by RNA polymerase II
downstream:
- target: Dysregulated developmental gene expression
description: >-
Altered CHD4 ATPase and remodeling activity disrupts NuRD-mediated
transcriptional repression, dysregulating developmental gene-expression
programs across multiple tissues.
causal_link_type: DIRECT
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: "Alterations in ATP hydrolysis and chromatin remodeling activities were observed in variants from different domains."
explanation: Functional assays show that SIHIWES variants alter CHD4 ATP hydrolysis and chromatin remodeling, the proximal molecular defect.
- target: Global developmental delay
description: >-
NuRD-dependent dysregulation of neurodevelopmental gene expression
manifests clinically as global developmental delay in the majority of
affected individuals.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Dysregulated developmental gene expression
- Impaired cortical neurogenesis
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The majority of participants had global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, and dysmorphic features."
explanation: Cohort data link CHD4/NuRD dysfunction to global developmental delay as a near-universal feature.
evidence:
- reference: PMID:38619323
reference_title: "CHD4 and SMYD1 repress common transcriptional programs in the developing heart."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "CHD4 is the catalytic core of the nucleosome remodeling and deacetylase (NuRD) complex, which represses gene transcription."
explanation: Establishes CHD4 as the catalytic core of the NuRD complex acting as a transcriptional repressor.
- reference: PMID:27616479
reference_title: "De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Chromodomain helicase DNA-binding protein 4 (CHD4) is an ATP-dependent chromatin remodeler involved in epigenetic regulation of gene transcription, DNA repair, and cell cycle progression."
explanation: The original SIHIWES report establishes CHD4 as an ATP-dependent chromatin remodeler whose disruption causes the syndrome.
- reference: PMID:27616479
reference_title: "De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: "Based on these findings, the mutations potentially alter the complex activity but not its formation."
explanation: Cell-based studies indicate CHD4 missense variants alter NuRD/HDAC complex activity rather than complex assembly, consistent with altered remodeling output.
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The majority of variants were nontruncating and affected the SNF2-like region of the protein."
explanation: Most SIHIWES variants are nontruncating and affect the SNF2-like ATPase region (reported from the 32-patient clinical cohort sequencing), consistent with altered remodeling activity rather than simple haploinsufficiency.
- name: Impaired NuRD-dependent cortical neurogenesis
description: >-
In the developing cortex, NuRD is a major chromatin-remodeling complex in
which a sequential switch of CHD subunits (CHD4, CHD5, CHD3) controls
distinct developmental steps. CHD4-containing NuRD promotes the early
proliferation of neural progenitor cells; loss or dysfunction of CHD4
disrupts progenitor proliferation and downstream neuronal differentiation
and migration, providing a cellular basis for the intellectual disability,
developmental delay, and brain anomalies of SIHIWES.
genes:
- preferred_term: CHD4
term:
id: hgnc:1919
label: CHD4
protein_complexes:
- preferred_term: NuRD complex
term:
id: GO:0016581
label: NuRD complex
cell_types:
- preferred_term: neural progenitor cell
term:
id: CL:0011020
label: neural progenitor cell
- preferred_term: cortical neuron
term:
id: CL:0000540
label: neuron
locations:
- preferred_term: cerebral cortex
term:
id: UBERON:0000956
label: cerebral cortex
biological_processes:
- preferred_term: cerebral cortex development
term:
id: GO:0021987
label: cerebral cortex development
- preferred_term: neural progenitor proliferation
term:
id: GO:0008283
label: cell population proliferation
downstream:
- target: Intellectual disability
description: >-
Disrupted CHD4/NuRD-dependent progenitor proliferation and neuronal
differentiation impairs cortical development, contributing to the
mild-to-moderate intellectual disability characteristic of SIHIWES.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Impaired neural progenitor proliferation
- Defective neuronal differentiation and migration
evidence:
- reference: PMID:27806305
reference_title: "A Functional Switch of NuRD Chromatin Remodeling Complex Subunits Regulates Mouse Cortical Development."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "Whereas CHD4 promotes the early proliferation of progenitors, CHD5 facilitates"
explanation: CHD4 promotes early proliferation of cortical progenitors, the cellular step whose disruption underlies cortical and cognitive phenotypes.
- reference: PMID:37738575
reference_title: "Divergent phenotypes in constitutive versus conditional mutant mouse models of Sifrim-Hitz-Weiss syndrome."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "Telencephalon-specific conditional knockouts had marked reductions in cortical growth, reduced numbers of upper-layer neurons, and exhibited alterations in anxiety and repetitive behaviors."
explanation: SIHIWES mouse models with Chd4 ATPase/helicase domain excision recapitulate reduced cortical growth and fewer upper-layer neurons, supporting impaired CHD4-dependent cortical neurogenesis.
- target: Brain structural anomalies
description: >-
Defective NuRD-dependent neuronal differentiation and migration disturbs
cortical cytoarchitecture, providing a mechanism for the brain anomalies
(including ventriculomegaly) seen in SIHIWES.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
intermediate_mechanisms:
- Defective neuronal differentiation and migration
evidence:
- reference: PMID:27806305
reference_title: "A Functional Switch of NuRD Chromatin Remodeling Complex Subunits Regulates Mouse Cortical Development."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "each CHD leads to defects of neuronal differentiation and migration, which"
explanation: Loss of CHD function causes neuronal differentiation and migration defects, a plausible substrate for SIHIWES brain anomalies.
evidence:
- reference: PMID:27806305
reference_title: "A Functional Switch of NuRD Chromatin Remodeling Complex Subunits Regulates Mouse Cortical Development."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "a sequential switch of CHDs within the complex results in the combinatorial assembly of NuRD complexes"
explanation: Establishes the CHD-subunit switch within NuRD as a regulator of cortical development.
- reference: PMID:37738575
reference_title: "Divergent phenotypes in constitutive versus conditional mutant mouse models of Sifrim-Hitz-Weiss syndrome."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "We utilized mouse genetics to excise the Chd4 ATPase/helicase domain-either constitutively, or conditionally in the developing telencephalon."
explanation: A disease-specific SIHIWES mouse model targeting the Chd4 ATPase/helicase domain in the developing telencephalon directly probes cortical neurodevelopment.
- name: NuRD-dependent cardiac transcriptional repression defect
description: >-
During heart development CHD4, as the catalytic core of NuRD, represses
cardiac gene-expression programs; it interacts with the striated
muscle-restricted histone methyltransferase SMYD1 to co-repress common
targets (including glycolysis, hypoxia-response, and angiogenesis
pathways). Disruption of CHD4-dependent cardiomyocyte transcriptional
control provides a developmental mechanism for the congenital heart defects
that are frequent in SIHIWES.
genes:
- preferred_term: CHD4
term:
id: hgnc:1919
label: CHD4
protein_complexes:
- preferred_term: NuRD complex
term:
id: GO:0016581
label: NuRD complex
cell_types:
- preferred_term: cardiomyocyte
term:
id: CL:0000746
label: cardiac muscle cell
locations:
- preferred_term: heart
term:
id: UBERON:0000948
label: heart
biological_processes:
- preferred_term: heart development
term:
id: GO:0007507
label: heart development
- preferred_term: transcriptional repression
term:
id: GO:0000122
label: negative regulation of transcription by RNA polymerase II
downstream:
- target: Congenital heart defects
description: >-
Loss of CHD4/NuRD-mediated repression of cardiac transcriptional
programs in developing cardiomyocytes disrupts cardiac morphogenesis,
producing structural congenital heart defects such as septal defects.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Derepression of CHD4/SMYD1 target genes in cardiomyocytes
- Disrupted cardiac morphogenesis
evidence:
- reference: PMID:38619323
reference_title: "CHD4 and SMYD1 repress common transcriptional programs in the developing heart."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "SMYD1 and CHD4 repress a group of common genes and pathways involved in glycolysis, response to hypoxia, and angiogenesis."
explanation: Identifies CHD4-dependent cardiac transcriptional repression whose loss perturbs heart development, the developmental basis for congenital heart defects.
evidence:
- reference: PMID:38619323
reference_title: "CHD4 and SMYD1 repress common transcriptional programs in the developing heart."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "Using quantitative proteomics, we identified that CHD4 interacts with SMYD1, a striated muscle-restricted histone methyltransferase that is essential for cardiomyocyte differentiation and cardiac morphogenesis."
explanation: Establishes CHD4's interaction with SMYD1 in cardiomyocyte differentiation and cardiac morphogenesis.
- reference: PMID:27479907
reference_title: "Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We identified three genome-wide significant S-CHD disorders caused by DNMs in CHD4, CDK13 and PRKD1."
explanation: The original exome study that established de novo CHD4 variants as a cause of syndromic congenital heart disease, defining SIHIWES.
phenotypes:
- name: Global developmental delay
description: >-
Global developmental delay is one of the most consistent manifestations of
Sifrim-Hitz-Weiss syndrome.
frequency: FREQUENT
phenotype_term:
preferred_term: Global developmental delay
term:
id: HP:0001263
label: Global developmental delay
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The majority of participants had global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, and dysmorphic features."
explanation: >-
Documents global developmental delay in the majority of patients. Author
wording "majority" maps to FREQUENT under the dismech qualitative mapping
(see docs/frequency-evidence-guidelines.md); no quantitative percentage
was reported.
- name: Intellectual disability
description: >-
Mild-to-moderate intellectual disability is characteristic, although
severity varies and a few individuals have normal intelligence.
frequency: FREQUENT
phenotype_term:
preferred_term: Intellectual disability
term:
id: HP:0001249
label: Intellectual disability
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The majority of participants had global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, and dysmorphic features."
explanation: >-
Cohort documents mild-to-moderate intellectual disability in the majority
of patients. Author wording "majority" maps to FREQUENT under the dismech
qualitative mapping (see docs/frequency-evidence-guidelines.md); no
quantitative percentage was reported.
- reference: PMID:32881470
reference_title: "CHD4 Neurodevelopmental Disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "CHD4 neurodevelopmental disorder (CHD4-NDD) is associated with developmental delay, speech delay, and usually mild-to-moderate intellectual disability."
explanation: GeneReviews documents mild-to-moderate intellectual disability as a core feature.
- name: Delayed speech and language development
description: >-
Speech delay is a consistent feature of the CHD4 neurodevelopmental
disorder.
phenotype_term:
preferred_term: Delayed speech and language development
term:
id: HP:0000750
label: Delayed speech and language development
evidence:
- reference: PMID:32881470
reference_title: "CHD4 Neurodevelopmental Disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "CHD4 neurodevelopmental disorder (CHD4-NDD) is associated with developmental delay, speech delay, and usually mild-to-moderate intellectual disability."
explanation: GeneReviews lists speech delay among the core manifestations.
- name: Hypotonia
description: >-
Hypotonia is a common feature of Sifrim-Hitz-Weiss syndrome.
phenotype_term:
preferred_term: Hypotonia
term:
id: HP:0001252
label: Hypotonia
notes: >-
Hypotonia is reported as a common feature in the MONDO/Orphanet definition
of Sifrim-Hitz-Weiss syndrome and in clinical series. A direct quantitative
abstract snippet supporting a frequency band was not available, so frequency
is intentionally omitted and no evidence item is attached pending a
quotable source.
- name: Congenital heart defect
description: >-
Congenital heart defects, including septal defects, are frequent in
Sifrim-Hitz-Weiss syndrome and were central to the disorder's initial
delineation in congenital heart disease cohorts.
frequency: FREQUENT
phenotype_term:
preferred_term: Congenital heart defect
term:
id: HP:0001627
label: Abnormal heart morphology
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The majority of participants had global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, and dysmorphic features."
explanation: >-
Cohort documents congenital heart defects as a major feature. Author
wording "majority" maps to FREQUENT under the dismech qualitative mapping
(see docs/frequency-evidence-guidelines.md); no quantitative percentage
was reported.
- name: Brain structural anomaly
description: >-
Brain anomalies, including ventriculomegaly, are a recognized feature of
Sifrim-Hitz-Weiss syndrome.
frequency: FREQUENT
phenotype_term:
preferred_term: Ventriculomegaly
term:
id: HP:0002119
label: Ventriculomegaly
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The majority of participants had global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, and dysmorphic features."
explanation: >-
Cohort documents brain anomalies as a major feature in the majority of
patients; ventriculomegaly is a characteristic brain anomaly of SIHIWES.
Author wording "majority" maps to FREQUENT under the dismech qualitative
mapping (see docs/frequency-evidence-guidelines.md); no quantitative
percentage was reported.
- reference: PMID:27616479
reference_title: "De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "These individuals have overlapping phenotypes including developmental delay, intellectual disability, hearing loss, macrocephaly, distinct facial dysmorphisms, palatal abnormalities, ventriculomegaly, and hypogonadism as well as additional findings such as bone fusions."
explanation: The original SIHIWES case series explicitly documents ventriculomegaly as a recurrent brain anomaly.
- name: Macrocephaly
description: >-
Macrocephaly is a frequent but not universal finding, often accompanying
the mild dysmorphic facial features.
frequency: FREQUENT
phenotype_term:
preferred_term: Macrocephaly
term:
id: HP:0000256
label: Macrocephaly
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Macrocephaly was a frequent but not universal finding."
explanation: >-
Cohort directly documents macrocephaly as a frequent but not universal
finding. Author wording "frequent" maps to FREQUENT under the dismech
qualitative mapping (see docs/frequency-evidence-guidelines.md); no
quantitative percentage was reported.
- reference: PMID:27616479
reference_title: "De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "These individuals have overlapping phenotypes including developmental delay, intellectual disability, hearing loss, macrocephaly, distinct facial dysmorphisms, palatal abnormalities, ventriculomegaly, and hypogonadism as well as additional findings such as bone fusions."
explanation: The original SIHIWES case series lists macrocephaly among the overlapping core phenotypes.
- name: Hearing impairment
description: >-
Hearing impairment (sensorineural and/or conductive) is a recognized but
less common feature of Sifrim-Hitz-Weiss syndrome.
phenotype_term:
preferred_term: Hearing impairment
term:
id: HP:0000365
label: Hearing impairment
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Additional common abnormalities included hypogonadism in males, skeletal and limb anomalies, hearing impairment, and ophthalmic abnormalities."
explanation: Cohort lists hearing impairment among additional common abnormalities.
- reference: PMID:27616479
reference_title: "De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "These individuals have overlapping phenotypes including developmental delay, intellectual disability, hearing loss, macrocephaly, distinct facial dysmorphisms, palatal abnormalities, ventriculomegaly, and hypogonadism as well as additional findings such as bone fusions."
explanation: The original SIHIWES case series lists hearing loss among the overlapping core phenotypes.
- name: Skeletal anomalies
description: >-
Skeletal and limb anomalies are among the additional common features of the
disorder.
phenotype_term:
preferred_term: Abnormality of the skeletal system
term:
id: HP:0000924
label: Abnormality of the skeletal system
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Additional common abnormalities included hypogonadism in males, skeletal and limb anomalies, hearing impairment, and ophthalmic abnormalities."
explanation: Cohort lists skeletal and limb anomalies among additional common abnormalities.
- name: Ophthalmic abnormalities
description: >-
Ophthalmic abnormalities, including refractive errors and strabismus, are
additional common features.
phenotype_term:
preferred_term: Abnormality of the eye
term:
id: HP:0000478
label: Abnormality of the eye
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Additional common abnormalities included hypogonadism in males, skeletal and limb anomalies, hearing impairment, and ophthalmic abnormalities."
explanation: Cohort lists ophthalmic abnormalities among additional common abnormalities.
- name: Hypogonadism in males
description: >-
Hypogonadism (including cryptorchidism) is a recognized feature in affected
males.
phenotype_term:
preferred_term: Hypogonadism
term:
id: HP:0000135
label: Hypogonadism
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Additional common abnormalities included hypogonadism in males, skeletal and limb anomalies, hearing impairment, and ophthalmic abnormalities."
explanation: Cohort documents hypogonadism in males as an additional common abnormality.
- reference: PMID:27616479
reference_title: "De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "These individuals have overlapping phenotypes including developmental delay, intellectual disability, hearing loss, macrocephaly, distinct facial dysmorphisms, palatal abnormalities, ventriculomegaly, and hypogonadism as well as additional findings such as bone fusions."
explanation: The original SIHIWES case series lists hypogonadism among the overlapping core phenotypes.
- name: Dysmorphic facial features
description: >-
Distinctive but usually mild, nonspecific dysmorphic facial features are a
core, recognized manifestation of Sifrim-Hitz-Weiss syndrome.
phenotype_term:
preferred_term: Dysmorphic facial features
term:
id: HP:0001999
label: Abnormal facial shape
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The majority of participants had global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, and dysmorphic features."
explanation: Cohort documents dysmorphic features in the majority of patients.
- reference: PMID:27616479
reference_title: "De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "These individuals have overlapping phenotypes including developmental delay, intellectual disability, hearing loss, macrocephaly, distinct facial dysmorphisms, palatal abnormalities, ventriculomegaly, and hypogonadism as well as additional findings such as bone fusions."
explanation: The original SIHIWES case series lists distinct facial dysmorphisms among the overlapping core phenotypes.
- name: Palatal abnormalities
description: >-
Palatal abnormalities are among the overlapping core phenotypes described in
the original delineation of Sifrim-Hitz-Weiss syndrome.
phenotype_term:
preferred_term: Palatal abnormalities
term:
id: HP:0000174
label: Abnormal palate morphology
evidence:
- reference: PMID:27616479
reference_title: "De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "These individuals have overlapping phenotypes including developmental delay, intellectual disability, hearing loss, macrocephaly, distinct facial dysmorphisms, palatal abnormalities, ventriculomegaly, and hypogonadism as well as additional findings such as bone fusions."
explanation: The original SIHIWES case series lists palatal abnormalities among the overlapping core phenotypes.
- name: Renal anomalies
description: >-
Renal anomalies are a recognized feature of CHD4 neurodevelopmental disorder
that are managed per standard care.
phenotype_term:
preferred_term: Renal anomalies
term:
id: HP:0000077
label: Abnormality of the kidney
evidence:
- reference: PMID:32881470
reference_title: "CHD4 Neurodevelopmental Disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Developmental delay / intellectual disability, cervical spine instability and risk of spinal cord compression, refractive errors and strabismus, hearing impairment, congenital heart defects, behavioral issues, growth delay, hypogonadism in males, and renal anomalies are managed per standard care."
explanation: GeneReviews lists renal anomalies among the manifestations managed per standard care.
- name: Cervical spine instability
description: >-
Cervical spine instability with risk of spinal cord compression is a
management-relevant feature of CHD4 neurodevelopmental disorder.
phenotype_term:
preferred_term: Cervical spine instability
term:
id: HP:0010646
label: Cervical spine instability
evidence:
- reference: PMID:32881470
reference_title: "CHD4 Neurodevelopmental Disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "cervical spine instability and risk of spinal cord compression"
explanation: GeneReviews identifies cervical spine instability and spinal cord compression risk as a management concern.
treatments:
- name: Developmental and educational support
description: >-
Developmental delay and intellectual disability are managed with
standard-of-care early intervention, special education, and developmental
therapies.
treatment_term:
preferred_term: rehabilitation
term:
id: NCIT:C15315
label: Rehabilitation
evidence:
- reference: PMID:32881470
reference_title: "CHD4 Neurodevelopmental Disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Developmental delay / intellectual disability, cervical spine instability and risk of spinal cord compression, refractive errors and strabismus, hearing impairment, congenital heart defects, behavioral issues, growth delay, hypogonadism in males, and renal anomalies are managed per standard care."
explanation: GeneReviews recommends standard-of-care management of developmental delay and intellectual disability.
- name: Avoidance of high-risk neck activities
description: >-
Because of the increased risk for cervical spine instability and spinal
cord compression, activities involving rapid neck motion or possible head
and neck trauma (e.g., contact sports, thrill rides) should be avoided.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
evidence:
- reference: PMID:32881470
reference_title: "CHD4 Neurodevelopmental Disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Activities that involve rapid neck motion and/or possible trauma to the head and neck region (e.g., contact sports or thrill rides at amusement parks) because of the possible increased risk for cervical spine instability and spinal cord compression."
explanation: GeneReviews recommends avoiding high-risk neck activities to reduce spinal cord compression risk.
- name: Genetic counseling
description: >-
Genetic counseling addresses the typically de novo, autosomal dominant
nature of the disorder, the low (~1%) sibling recurrence risk from possible
parental germline mosaicism, and options for prenatal and preimplantation
genetic testing.
treatment_term:
preferred_term: Genetic Counseling
term:
id: NCIT:C15240
label: Genetic Counseling
evidence:
- reference: PMID:32881470
reference_title: "CHD4 Neurodevelopmental Disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Once the CHD4 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible."
explanation: GeneReviews supports genetic counseling with prenatal and preimplantation testing options.
genetic:
- name: CHD4 de novo variants
notes: >-
Sifrim-Hitz-Weiss syndrome is caused by heterozygous, typically de novo
variants in CHD4 (chromodomain helicase DNA binding protein 4), which
encodes the catalytic ATPase subunit of the NuRD chromatin-remodeling
complex. Most disease-causing variants are nontruncating missense changes
clustered in the SNF2-like ATPase region.
gene_term:
preferred_term: CHD4
term:
id: hgnc:1919
label: CHD4
inheritance:
- name: Autosomal dominant inheritance
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Sifrim-Hitz-Weiss syndrome (SIHIWES) is a recently described multisystemic neurodevelopmental disorder caused by de novo variants inCHD4."
explanation: De novo CHD4 variants are consistent with autosomal dominant inheritance.
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Sifrim-Hitz-Weiss syndrome (SIHIWES) is a recently described multisystemic neurodevelopmental disorder caused by de novo variants inCHD4."
explanation: Establishes de novo CHD4 variants as the molecular cause.
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The majority of variants were nontruncating and affected the SNF2-like region of the protein."
explanation: Documents the predominance of nontruncating variants in the SNF2-like ATPase region.
- reference: PMID:27616479
reference_title: "De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Here we report five individuals with de novo missense substitutions in CHD4 identified through whole-exome sequencing and web-based gene matching."
explanation: The original report identified de novo CHD4 missense substitutions in five individuals, establishing the genetic basis of SIHIWES.
discussions:
- discussion_id: gap_shw_genotype_phenotype_correlation
prompt: >-
Are there genotype-phenotype correlations in Sifrim-Hitz-Weiss syndrome
based on the type or location of CHD4 variants, and do the
variant-specific differences in CHD4 ATP hydrolysis and chromatin
remodeling activity translate into distinct clinical subphenotypes?
kind: KNOWLEDGE_GAP
status: OPEN
attaches_to:
- pathophysiology#CHD4/NuRD chromatin remodeling dysfunction
- genetic#CHD4 de novo variants
rationale: >-
The largest published cohort (32 individuals) found variant-specific
alterations in CHD4 ATP hydrolysis and chromatin remodeling activity in
vitro, yet a similar phenotype in humans and no detectable
genotype-phenotype correlation by variant type or location. Resolving
whether finer functional or domain-level stratification predicts clinical
severity would clarify the genotype-phenotype map and inform prognosis.
evidence:
- reference: PMID:31388190
reference_title: "The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We did not identify genotype-phenotype correlations based on the type or location of variants."
explanation: >-
The cohort study explicitly reports the absence of a detectable
genotype-phenotype correlation, defining this as an open knowledge gap.
References & Deep Research
References
1Deep Research
1Question: You are an expert researcher providing comprehensive, well-cited information.
Provide detailed information focusing on: 1. Key concepts and definitions with current understanding 2. Recent developments and latest research (prioritize 2023-2024 sources) 3. Current applications and real-world implementations 4. Expert opinions and analysis from authoritative sources 5. Relevant statistics and data from recent studies
Format as a comprehensive research report with proper citations. Include URLs and publication dates where available. Always prioritize recent, authoritative sources and provide specific citations for all major claims.
Disease Characteristics Research Template
Target Disease
- Disease Name: Sifrim-Hitz-Weiss syndrome
- MONDO ID: (if available)
- Category: Mendelian
Research Objectives
Please provide a comprehensive research report on Sifrim-Hitz-Weiss syndrome covering all of the disease characteristics listed below. This report will be used to populate a disease knowledge base entry. Be thorough and cite primary literature (PMID preferred) for all claims.
For each section, suggested databases/resources are listed. These are the first places you should search for information on each topic.
1. Disease Information
Search first: OMIM, Orphanet, ICD-10/ICD-11, MeSH, PubMed
- What is the disease? Provide a concise overview.
- What are the key identifiers? (OMIM, Orphanet, ICD-10/ICD-11, MeSH, Mondo)
- What are the common synonyms and alternative names?
- Is the information derived from individual patients (e.g., EHR) or aggregated disease-level resources?
2. Etiology
- Disease Causal Factors: What are the primary causes? (genetic, environmental, infectious, mechanistic)
- Risk Factors:
Search first: PubMed, Cochrane Library, UpToDate, clinical guidelines, ClinVar, ClinGen, GWAS Catalog, PheGenI, CTD, CDC, WHO, epidemiological databases
- Genetic risk factors (causal variants, susceptibility loci, modifier genes)
- Environmental risk factors (toxins, lifestyle, occupational exposures, age, sex, family history)
- Protective Factors:
Search first: PubMed, Cochrane Library, clinical trial databases, GWAS Catalog, gnomAD, WHO, CDC, nutrition databases
- Genetic protective factors (protective variants, modifier alleles)
- Environmental protective factors (diet, lifestyle, exposures that reduce risk)
- Gene-Environment Interactions: How do genetic and environmental factors interact to influence disease?
Search first: CTD, PubMed, PheGenI, GxE databases
3. Phenotypes
Search first: HPO (Human Phenotype Ontology), OMIM, Orphanet, PubMed, clinicaltrials.gov, MedDRA, SNOMED CT, DECIPHER, LOINC
For each phenotype, provide: - Phenotype type: symptoms, clinical signs, physical manifestations, behavioral changes, or laboratory abnormalities
For symptoms/signs: HPO, OMIM, Orphanet, PubMed For behavioral changes: HPO, DSM, RDoC (Research Domain Criteria), PubMed For laboratory abnormalities: LOINC, SNOMED CT, LabTests Online, PubMed - Phenotype characteristics: Search first: OMIM, Orphanet, HPO, PubMed - Age of symptom onset (neonatal, childhood, adult-onset, late-onset) - Symptom severity (mild, moderate, severe, variable) - Symptom progression (stable, progressive, episodic, fluctuating) - Frequency among affected individuals (percentage or qualitative) - Quality of life impact: Effects on daily functioning and well-being (per-phenotype when possible) Search first: EQ-5D database, SF-36, WHO QOL databases, PubMed - Suggest HPO (Human Phenotype Ontology) terms for each phenotype
4. Genetic/Molecular Information
- Causal Genes: Gene mutations or chromosomal abnormalities responsible for disease (gene symbols, OMIM IDs)
Search first: OMIM, ClinVar, HGMD, Ensembl, NCBI Gene
- Pathogenic Variants:
- Affected genes (gene symbols, HGNC IDs) > Search first: OMIM, NCBI Gene, Ensembl, HGNC, UniProt, GeneCards
- Variant classification (pathogenic, likely pathogenic, VUS per ACMG/AMP guidelines) > Search first: ClinVar, ClinGen, ACMG/AMP guidelines, VarSome
- Variant type/class (missense, frameshift, nonsense, splice-site, structural)
- Allele frequency in population databases > Search first: gnomAD, 1000 Genomes, ExAC, TOPMed, dbSNP
- Somatic vs germline origin > Search first: COSMIC (somatic), ClinVar, ICGC, TCGA
- Functional consequences (loss of function, gain of function, dominant negative)
- Modifier Genes: Genes that modify disease severity or expression
- Epigenetic Information: DNA methylation, histone modifications, chromatin changes affecting disease
Search first: ENCODE, Roadmap Epigenomics, MethBase, DiseaseMeth
- Chromosomal Abnormalities: Large-scale genetic changes (aneuploidy, translocations, inversions)
Search first: DECIPHER, ClinVar, ECARUCA, UCSC Genome Browser
5. Environmental Information
- Environmental Factors: Non-genetic contributing factors (toxins, radiation, pollution, occupational exposure)
Search first: CTD (Comparative Toxicogenomics Database), TOXNET, PubMed, EPA databases
- Lifestyle Factors: Behavioral factors (smoking, diet, exercise, alcohol consumption)
Search first: CDC databases, WHO, PubMed, NHANES
- Infectious Agents: If applicable, pathogens causing or triggering disease (bacteria, viruses, fungi, parasites)
Search first: NCBI Taxonomy, ViPR, BV-BRC, MicrobeDB, GIDEON
6. Mechanism / Pathophysiology
- Molecular Pathways: Specific signaling cascades or biochemical pathways involved (Wnt, MAPK, mTOR, PI3K-AKT, etc.)
Search first: KEGG, Reactome, WikiPathways, PathBank, BioCyc
- Cellular Processes: Cell-level mechanisms (apoptosis, autophagy, cell cycle dysregulation, inflammation, etc.)
Search first: Gene Ontology (GO), Reactome, KEGG, PubMed
- Protein Dysfunction: How protein structure or function is altered (misfolding, aggregation, loss of function, gain of function)
Search first: UniProt, PDB (Protein Data Bank), InterPro, Pfam, AlphaFold
- Metabolic Changes: Alterations in metabolic processes (energy metabolism, lipid metabolism, amino acid metabolism)
Search first: KEGG, BioCyc, HMDB (Human Metabolome Database), BRENDA
- Immune System Involvement: Role of immune response (autoimmunity, immunodeficiency, chronic inflammation)
Search first: ImmPort, Immunome Database, IEDB, Gene Ontology
- Tissue Damage Mechanisms: How tissues/ are injured (oxidative stress, ischemia, fibrosis, necrosis)
Search first: PubMed, Gene Ontology, Reactome
- Biochemical Abnormalities: Specific molecular defects (enzyme deficiencies, receptor dysfunction, ion channel defects)
Search first: BRENDA, UniProt, KEGG, OMIM, PubMed
- Epigenetic Changes: DNA methylation, histone modifications affecting gene expression in disease
Search first: ENCODE, Roadmap Epigenomics, MethBase, DiseaseMeth
- Molecular Profiling (if available):
- Transcriptomics/gene expression changes > Search first: GEO (Gene Expression Omnibus), ArrayExpress, GTEx, Human Cell Atlas, SRA
- Proteomics findings > Search first: PRIDE, ProteomeXchange, Human Protein Atlas, STRING, BioGRID
- Metabolomics signatures > Search first: MetaboLights, Metabolomics Workbench, HMDB, METLIN
- Lipidomics alterations > Search first: LIPID MAPS, SwissLipids, LipidHome, Metabolomics Workbench
- Genomic structural features > Search first: UCSC Genome Browser, Ensembl, NCBI, dbVar, DGV
- Advanced Technologies (if applicable):
- Single-cell analysis findings (cell-type specific mechanisms, cellular heterogeneity) > Search first: Human Cell Atlas, Single Cell Portal, GEO, CELLxGENE
- Spatial transcriptomics findings > Search first: GEO, Spatial Research, Vizgen, 10x Genomics data
- Multi-omics integration results > Search first: TCGA, ICGC, cBioPortal, LinkedOmics, PubMed
- Functional genomics screens (CRISPR, RNAi) > Search first: DepMap, GenomeRNAi, PubMed, BioGRID ORCS
For each mechanism, describe: - The causal chain from initial trigger to clinical manifestation - Which mechanisms are upstream vs downstream - What cell types and biological processes are involved - Suggest GO terms for biological processes and CL terms for cell types
7. Anatomical Structures Affected
- Organ Level:
- Primary organs directly affected
- Secondary organ involvement (complications, secondary effects)
- Body systems involved (cardiovascular, nervous, digestive, respiratory, endocrine, etc.)
Search first: Uberon, FMA (Foundational Model of Anatomy), OMIM, HPO, ICD-11, MeSH, SNOMED CT
- Tissue and Cell Level:
- Specific tissue types affected (epithelial, connective, muscle, nervous)
- Specific cell populations targeted (with Cell Ontology terms)
Search first: Uberon, Human Protein Atlas, Cell Ontology, Human Cell Atlas, CellMarker, PanglaoDB
- Subcellular Level:
- Cellular compartments involved (mitochondria, nucleus, ER, lysosomes) (with GO Cellular Component terms)
Search first: Gene Ontology (Cellular Component), UniProt, Human Protein Atlas
- Localization:
- Specific anatomical sites (with UBERON terms) > Search first: FMA, Uberon, NeuroNames (for brain), SNOMED CT
- Lateralization (unilateral, bilateral, asymmetric) > Search first: HPO, clinical literature, imaging databases
8. Temporal Development
- Onset:
- Typical age of onset (congenital, pediatric, adult, geriatric)
- Onset pattern (acute, subacute, chronic, insidious)
Search first: OMIM, Orphanet, HPO, PubMed
- Progression:
- Disease stages (early, intermediate, advanced, end-stage) > Search first: Cancer Staging Manual (AJCC), WHO classifications, PubMed
- Progression rate (rapid, slow, variable)
- Disease course pattern (episodic, relapsing-remitting, progressive, stable)
- Disease duration (self-limited, chronic lifelong)
Search first: Disease registries, longitudinal cohort databases, natural history studies, PubMed, Orphanet, OMIM
- Patterns:
- Remission patterns (spontaneous, treatment-induced) > Search first: Clinical trial databases, disease registries, PubMed
- Critical periods (time windows of vulnerability or opportunity for intervention) > Search first: PubMed, developmental biology databases, clinical guidelines
9. Inheritance and Population
- Epidemiology:
- Prevalence (cases per 100,000 at given time)
- Incidence (new cases per 100,000 per year)
Search first: Orphanet, CDC, WHO, GBD (Global Burden of Disease), national registries, SEER, disease registries
- For Genetic Etiology:
- Inheritance pattern (AD, AR, X-linked, mitochondrial, multifactorial, polygenic) > Search first: OMIM, Orphanet, ClinVar, GTR (Genetic Testing Registry)
- Penetrance (complete, incomplete, age-dependent) > Search first: ClinVar, OMIM, PubMed, ClinGen
- Expressivity (variable, consistent) > Search first: OMIM, ClinVar, PubMed
- Genetic anticipation (increasing severity in successive generations) > Search first: OMIM, PubMed (especially for repeat expansion disorders)
- Germline mosaicism > Search first: ClinVar, OMIM, genetic counseling literature, PubMed
- Founder effects (population-specific mutations) > Search first: gnomAD, population genetics databases, PubMed
- Consanguinity role > Search first: OMIM, population studies, genetic counseling resources
- Carrier frequency > Search first: gnomAD, carrier screening databases, GeneReviews, GTR
- Population Demographics:
- Affected populations (ethnic or demographic groups with higher prevalence) > Search first: gnomAD, 1000 Genomes, PAGE Study, PubMed, population registries
- Geographic distribution (endemic areas, regional variation) > Search first: WHO, CDC, GBD, Orphanet, geographic epidemiology databases
- Geographic distribution of specific variants
- Sex ratio (male:female) > Search first: Disease registries, OMIM, PubMed, epidemiological databases
- Age distribution of affected individuals > Search first: CDC, disease registries, SEER, Orphanet
10. Diagnostics
- Clinical Tests:
- Laboratory tests (blood, urine, tissue chemistry, specific enzyme assays) > Search first: LOINC, LabTests Online, PubMed
- Biomarkers (proteins, metabolites, genetic markers, circulating biomarkers) > Search first: FDA Biomarker List, BEST (Biomarkers, EndpointS, and other Tools), PubMed
- Imaging studies (X-ray, CT, MRI, PET, ultrasound) > Search first: RadLex, DICOM, Radiopaedia, imaging databases
- Functional tests (pulmonary function, cardiac stress tests) > Search first: LOINC, clinical guidelines, PubMed
- Electrophysiology (EEG, EMG, ECG, nerve conduction studies) > Search first: LOINC, clinical neurophysiology databases, PubMed
- Biopsy findings (histopathology, immunohistochemistry) > Search first: SNOMED CT, College of American Pathologists resources, PubMed
- Pathology findings (microscopic examination) > Search first: SNOMED CT, Digital Pathology databases, PubMed
- Genetic Testing:
Search first: GTR (Genetic Testing Registry), GeneReviews, ClinGen
- Overview of recommended genetic testing approach
- Whole genome sequencing (WGS) utility > Search first: GTR, ClinVar, GEL (Genomics England), gnomAD
- Whole exome sequencing (WES) utility > Search first: GTR, ClinVar, OMIM, GeneMatcher
- Gene panels (which panels, which genes) > Search first: GTR, ClinVar, laboratory-specific databases
- Single gene testing > Search first: GTR, ClinVar, OMIM, GeneReviews
- Chromosomal microarray (CMA) > Search first: DECIPHER, ClinVar, dbVar, ECARUCA
- Karyotyping > Search first: Chromosome Abnormality Database, ClinVar, cytogenetics resources
- FISH > Search first: ClinVar, cytogenetics databases, PubMed
- Mitochondrial DNA testing > Search first: MITOMAP, MSeqDR, ClinVar, GTR
- Repeat expansion testing > Search first: GTR, ClinVar, repeat expansion databases, PubMed
- Omics-Based Diagnostics (if applicable):
- RNA sequencing / transcriptomics > Search first: GEO, ArrayExpress, GTEx, RNA-seq databases
- Proteomics > Search first: PRIDE, ProteomeXchange, FDA Biomarker database
- Metabolomics > Search first: MetaboLights, Metabolomics Workbench, HMDB
- Epigenomics > Search first: GEO, ENCODE, Roadmap Epigenomics, MethBase
- Liquid biopsy > Search first: COSMIC, ClinVar, liquid biopsy databases, PubMed
- Clinical Criteria:
- Standardized diagnostic criteria (DSM, ICD, society guidelines) > Search first: DSM-5, ICD-11, clinical society guidelines, UpToDate
- Differential diagnosis (other conditions to rule out, with distinguishing features) > Search first: DynaMed, UpToDate, clinical decision support systems
- Screening:
- Screening methods for asymptomatic individuals (newborn screening, carrier screening, cascade screening) > Search first: ACMG recommendations, CDC newborn screening, GTR
11. Outcome/Prognosis
- Survival and Mortality:
- Survival rate (5-year, 10-year, overall) > Search first: SEER, cancer registries, disease-specific registries, PubMed
- Life expectancy (with and without treatment if applicable) > Search first: Orphanet, disease registries, actuarial databases, PubMed
- Mortality rate > Search first: CDC, WHO, GBD, national mortality databases
- Disease-specific mortality (deaths directly attributable to disease) > Search first: Disease registries, CDC Wonder, GBD, PubMed
- Morbidity and Function:
- Morbidity (disease-related disability and health impacts) > Search first: GBD, WHO, disability databases, PubMed
- Disability outcomes (long-term functional impairments) > Search first: ICF (International Classification of Functioning), disability registries
- Quality of life measures (EQ-5D, SF-36, PROMIS, disease-specific tools) > Search first: EQ-5D database, SF-36, PROMIS, PubMed
- Disease Course:
- Complications (secondary problems: infections, organ failure, etc.) > Search first: ICD codes, disease registries, clinical databases, PubMed
- Recovery potential (likelihood and extent of recovery, with vs without treatment) > Search first: Natural history studies, rehabilitation databases, PubMed
- Prediction:
- Prognostic factors (age, disease severity, biomarkers, treatment response) > Search first: Prognostic models databases, clinical calculators, PubMed
- Prognostic biomarkers (molecular markers predicting disease course) > Search first: FDA Biomarker database, PubMed, cancer prognostic databases
12. Treatment
- Pharmacotherapy:
- Pharmacological treatments (drug names, drug classes, mechanisms of action) > Search first: DrugBank, RxNorm, ATC classification, DailyMed, FDA databases
- Pharmacogenomics (how genetic variants affect drug metabolism, efficacy, toxicity) > Search first: PharmGKB, CPIC (Clinical Pharmacogenetics), FDA Table of PGx Biomarkers
- Advanced Therapeutics:
- Gene therapy (viral vectors, CRISPR, gene replacement, gene editing) > Search first: ClinicalTrials.gov, FDA gene therapy database, ASGCT resources
- Cell therapy (stem cell transplant, CAR-T, cellular therapeutics) > Search first: ClinicalTrials.gov, FDA cell therapy database, FACT standards
- RNA-based therapies (ASOs, siRNA, mRNA therapies) > Search first: ClinicalTrials.gov, FDA approvals, PubMed
- Targeted therapies (treatments directed at specific molecular targets) > Search first: My Cancer Genome, OncoKB, ClinicalTrials.gov, FDA approvals
- Immunotherapies (checkpoint inhibitors, monoclonal antibodies) > Search first: Cancer Immunotherapy Database, FDA approvals, ClinicalTrials.gov
- Surgical and Interventional:
- Surgical interventions (types of surgery, timing, outcomes) > Search first: CPT codes, surgical registries, clinical guidelines, PubMed
- Supportive and Rehabilitative:
- Supportive care (symptom management, pain control, nutrition) > Search first: Clinical guidelines, Cochrane Library, PubMed
- Rehabilitation (physical therapy, occupational therapy, speech therapy) > Search first: Rehabilitation medicine databases, clinical guidelines, PubMed
- Experimental:
- Experimental treatments in clinical trials (with NCT identifiers if available) > Search first: ClinicalTrials.gov, EU Clinical Trials Register, WHO ICTRP
- Treatment Outcomes:
- Treatment response rates > Search first: Clinical trial databases, FDA reviews, systematic reviews, PubMed
- Side effects and adverse events > Search first: FDA Adverse Event Reporting System (FAERS), MedWatch, PubMed
- Treatment Strategy:
- Treatment algorithms (clinical pathways, decision trees) > Search first: Clinical practice guidelines, NCCN Guidelines, UpToDate
- Combination therapies > Search first: ClinicalTrials.gov, treatment guidelines, PubMed
- Personalized medicine approaches (genotype-guided treatment) > Search first: My Cancer Genome, CIViC, PharmGKB, precision medicine databases
For each treatment, suggest MAXO (Medical Action Ontology) terms where applicable.
13. Prevention
- Prevention Levels:
- Primary prevention (preventing disease occurrence: vaccination, risk factor modification) > Search first: CDC, WHO, USPSTF recommendations, Cochrane Library
- Secondary prevention (early detection and treatment: screening programs, early intervention) > Search first: USPSTF, CDC screening guidelines, WHO
- Tertiary prevention (preventing complications in those with disease) > Search first: Clinical guidelines, disease management protocols, PubMed
- Immunization: Vaccine strategies (if applicable)
Search first: CDC vaccine schedules, WHO immunization, FDA vaccine database
- Screening and Early Detection:
- Screening programs (population-based: newborn screening, cancer screening) > Search first: CDC screening programs, USPSTF, cancer screening databases
- Genetic screening (carrier screening, preimplantation genetic diagnosis, prenatal testing) > Search first: ACMG recommendations, ACOG guidelines, GTR
- Risk stratification (identifying high-risk individuals for targeted prevention) > Search first: Risk prediction models, clinical calculators, PubMed
- Behavioral Interventions: Lifestyle modifications to reduce risk
Search first: CDC, WHO, behavioral intervention databases, Cochrane Library
- Counseling: Genetic counseling (risk assessment, family planning guidance)
Search first: NSGC resources, ACMG guidelines, GeneReviews
- Public Health:
- Public health interventions (sanitation, vector control, health education) > Search first: CDC, WHO, public health databases, PubMed
- Environmental interventions (reducing environmental risk factors) > Search first: EPA databases, WHO environmental health, PubMed
- Prophylaxis: Preventive medications or procedures
Search first: Clinical guidelines, FDA approvals, PubMed
14. Other Species / Natural Disease
- Taxonomy: Species affected (with NCBI Taxon identifiers)
Search first: NCBI Taxonomy
- Breed: Specific breeds affected (with VBO identifiers if applicable)
Search first: VBO (Vertebrate Breed Ontology)
- Gene: Orthologous genes in other species (with NCBI Gene IDs)
Search first: NCBI Gene
- Natural Disease:
- Naturally occurring disease in other species (companion animals, wildlife) > Search first: OMIA (Online Mendelian Inheritance in Animals), VetCompass, PubMed
- Veterinary relevance and importance in animal health > Search first: OMIA, veterinary databases, PubMed
- Comparative Biology:
- Comparative pathology (similarities and differences across species) > Search first: OMIA, comparative pathology databases, PubMed
- Evolutionary conservation of disease mechanisms > Search first: HomoloGene, OrthoMCL, Alliance of Genome Resources
- Transmission (if applicable):
- Zoonotic potential > Search first: CDC zoonotic diseases, WHO zoonoses, GIDEON
- Cross-species susceptibility > Search first: NCBI Taxonomy, veterinary databases, PubMed
15. Model Organisms
- Model Types:
- Model organism type (mammalian, invertebrate, cellular, in vitro) > Search first: Alliance of Genome Resources, model organism databases
- Specific model systems (mouse, rat, zebrafish, Drosophila, C. elegans, yeast, cell lines, organoids, iPSCs) > Search first: MGI, RGD, ZFIN, FlyBase, WormBase, SGD, ATCC, Cellosaurus
- Induced models (drug treatment, surgical intervention, environmental manipulation) > Search first: MGI, model organism databases, PubMed
- Genetic Models:
- Types available (knockout, knock-in, transgenic, conditional, humanized) > Search first: MGI, IMPC, KOMP, EuMMCR, IMSR
- Model Characteristics:
- Phenotype recapitulation (how well model reproduces human disease features) > Search first: Model organism databases, comparative studies, PubMed
- Model limitations (aspects of human disease not captured) > Search first: Model organism databases, PubMed, review articles
- Applications:
- Research applications (what aspects of disease can be studied) > Search first: Model organism databases, PubMed
- Resources:
- Model databases > Search first: MGI, RGD, ZFIN, FlyBase, WormBase, IMSR, EMMA, MMRRC
Citation Requirements
- Cite primary literature (PMID preferred) for all mechanistic and clinical claims
- Prioritize recent reviews and landmark papers
- Include direct quotes from abstracts where possible to support key statements
- Distinguish evidence source types: human clinical, model organism, in vitro, computational
Output Format
Structure your response as a comprehensive narrative organized by the sections above. For each section, provide: - Factual content with specific details (numbers, percentages, gene names, variant nomenclature) - Ontology term suggestions (HPO, GO, CL, UBERON, CHEBI, MAXO, MONDO) where applicable - Evidence citations with PMIDs - Direct quotes from abstracts to support key claims - Clear indication when information is not available or not applicable for this disease
This report will be used to populate a disease knowledge base entry with: - Pathophysiology descriptions with causal chains - Gene/protein annotations (HGNC, GO terms) - Phenotype associations (HP terms) with frequencies - Cell type involvement (CL terms) - Anatomical locations (UBERON terms) - Chemical entities (CHEBI terms) - Treatment annotations (MAXO terms) - Evidence items with PMIDs and exact abstract quotes - Epidemiology, prognosis, diagnostic, and prevention information - Animal model descriptions with phenotype recapitulation details
Comprehensive Research Report: Sifrim–Hitz–Weiss syndrome (SIHIWES; CHD4-related syndrome)
Executive summary (current understanding)
Sifrim–Hitz–Weiss syndrome (SIHIWES), also called CHD4-related syndrome, is a rare, autosomal dominant, multisystem neurodevelopmental disorder caused primarily by heterozygous, usually de novo, CHD4 variants that disrupt ATP-dependent chromatin remodeling. The best-curated cohort-level clinical data available in this evidence set come from a 32-individual Genetics in Medicine series that reported high rates of developmental delay, brain structural abnormalities, congenital heart defects, and sensory (hearing/vision) involvement; management is currently supportive and surveillance-based, with no disease-specific approved therapy identified. (weiss2020thechd4relatedsyndrome pages 1-2, weiss2020thechd4relatedsyndrome pages 3-5)
1. Disease information
1.1 Definition / overview
- Disease concept: SIHIWES is a CHD4-related, multisystem neurodevelopmental disorder characterized by early-onset global developmental delay/intellectual disability with congenital anomalies (notably brain and heart) and characteristic craniofacial gestalt. (weiss2020thechd4relatedsyndrome pages 1-2, weiss2020thechd4relatedsyndrome pages 3-5)
- The original description (pre-eponym) framed it as an “intellectual disability syndrome with distinctive dysmorphisms” due to de novo CHD4 mutations. (weiss2016denovomutations pages 2-3, weiss2016denovomutations pages 1-2)
1.2 Key identifiers (as available in retrieved primary literature)
- OMIM/MIM: 617159 (explicitly cited for SIHIWES). (weiss2020thechd4relatedsyndrome pages 1-2)
- Other identifiers requested (MONDO, Orphanet, MeSH, ICD-10/ICD-11): Not found in the retrieved full-text evidence set; these identifiers likely exist in external disease ontologies, but cannot be asserted here without direct sourced evidence. (weiss2020thechd4relatedsyndrome pages 1-2, weiss2020thechd4relatedsyndrome pages 3-5)
1.3 Synonyms / alternative names
- Sifrim–Hitz–Weiss syndrome (SIHIWES)
- CHD4-related syndrome (used as a preferred descriptive name in cohort study literature) (weiss2020thechd4relatedsyndrome pages 1-2, weiss2020thechd4relatedsyndrome pages 3-5)
- Earlier descriptive label: “intellectual disability syndrome with distinctive dysmorphisms” caused by de novo CHD4 variants. (weiss2016denovomutations pages 2-3, weiss2016denovomutations pages 1-2)
1.4 Evidence sources (patient-level vs aggregated)
- The clinical knowledge base for SIHIWES is primarily derived from aggregated case series/cohorts (e.g., 32-individual cohort) and individual case reports expanding the phenotypic spectrum. (weiss2020thechd4relatedsyndrome pages 3-5, silva2022anovelframeshift pages 2-4)
2. Etiology
2.1 Disease causal factors
- Primary cause: Pathogenic heterozygous CHD4 variants (predominantly missense/in-frame indels) that alter CHD4 chromatin remodeling activity. (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 6-7)
- Mechanistic class: A “chromatinopathy”—a disorder of chromatin regulation—via disruption of ATP-dependent nucleosome remodeling in complexes such as NuRD and ChAHP. (boulasiki2023thenurdcomplex pages 1-2, reid2024howdoeschd4 pages 1-2)
2.2 Risk factors
- Genetic risk factor: De novo occurrence is predominant; thus, parental age-related de novo mutational burden may be a general risk concept, but SIHIWES-specific epidemiologic risk quantification was not available in the retrieved evidence. (weiss2020thechd4relatedsyndrome pages 2-3)
- Environmental risk factors: No disease-specific environmental risk factors were identified in the retrieved evidence; SIHIWES is primarily a monogenic disorder. (weiss2020thechd4relatedsyndrome pages 2-3)
2.3 Protective factors / gene–environment interactions
- No protective factors or gene–environment interaction data specific to SIHIWES were found in the retrieved evidence. (weiss2020thechd4relatedsyndrome pages 3-5)
3. Phenotypes
3.1 Core phenotype spectrum and frequencies (cohort-level)
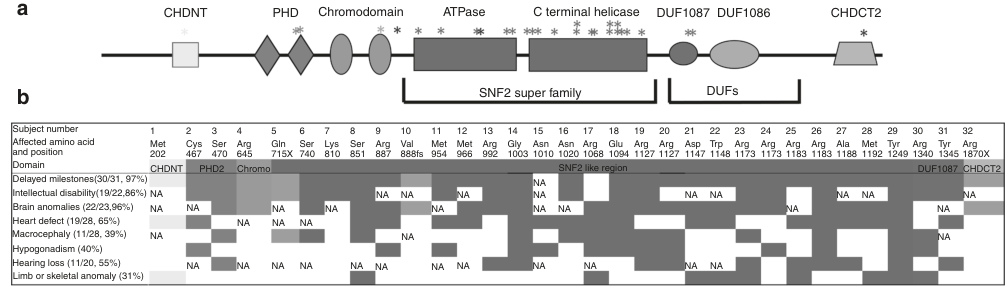
The most detailed quantitative phenotype data in this evidence set come from Weiss et al. (Genetics in Medicine, 2020; n=32). Key frequencies include developmental delay (97%), speech delay (93%), motor delay (83%), intellectual disability (86% among assessed), brain MRI anomalies (96% among imaged), congenital heart defects (65% among evaluated), hearing loss (55%), ophthalmic abnormalities (73%), macrocephaly (39%), and skeletal anomalies (31%). (weiss2020thechd4relatedsyndrome pages 3-5)
| System/Phenotype | Frequency (n/N) | Percent | Notes/Typical findings | Suggested HPO term(s) |
|---|---|---|---|---|
| Neurodevelopment: developmental delay | 30/31 | 97% | Near-universal developmental delay/delayed milestones in reported cohort; onset in infancy/early childhood (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 3-5) | HP:0001263 Global developmental delay |
| Neurodevelopment: speech delay | 29/31 | 93% | Marked speech/language delay was one of the most frequent features (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 3-5) | HP:0000750 Delayed speech and language development |
| Neurodevelopment: motor delay | 26/31 | 83% | Delayed motor milestones; mean independent ambulation ~30 months, with some walking after age 5 years (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 3-5) | HP:0001270 Motor delay; HP:0001252 Muscular hypotonia |
| Neurodevelopment: intellectual disability | 19/22 | 86% | Usually mild-to-moderate ID among assessed individuals; a minority had normal IQ (weiss2020thechd4relatedsyndrome pages 3-5) | HP:0001249 Intellectual disability |
| Neuroimaging abnormalities | 22/23 | 96% | Brain MRI findings included ventriculomegaly, hydrocephalus, thin corpus callosum, white-matter changes, Arnold–Chiari I, and moyamoya-type changes (weiss2020thechd4relatedsyndrome pages 3-5, weiss2020thechd4relatedsyndrome pages 6-7) | HP:0001276 Ventriculomegaly; HP:0001273 Abnormality of the corpus callosum; HP:0001083 Hydrocephalus; HP:0001272 Cerebellar tonsillar ectopia |
| Cardiovascular: congenital heart defects | 19/29 | 65% | Septal, conotruncal, valvular, and other structural cardiac anomalies were reported; echocardiography recommended (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 3-5, weiss2020thechd4relatedsyndrome pages 6-7) | HP:0001627 Abnormality of the cardiovascular system; HP:0001631 Atrial septal defect; HP:0001629 Ventricular septal defect |
| Growth/head size: macrocephaly | 11/28 | 39% | Common but not universal; macrocephaly is part of the recognizable phenotype (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 3-5) | HP:0000256 Macrocephaly |
| Hearing loss | 11/20 | 55% | Conductive and/or sensorineural hearing loss occurred frequently; audiology assessment advised (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 3-5, weiss2020thechd4relatedsyndrome pages 6-7) | HP:0000365 Hearing impairment; HP:0000407 Sensorineural hearing impairment; HP:0000405 Conductive hearing impairment |
| Ophthalmic abnormalities | 14/19 | 73% | Frequent eye/vision abnormalities, though specific ophthalmic findings varied across individuals (weiss2020thechd4relatedsyndrome pages 3-5, weiss2020thechd4relatedsyndrome pages 6-7) | HP:0000478 Abnormality of the eye; HP:0000501 Visual impairment |
| Skeletal/limb anomalies | 10/32 | 31% | Included vertebral fusion, carpal/tarsal coalition, syndactyly, and polydactyly (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 3-5, weiss2020thechd4relatedsyndrome pages 6-7) | HP:0000924 Abnormality of the skeletal system; HP:0002949 Vertebral fusion; HP:0006101 Finger syndactyly; HP:0010442 Polydactyly |
| Male genital anomalies: cryptorchidism/microphallus | 13/20 (males) | 65% of males | High prevalence of male genital anomalies; supports endocrine/reproductive evaluation (weiss2020thechd4relatedsyndrome pages 3-5, weiss2020thechd4relatedsyndrome pages 6-7) | HP:0000028 Cryptorchidism; HP:0000054 Micropenis |
| Endocrine/reproductive: hypogonadism | 13/32 | 40% | Hypogonadotropic hypogonadism reported; low gonadotropins documented in some males (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 3-5, weiss2020thechd4relatedsyndrome pages 6-7) | HP:0000135 Hypogonadism; HP:0000044 Hypogonadotropic hypogonadism |
| Endocrine/growth: growth hormone deficiency | 3 cases | NR | Three individuals had growth hormone deficiency; one was treated (weiss2020thechd4relatedsyndrome pages 3-5) | HP:0000824 Growth hormone deficiency; HP:0001510 Growth delay |
| Additional commonly reported but not frequency-specified feature: facial gestalt | NR | NR | High broad forehead, squared face, periorbital fullness, hypertelorism/widely spaced eyes, short nose, and small/dysmorphic ears (weiss2020thechd4relatedsyndrome pages 3-5, weiss2020thechd4relatedsyndrome pages 6-7) | HP:0002007 Frontal bossing; HP:0000316 Hypertelorism; HP:0000586 Fullness of the eyelids; HP:0003196 Short nose; HP:0000377 Abnormality of the pinna |
| Additional vascular complication | 2 cases | NR | Moyamoya angiopathy/stroke reported in a small subset; supports consideration of neurovascular imaging when clinically indicated (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 6-7) | HP:0002527 Stroke; HP:0010885 Moyamoya disease |
Table: This table summarizes the key cohort-level clinical features reported for Sifrim–Hitz–Weiss syndrome (CHD4-related syndrome) in Weiss et al. 2020, including frequencies, typical manifestations, and suggested HPO terms for knowledge-base annotation.
3.2 Phenotype characteristics (onset, severity, progression)
- Onset: Most features are apparent in infancy/childhood (developmental delays; congenital anomalies identified early with imaging/cardiac evaluation). (weiss2020thechd4relatedsyndrome pages 3-5)
- Severity: Intellectual disability is often mild-to-moderate but variable; some individuals have normal IQ. (weiss2020thechd4relatedsyndrome pages 3-5, weiss2020thechd4relatedsyndrome pages 6-7)
- Progression/course: Data are limited; however, severe complications (e.g., hydrocephalus requiring shunting; cervical spine instability) can occur and may affect long-term outcomes. (weiss2020thechd4relatedsyndrome pages 3-5)
3.3 Quality of life impact
Formal QoL instruments (e.g., PROMIS, EQ-5D) were not reported in the retrieved evidence. Functional impacts are inferred from frequent neurodevelopmental impairment and medical comorbidities. (weiss2020thechd4relatedsyndrome pages 3-5)
3.4 Visual evidence (phenotype summary tables)
Figures from the 2020 cohort paper contain phenotype-frequency tables and variant-distribution schematics. (weiss2020thechd4relatedsyndrome media ccfeaff1, weiss2020thechd4relatedsyndrome media bfb3d2d5)
4. Genetic / molecular information
4.1 Causal gene
- CHD4 (Chromodomain Helicase DNA Binding Protein 4), encoding an ATP-dependent chromatin remodeler. (weiss2016denovomutations pages 1-2)
4.2 Inheritance
- Autosomal dominant, predominantly de novo heterozygous variants in reported cohorts. (weiss2020thechd4relatedsyndrome pages 7-8, weiss2020thechd4relatedsyndrome pages 2-3)
4.3 Pathogenic variant spectrum
- Variant types: Predominantly missense or in-frame indels; a minority of truncating variants are reported, with uncertainty regarding mechanism and/or phenotypic severity in some cases. (weiss2020thechd4relatedsyndrome pages 6-7, weiss2020thechd4relatedsyndrome pages 3-5)
- Domain enrichment / hotspot: Variants are enriched in the SNF2-like ATPase/helicase region; ~50% reported in a hotspot between amino acids 1127–1192. (weiss2020thechd4relatedsyndrome pages 6-7)
- Recurrent residues: Arg1127/Arg1173/Arg1183 recur and show variable expressivity; robust genotype–phenotype correlation was not established. (weiss2020thechd4relatedsyndrome pages 6-7)
- Examples of de novo missense variants (original case series): p.Arg1127Gln, p.Trp1148Leu, p.Arg1173Leu, p.Gly1003Asp. (weiss2016denovomutations pages 1-2)
- Example of frameshift case consistent with SIHIWES: CHD4 c.4442del, p.(Gly1481Valfs*21). (silva2022anovelframeshift pages 1-2)
4.4 Functional consequences (conceptual)
- Non-truncating variants often encode stable mutant protein that can plausibly incorporate into remodeling complexes; disease mechanism is hypothesized to involve dominant-negative and/or gain-of-function effects altering chromatin remodeling output. (weiss2020thechd4relatedsyndrome pages 7-8, weiss2016denovomutations pages 1-2)
4.5 Modifier genes / epigenetics
- Modifier genes: Not identified in the retrieved evidence.
- Epigenetics: SIHIWES is a chromatinopathy; disease-specific episignature details were not available in the retrieved evidence set, though episignature-based diagnostics are discussed as a clinical approach for neurodevelopmental disorders more broadly. (alegretgarcia2025analysismethodsfor pages 23-25, alegretgarcia2025analysismethodsfor pages 1-2)
5. Environmental information
No disease-specific environmental or infectious contributors were identified in the retrieved evidence; SIHIWES is primarily monogenic. (weiss2020thechd4relatedsyndrome pages 3-5)
6. Mechanism / pathophysiology
6.1 Molecular pathways and causal chain (current model)
- Trigger: Heterozygous pathogenic CHD4 variant (usually de novo). (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 7-8)
- Primary molecular defect: Altered ATP hydrolysis and/or nucleosome sliding/remodeling, in a variant-specific manner. (weiss2020thechd4relatedsyndrome pages 2-3, weiss2020thechd4relatedsyndrome pages 7-8)
- Cellular consequence: Dysregulated chromatin accessibility and transcriptional programs during development (cell fate decisions, proliferation/differentiation timing), mediated via remodeling complexes including NuRD and ChAHP. (larrigan2023divergentphenotypesina pages 1-2, boulasiki2023thenurdcomplex pages 1-2)
- Tissue-level consequence: Abnormal neurodevelopment (cortical growth/layer production; behavioral phenotypes) and congenital anomalies (brain structural changes; heart defects). (larrigan2023divergentphenotypesina pages 1-2, weiss2020thechd4relatedsyndrome pages 3-5)
6.2 NuRD/CHD4 concepts and definitions
- The NuRD complex uniquely couples nucleosome remodeling (“opening” chromatin via sliding) with histone deacetylase activity, with CHD-family ATPases as core motors. (boulasiki2023thenurdcomplex pages 1-2)
- A 2024 mechanistic review highlights how cryo-EM and single-molecule studies clarified nucleosome sliding mechanisms, and emphasizes that auxiliary domains and complex context (NuRD vs ChAHP) modulate CHD4 activity. (reid2024howdoeschd4 pages 1-2)
6.3 Model organism evidence (recent developments; 2023)
- Mouse genetics excising the Chd4 ATPase/helicase domain shows that spatial/temporal context of CHD4 disruption produces divergent neurodevelopmental outcomes: telencephalon-specific knockout reduced cortical growth and upper-layer neuron numbers and altered anxiety/repetitive behaviors; whole-body heterozygotes showed growth defects and female-specific learning/memory changes. (larrigan2023divergentphenotypesina pages 1-2)
6.4 Suggested ontology terms (examples)
- GO biological processes: chromatin remodeling; regulation of transcription; neurogenesis; cell proliferation; cell fate commitment (supported conceptually by CHD4’s chromatin remodeler function and mouse neurodevelopmental phenotypes). (larrigan2023divergentphenotypesina pages 1-2, reid2024howdoeschd4 pages 1-2)
- CL cell types (brain): cortical progenitor cells; excitatory neurons; glia (supported by CHD4 expression across progenitors and differentiated neurons/glia in mouse cortex model). (larrigan2023divergentphenotypesina pages 1-2)
7. Anatomical structures affected
Based on cohort-level and mechanistic evidence, primary affected systems include: - Central nervous system (abnormal brain imaging; neurodevelopmental impairment). (weiss2020thechd4relatedsyndrome pages 3-5) - Cardiovascular system (congenital heart defects). (weiss2020thechd4relatedsyndrome pages 3-5) - Auditory system (hearing loss). (weiss2020thechd4relatedsyndrome pages 3-5) - Visual system (ophthalmic abnormalities). (weiss2020thechd4relatedsyndrome pages 3-5) - Skeletal system (vertebral fusion, coalitions, limb anomalies). (weiss2020thechd4relatedsyndrome pages 3-5) - Endocrine/reproductive system (hypogonadism in males; growth hormone deficiency in a minority). (weiss2020thechd4relatedsyndrome pages 3-5)
Suggested UBERON mappings (examples; ontology IDs not asserted here without direct ontology sources): brain, heart, eye, inner ear, vertebral column, gonad/pituitary axis.
8. Temporal development (onset and progression)
- Typical onset: congenital/early childhood, recognized via developmental delay and congenital anomalies. (weiss2020thechd4relatedsyndrome pages 3-5)
- Course: generally lifelong neurodevelopmental disability with variable severity; medical complications may occur (hydrocephalus; cervical spine instability; moyamoya/stroke in rare cases). (weiss2020thechd4relatedsyndrome pages 6-7, weiss2020thechd4relatedsyndrome pages 3-5)
9. Inheritance and population
9.1 Epidemiology
- Prevalence/incidence: Not available in the retrieved evidence set; published literature is case-based and does not provide population prevalence estimates. (weiss2020thechd4relatedsyndrome pages 3-5)
9.2 Inheritance, penetrance, expressivity
- Inheritance: Autosomal dominant; reported cohorts are predominantly de novo. (weiss2020thechd4relatedsyndrome pages 7-8, weiss2020thechd4relatedsyndrome pages 2-3)
- Expressivity: Variable (including discordant outcomes for recurrent variants). (weiss2020thechd4relatedsyndrome pages 6-7)
- Penetrance: Not quantified in the retrieved evidence.
10. Diagnostics
10.1 Genetic testing (current clinical implementation)
- SIHIWES is commonly identified via gene panels or exome sequencing; case reports demonstrate WES-based diagnosis in complex phenotypes and support broad genomic testing. (weiss2020thechd4relatedsyndrome pages 6-7, zeka2023casereportdiagnosis pages 1-2)
- In complex neurodevelopmental disorders, exome sequencing is described as a first-line approach (general NDD context), with added value from reanalysis and multi-omics support. (krgovic2026resolvingdiagnosticuncertainty pages 1-2)
10.2 Recommended baseline evaluations / surveillance (cohort-derived expert practice)
From the cohort authors’ clinical recommendations: - Brain and cervical spine imaging (hydrocephalus/Chiari/cervical spine anomalies). (weiss2020thechd4relatedsyndrome pages 6-7) - Echocardiogram (high CHD rate). (weiss2020thechd4relatedsyndrome pages 6-7) - Abdominal ultrasound, skeletal survey, audiologic and ophthalmologic assessments. (weiss2020thechd4relatedsyndrome pages 6-7) - Endocrine evaluation at diagnosis and puberty (hypogonadotropic hypogonadism; occasional low growth hormone). (weiss2020thechd4relatedsyndrome pages 6-7)
10.3 Differential diagnosis
Specific differentials were not enumerated in the retrieved evidence. However, the syndrome is discussed in the context of other chromatin remodeler-related neurodevelopmental disorders (conceptual overlap). (boulasiki2023thenurdcomplex pages 1-2, reid2024howdoeschd4 pages 1-2)
10.4 Omics-based adjunct diagnostics (episignatures)
- Episignature-based methylation testing (e.g., EpiSign, EpigenCentral) is described as a diagnostic adjunct for neurodevelopmental disorders, particularly to support VUS reclassification; SIHIWES-specific episignature performance was not available in the retrieved pages. (alegretgarcia2025analysismethodsfor pages 23-25, alegretgarcia2025analysismethodsfor pages 1-2)
11. Outcomes / prognosis
- Mortality: In the 32-individual cohort, two deaths were reported: one neonate died due to complications of congenital heart malformation, and one adult (age 21) died related to cervical vertebrae instability and long-term tracheostomy. (weiss2020thechd4relatedsyndrome pages 3-5)
- Morbidity: High burden of neurodevelopmental impairment and multisystem medical complications, including potential neurovascular events (moyamoya/stroke) in a small subset. (weiss2020thechd4relatedsyndrome pages 6-7)
- Life expectancy: Not quantified in the retrieved evidence.
12. Treatment
12.1 Current management paradigm
No disease-modifying therapy is established; management is supportive and complication-directed with proactive surveillance (cardiac, neuroimaging, hearing/vision, endocrine, skeletal). (weiss2020thechd4relatedsyndrome pages 6-7, weiss2020thechd4relatedsyndrome pages 3-5)
12.2 Reported interventions (case-based)
- Growth hormone (rhGH): Growth hormone deficiency was reported in 3 individuals in the cohort, with at least one treated. (weiss2020thechd4relatedsyndrome pages 3-5)
- A later case report described rhGH therapy with improved growth velocity over follow-up, but emphasized that long-term efficacy/safety requires larger studies. (zhang2026clinicalinsightsfrom pages 4-5, zhang2026clinicalinsightsfrom pages 1-2)
- Antiseizure medications: In a complex case with multiple diagnoses including SIHIWES, polytherapy (valproic acid, levetiracetam, phenobarbital, clonazepam) provided only partial seizure control. (zeka2023casereportdiagnosis pages 1-2)
12.3 Experimental / clinical trials
- A ClinicalTrials.gov search performed in this run did not identify SIHIWES-targeted interventional trials in the retrieved results. (zeka2023casereportdiagnosis pages 1-2)
12.4 Suggested MAXO terms (examples)
- Genetic testing / exome sequencing; echocardiography; audiologic evaluation; ophthalmologic evaluation; endocrine evaluation; growth hormone therapy; developmental therapy/early intervention (supportive recommendations inferred from surveillance practices and reported rhGH use). (weiss2020thechd4relatedsyndrome pages 6-7, zhang2026clinicalinsightsfrom pages 4-5)
13. Prevention
- Primary prevention: Not applicable in the typical sense for a predominantly de novo Mendelian disorder.
- Secondary/tertiary prevention: Emphasis is on early genetic diagnosis and proactive surveillance to prevent complications (e.g., manage CHD, monitor hydrocephalus/cervical spine issues, address hearing/vision problems, endocrine management). (weiss2020thechd4relatedsyndrome pages 6-7)
- Genetic counseling: Implied by autosomal dominant genetics and de novo occurrence; specific counseling guidance text was not present in retrieved evidence. (weiss2020thechd4relatedsyndrome pages 7-8)
14. Other species / natural disease
No naturally occurring veterinary disease analogs were identified in the retrieved evidence set.
15. Model organisms
- Mouse models (neurodevelopment): Conditional and germline manipulations of Chd4 ATPase/helicase region demonstrate roles in cortical development and behavior and illustrate context dependence of phenotypes. (larrigan2023divergentphenotypesina pages 1-2)
Recent developments and latest research (prioritizing 2023–2024)
2023
- Mouse-model mechanistic dissection: Divergent phenotypes in germline versus conditional/telencephalon-specific CHD4 loss-of-function manipulations highlight spatiotemporal dependence of CHD4 roles in neurodevelopment and behavior. (larrigan2023divergentphenotypesina pages 1-2)
- NuRD-focused synthesis: Review articles emphasize NuRD composition/function in neurodevelopment and how NuRD subunit mutations, including CHD family members, lead to neurodevelopmental disorders. (boulasiki2023thenurdcomplex pages 1-2)
2024
- Molecular mechanism of CHD4 nucleosome sliding: A 2024 review summarizes how cryo-EM and single-molecule biophysics have recently clarified core remodeling mechanisms and the influence of auxiliary domains/complex context (NuRD vs ChAHP). (reid2024howdoeschd4 pages 1-2)
Limitations of this report (evidence gaps)
- Formal identifiers beyond OMIM/MIM 617159 (e.g., MONDO, Orphanet, MeSH, ICD) were not retrievable in the provided full-text evidence set.
- Robust epidemiology (incidence/prevalence), penetrance estimates, and validated QoL statistics were not available in the retrieved literature excerpts.
- Treatment evidence is limited to supportive care recommendations and individual case reports (e.g., rhGH; antiseizure therapy in a dual-diagnosis case) without controlled trial data. (weiss2020thechd4relatedsyndrome pages 3-5, zhang2026clinicalinsightsfrom pages 4-5, zeka2023casereportdiagnosis pages 1-2)
Key primary sources (with publication dates and URLs)
- Weiss K. et al. “De Novo Mutations in CHD4… Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms.” Am J Hum Genet. Oct 2016. https://doi.org/10.1016/j.ajhg.2016.08.001 (weiss2016denovomutations pages 2-3)
- Weiss K. et al. “The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis.” Genetics in Medicine Feb 2020. https://doi.org/10.1038/s41436-019-0612-0 (weiss2020thechd4relatedsyndrome pages 1-2)
- Larrigan S. et al. “Divergent phenotypes… mouse models of Sifrim-Hitz-Weiss syndrome.” Hum Mol Genet Sep 2023. https://doi.org/10.1093/hmg/ddad157 (larrigan2023divergentphenotypesina pages 1-2)
- Boulasiki P. et al. “The NuRD Complex in Neurodevelopment and Disease: A Case of Sliding Doors.” Cells Apr 2023. https://doi.org/10.3390/cells12081179 (boulasiki2023thenurdcomplex pages 1-2)
- Reid XJ. et al. “How does CHD4 slide nucleosomes?” Biochem Soc Trans Sep 2024. https://doi.org/10.1042/bst20230070 (reid2024howdoeschd4 pages 1-2)
- Zeka N. et al. Case report: SIHIWES + KCNT1 + ACADM diagnosis via exome sequencing. Front Pediatr Sep 2023. https://doi.org/10.3389/fped.2023.1230056 (zeka2023casereportdiagnosis pages 1-2)
References
-
(weiss2020thechd4relatedsyndrome pages 1-2): Karin Weiss, Hayley P. Lazar, Alina Kurolap, Ariel F. Martinez, Tamar Paperna, Lior Cohen, Marie F. Smeland, Sandra Whalen, Solveig Heide, Boris Keren, Pauline Terhal, Melita Irving, Motoki Takaku, John D. Roberts, Robert M. Petrovich, Samantha A. Schrier Vergano, Amy Kenney, Hanne Hove, Elizabeth DeChene, Shane C. Quinonez, Estelle Colin, Alban Ziegler, Melissa Rumple, Mahim Jain, Danielle Monteil, Elizabeth R. Roeder, Kimberly Nugent, Arie van Haeringen, Michael Gambello, Avni Santani, Līvija Medne, Bryan Krock, Cara M. Skraban, Elaine H. Zackai, Holly A. Dubbs, Thomas Smol, Jamal Ghoumid, Michael J. Parker, Michael Wright, Peter Turnpenny, Jill Clayton-Smith, Kay Metcalfe, Hitoshi Kurumizaka, Bruce D. Gelb, Hagit Baris Feldman, Philippe M. Campeau, Maximilian Muenke, Paul A. Wade, and Katherine Lachlan. The chd4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis. Genetics in Medicine, 22:389-397, Feb 2020. URL: https://doi.org/10.1038/s41436-019-0612-0, doi:10.1038/s41436-019-0612-0. This article has 91 citations and is from a highest quality peer-reviewed journal.
-
(weiss2020thechd4relatedsyndrome pages 3-5): Karin Weiss, Hayley P. Lazar, Alina Kurolap, Ariel F. Martinez, Tamar Paperna, Lior Cohen, Marie F. Smeland, Sandra Whalen, Solveig Heide, Boris Keren, Pauline Terhal, Melita Irving, Motoki Takaku, John D. Roberts, Robert M. Petrovich, Samantha A. Schrier Vergano, Amy Kenney, Hanne Hove, Elizabeth DeChene, Shane C. Quinonez, Estelle Colin, Alban Ziegler, Melissa Rumple, Mahim Jain, Danielle Monteil, Elizabeth R. Roeder, Kimberly Nugent, Arie van Haeringen, Michael Gambello, Avni Santani, Līvija Medne, Bryan Krock, Cara M. Skraban, Elaine H. Zackai, Holly A. Dubbs, Thomas Smol, Jamal Ghoumid, Michael J. Parker, Michael Wright, Peter Turnpenny, Jill Clayton-Smith, Kay Metcalfe, Hitoshi Kurumizaka, Bruce D. Gelb, Hagit Baris Feldman, Philippe M. Campeau, Maximilian Muenke, Paul A. Wade, and Katherine Lachlan. The chd4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis. Genetics in Medicine, 22:389-397, Feb 2020. URL: https://doi.org/10.1038/s41436-019-0612-0, doi:10.1038/s41436-019-0612-0. This article has 91 citations and is from a highest quality peer-reviewed journal.
-
(weiss2016denovomutations pages 2-3): Karin Weiss, Paulien A. Terhal, Lior Cohen, Michael Bruccoleri, Melita Irving, Ariel F. Martinez, Jill A. Rosenfeld, Keren Machol, Yaping Yang, Pengfei Liu, Magdalena Walkiewicz, Joke Beuten, Natalia Gomez-Ospina, Katrina Haude, Chin-To Fong, Gregory M. Enns, Jonathan A. Bernstein, Judith Fan, Garrett Gotway, Mohammad Ghorbani, Koen van Gassen, Glen R. Monroe, Gijs van Haaften, Lina Basel-Vanagaite, Xiang-Jiao Yang, Philippe M. Campeau, and Maximilian Muenke. De novo mutations in chd4, an atp-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. American journal of human genetics, 99 4:934-941, Oct 2016. URL: https://doi.org/10.1016/j.ajhg.2016.08.001, doi:10.1016/j.ajhg.2016.08.001. This article has 175 citations and is from a highest quality peer-reviewed journal.
-
(weiss2016denovomutations pages 1-2): Karin Weiss, Paulien A. Terhal, Lior Cohen, Michael Bruccoleri, Melita Irving, Ariel F. Martinez, Jill A. Rosenfeld, Keren Machol, Yaping Yang, Pengfei Liu, Magdalena Walkiewicz, Joke Beuten, Natalia Gomez-Ospina, Katrina Haude, Chin-To Fong, Gregory M. Enns, Jonathan A. Bernstein, Judith Fan, Garrett Gotway, Mohammad Ghorbani, Koen van Gassen, Glen R. Monroe, Gijs van Haaften, Lina Basel-Vanagaite, Xiang-Jiao Yang, Philippe M. Campeau, and Maximilian Muenke. De novo mutations in chd4, an atp-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. American journal of human genetics, 99 4:934-941, Oct 2016. URL: https://doi.org/10.1016/j.ajhg.2016.08.001, doi:10.1016/j.ajhg.2016.08.001. This article has 175 citations and is from a highest quality peer-reviewed journal.
-
(silva2022anovelframeshift pages 2-4): Jorge Diogo Da Silva, Natália Oliva-Teles, Nataliya Tkachenko, Joana Fino, Mariana Marques, Ana Maria Fortuna, and Dezso David. A novel frameshift chd4 variant leading to sifrim-hitz-weiss syndrome in a proband with a subclinical familial t(17;19) and a large dup(2)(q14.3q21.1). Biomedicines, 11:12, Dec 2022. URL: https://doi.org/10.3390/biomedicines11010012, doi:10.3390/biomedicines11010012. This article has 2 citations.
-
(weiss2020thechd4relatedsyndrome pages 2-3): Karin Weiss, Hayley P. Lazar, Alina Kurolap, Ariel F. Martinez, Tamar Paperna, Lior Cohen, Marie F. Smeland, Sandra Whalen, Solveig Heide, Boris Keren, Pauline Terhal, Melita Irving, Motoki Takaku, John D. Roberts, Robert M. Petrovich, Samantha A. Schrier Vergano, Amy Kenney, Hanne Hove, Elizabeth DeChene, Shane C. Quinonez, Estelle Colin, Alban Ziegler, Melissa Rumple, Mahim Jain, Danielle Monteil, Elizabeth R. Roeder, Kimberly Nugent, Arie van Haeringen, Michael Gambello, Avni Santani, Līvija Medne, Bryan Krock, Cara M. Skraban, Elaine H. Zackai, Holly A. Dubbs, Thomas Smol, Jamal Ghoumid, Michael J. Parker, Michael Wright, Peter Turnpenny, Jill Clayton-Smith, Kay Metcalfe, Hitoshi Kurumizaka, Bruce D. Gelb, Hagit Baris Feldman, Philippe M. Campeau, Maximilian Muenke, Paul A. Wade, and Katherine Lachlan. The chd4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis. Genetics in Medicine, 22:389-397, Feb 2020. URL: https://doi.org/10.1038/s41436-019-0612-0, doi:10.1038/s41436-019-0612-0. This article has 91 citations and is from a highest quality peer-reviewed journal.
-
(weiss2020thechd4relatedsyndrome pages 6-7): Karin Weiss, Hayley P. Lazar, Alina Kurolap, Ariel F. Martinez, Tamar Paperna, Lior Cohen, Marie F. Smeland, Sandra Whalen, Solveig Heide, Boris Keren, Pauline Terhal, Melita Irving, Motoki Takaku, John D. Roberts, Robert M. Petrovich, Samantha A. Schrier Vergano, Amy Kenney, Hanne Hove, Elizabeth DeChene, Shane C. Quinonez, Estelle Colin, Alban Ziegler, Melissa Rumple, Mahim Jain, Danielle Monteil, Elizabeth R. Roeder, Kimberly Nugent, Arie van Haeringen, Michael Gambello, Avni Santani, Līvija Medne, Bryan Krock, Cara M. Skraban, Elaine H. Zackai, Holly A. Dubbs, Thomas Smol, Jamal Ghoumid, Michael J. Parker, Michael Wright, Peter Turnpenny, Jill Clayton-Smith, Kay Metcalfe, Hitoshi Kurumizaka, Bruce D. Gelb, Hagit Baris Feldman, Philippe M. Campeau, Maximilian Muenke, Paul A. Wade, and Katherine Lachlan. The chd4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis. Genetics in Medicine, 22:389-397, Feb 2020. URL: https://doi.org/10.1038/s41436-019-0612-0, doi:10.1038/s41436-019-0612-0. This article has 91 citations and is from a highest quality peer-reviewed journal.
-
(boulasiki2023thenurdcomplex pages 1-2): Paraskevi Boulasiki, Xiao Wei Tan, Matteo Spinelli, and Antonella Riccio. The nurd complex in neurodevelopment and disease: a case of sliding doors. Cells, 12:1179, Apr 2023. URL: https://doi.org/10.3390/cells12081179, doi:10.3390/cells12081179. This article has 30 citations.
-
(reid2024howdoeschd4 pages 1-2): Xavier J. Reid, Yichen Zhong, and Joel P. Mackay. How does chd4 slide nucleosomes? Biochemical Society Transactions, 52:1995-2008, Sep 2024. URL: https://doi.org/10.1042/bst20230070, doi:10.1042/bst20230070. This article has 9 citations and is from a peer-reviewed journal.
-
(weiss2020thechd4relatedsyndrome media ccfeaff1): Karin Weiss, Hayley P. Lazar, Alina Kurolap, Ariel F. Martinez, Tamar Paperna, Lior Cohen, Marie F. Smeland, Sandra Whalen, Solveig Heide, Boris Keren, Pauline Terhal, Melita Irving, Motoki Takaku, John D. Roberts, Robert M. Petrovich, Samantha A. Schrier Vergano, Amy Kenney, Hanne Hove, Elizabeth DeChene, Shane C. Quinonez, Estelle Colin, Alban Ziegler, Melissa Rumple, Mahim Jain, Danielle Monteil, Elizabeth R. Roeder, Kimberly Nugent, Arie van Haeringen, Michael Gambello, Avni Santani, Līvija Medne, Bryan Krock, Cara M. Skraban, Elaine H. Zackai, Holly A. Dubbs, Thomas Smol, Jamal Ghoumid, Michael J. Parker, Michael Wright, Peter Turnpenny, Jill Clayton-Smith, Kay Metcalfe, Hitoshi Kurumizaka, Bruce D. Gelb, Hagit Baris Feldman, Philippe M. Campeau, Maximilian Muenke, Paul A. Wade, and Katherine Lachlan. The chd4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis. Genetics in Medicine, 22:389-397, Feb 2020. URL: https://doi.org/10.1038/s41436-019-0612-0, doi:10.1038/s41436-019-0612-0. This article has 91 citations and is from a highest quality peer-reviewed journal.
-
(weiss2020thechd4relatedsyndrome media bfb3d2d5): Karin Weiss, Hayley P. Lazar, Alina Kurolap, Ariel F. Martinez, Tamar Paperna, Lior Cohen, Marie F. Smeland, Sandra Whalen, Solveig Heide, Boris Keren, Pauline Terhal, Melita Irving, Motoki Takaku, John D. Roberts, Robert M. Petrovich, Samantha A. Schrier Vergano, Amy Kenney, Hanne Hove, Elizabeth DeChene, Shane C. Quinonez, Estelle Colin, Alban Ziegler, Melissa Rumple, Mahim Jain, Danielle Monteil, Elizabeth R. Roeder, Kimberly Nugent, Arie van Haeringen, Michael Gambello, Avni Santani, Līvija Medne, Bryan Krock, Cara M. Skraban, Elaine H. Zackai, Holly A. Dubbs, Thomas Smol, Jamal Ghoumid, Michael J. Parker, Michael Wright, Peter Turnpenny, Jill Clayton-Smith, Kay Metcalfe, Hitoshi Kurumizaka, Bruce D. Gelb, Hagit Baris Feldman, Philippe M. Campeau, Maximilian Muenke, Paul A. Wade, and Katherine Lachlan. The chd4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis. Genetics in Medicine, 22:389-397, Feb 2020. URL: https://doi.org/10.1038/s41436-019-0612-0, doi:10.1038/s41436-019-0612-0. This article has 91 citations and is from a highest quality peer-reviewed journal.
-
(weiss2020thechd4relatedsyndrome pages 7-8): Karin Weiss, Hayley P. Lazar, Alina Kurolap, Ariel F. Martinez, Tamar Paperna, Lior Cohen, Marie F. Smeland, Sandra Whalen, Solveig Heide, Boris Keren, Pauline Terhal, Melita Irving, Motoki Takaku, John D. Roberts, Robert M. Petrovich, Samantha A. Schrier Vergano, Amy Kenney, Hanne Hove, Elizabeth DeChene, Shane C. Quinonez, Estelle Colin, Alban Ziegler, Melissa Rumple, Mahim Jain, Danielle Monteil, Elizabeth R. Roeder, Kimberly Nugent, Arie van Haeringen, Michael Gambello, Avni Santani, Līvija Medne, Bryan Krock, Cara M. Skraban, Elaine H. Zackai, Holly A. Dubbs, Thomas Smol, Jamal Ghoumid, Michael J. Parker, Michael Wright, Peter Turnpenny, Jill Clayton-Smith, Kay Metcalfe, Hitoshi Kurumizaka, Bruce D. Gelb, Hagit Baris Feldman, Philippe M. Campeau, Maximilian Muenke, Paul A. Wade, and Katherine Lachlan. The chd4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype–phenotype correlations, and molecular basis. Genetics in Medicine, 22:389-397, Feb 2020. URL: https://doi.org/10.1038/s41436-019-0612-0, doi:10.1038/s41436-019-0612-0. This article has 91 citations and is from a highest quality peer-reviewed journal.
-
(silva2022anovelframeshift pages 1-2): Jorge Diogo Da Silva, Natália Oliva-Teles, Nataliya Tkachenko, Joana Fino, Mariana Marques, Ana Maria Fortuna, and Dezso David. A novel frameshift chd4 variant leading to sifrim-hitz-weiss syndrome in a proband with a subclinical familial t(17;19) and a large dup(2)(q14.3q21.1). Biomedicines, 11:12, Dec 2022. URL: https://doi.org/10.3390/biomedicines11010012, doi:10.3390/biomedicines11010012. This article has 2 citations.
-
(alegretgarcia2025analysismethodsfor pages 23-25): Albert Alegret-García, Alejandro Cáceres, Marta Sevilla-Porras, Luís A. Pérez-Jurado, and Juan R. González. Analysis methods for diagnosing rare neurodevelopmental diseases with episignatures: a systematic review of the literature. Biomedicines, 13:3043, Dec 2025. URL: https://doi.org/10.3390/biomedicines13123043, doi:10.3390/biomedicines13123043. This article has 0 citations.
-
(alegretgarcia2025analysismethodsfor pages 1-2): Albert Alegret-García, Alejandro Cáceres, Marta Sevilla-Porras, Luís A. Pérez-Jurado, and Juan R. González. Analysis methods for diagnosing rare neurodevelopmental diseases with episignatures: a systematic review of the literature. Biomedicines, 13:3043, Dec 2025. URL: https://doi.org/10.3390/biomedicines13123043, doi:10.3390/biomedicines13123043. This article has 0 citations.
-
(larrigan2023divergentphenotypesina pages 1-2): Sarah Larrigan, Shrilaxmi V Joshi, and Pierre Mattar. Divergent phenotypes in constitutive versus conditional mutant mouse models of sifrim-hitz-weiss syndrome. Human molecular genetics, 32:3361-3373, Sep 2023. URL: https://doi.org/10.1093/hmg/ddad157, doi:10.1093/hmg/ddad157. This article has 5 citations and is from a domain leading peer-reviewed journal.
-
(zeka2023casereportdiagnosis pages 1-2): Naim Zeka, Eris Zeka, Esra Zhubi, and Ilir Hoxha. Case report: diagnosis of a patient with sifrim–hitz–weiss syndrome, development and epileptic encephalopathy-14, and medium chain acyl-coa dehydrogenase deficiency. Frontiers in Pediatrics, Sep 2023. URL: https://doi.org/10.3389/fped.2023.1230056, doi:10.3389/fped.2023.1230056. This article has 4 citations.
-
(krgovic2026resolvingdiagnosticuncertainty pages 1-2): Danijela Krgovic, Peter Gradisnik, Andreja Osterc Koprivsek, Ana Kogovsek, Nadja Kokalj Vokac, and Spela Stangler Herodez. Resolving diagnostic uncertainty in neurodevelopmental disorders using exome sequencing supported by literature-based multi-omics evidence. Biomolecules, 16:399, Mar 2026. URL: https://doi.org/10.3390/biom16030399, doi:10.3390/biom16030399. This article has 0 citations.
-
(zhang2026clinicalinsightsfrom pages 4-5): Jianmei Zhang, Shuangzhong Chen, Guanping Dong, Suhong Yang, Ping Wang, Qiong Zhou, and Pingping Wang. Clinical insights from a case of sifrim‐hitz‐weiss syndrome with a
chd4 variant: expanding the phenotypic spectrum and its response to growth hormone therapy. American Journal of Medical Genetics Part A, 200:1403-1410, Feb 2026. URL: https://doi.org/10.1002/ajmg.a.70084, doi:10.1002/ajmg.a.70084. This article has 0 citations. -
(zhang2026clinicalinsightsfrom pages 1-2): Jianmei Zhang, Shuangzhong Chen, Guanping Dong, Suhong Yang, Ping Wang, Qiong Zhou, and Pingping Wang. Clinical insights from a case of sifrim‐hitz‐weiss syndrome with a
chd4 variant: expanding the phenotypic spectrum and its response to growth hormone therapy. American Journal of Medical Genetics Part A, 200:1403-1410, Feb 2026. URL: https://doi.org/10.1002/ajmg.a.70084, doi:10.1002/ajmg.a.70084. This article has 0 citations.