Rubinstein-Taybi Syndrome
Rubinstein-Taybi syndrome (RSTS) is a rare autosomal dominant multiple-congenital-anomaly / intellectual disability syndrome caused by haploinsufficiency of CREBBP (RSTS1, ~70% of cases) or, less commonly, EP300 (RSTS2, ~5-10% of cases). CREBBP and EP300 encode the paralogous histone acetyltransferases CBP and p300, which act as transcriptional coactivators. Loss of one functional copy reduces histone acetyltransferase activity and disrupts chromatin-mediated transcriptional regulation during development, producing broad and often angulated thumbs and halluces, distinctive facial features, postnatal growth retardation, short stature, moderate-to-severe intellectual disability, and an increased incidence of certain benign tumors.
Ask OpenScientist
Ask a research question about Rubinstein-Taybi Syndrome. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (2 references)
Subtypes
2Show evidence (1 reference)
Show evidence (2 references)
Pathophysiology
3Show evidence (4 references)
Show evidence (1 reference)
Show evidence (2 references)
Pathograph
Phenotypes
22Cardiovascular 1
Show evidence (1 reference)
Digestive 2
Show evidence (1 reference)
Show evidence (1 reference)
Ear 1
Show evidence (1 reference)
Genitourinary 2
Show evidence (1 reference)
Show evidence (1 reference)
Head and Neck 3
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Immune 1
Show evidence (2 references)
Integument 1
Show evidence (1 reference)
Limbs 2
Show evidence (2 references)
Show evidence (2 references)
Nervous System 2
Show evidence (1 reference)
Show evidence (2 references)
Respiratory 1
Show evidence (1 reference)
Growth 2
Show evidence (1 reference)
Show evidence (1 reference)
Neoplasm 1
Show evidence (1 reference)
Other 3
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Genetic Associations
2Show evidence (2 references)
Show evidence (2 references)
Medical Actions
4Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Clinical Trials
1Show evidence (2 references)
Source YAML
click to showname: Rubinstein-Taybi Syndrome
creation_date: "2026-06-03T00:00:00Z"
description: >
Rubinstein-Taybi syndrome (RSTS) is a rare autosomal dominant
multiple-congenital-anomaly / intellectual disability syndrome caused by
haploinsufficiency of CREBBP (RSTS1, ~70% of cases) or, less commonly, EP300
(RSTS2, ~5-10% of cases). CREBBP and EP300 encode the paralogous histone
acetyltransferases CBP and p300, which act as transcriptional coactivators.
Loss of one functional copy reduces histone acetyltransferase activity and
disrupts chromatin-mediated transcriptional regulation during development,
producing broad and often angulated thumbs and halluces, distinctive facial
features, postnatal growth retardation, short stature, moderate-to-severe
intellectual disability, and an increased incidence of certain benign tumors.
category: Mendelian
parents:
- Autosomal dominant syndromic intellectual disability
disease_term:
preferred_term: Rubinstein-Taybi syndrome
term:
id: MONDO:0019188
label: Rubinstein-Taybi syndrome
references:
- reference: PMID:20301699
title: "Rubinstein-Taybi Syndrome."
tags:
- GeneReviews
- reference: PMID:38471765
title: "Diagnosis and management in Rubinstein-Taybi syndrome: first international consensus statement."
has_subtypes:
- name: RSTS1
display_name: Rubinstein-Taybi syndrome type 1 (CREBBP)
description: >
The most common form of Rubinstein-Taybi syndrome, caused by heterozygous
pathogenic variants or microdeletions involving CREBBP (16p13.3), accounting
for approximately 70% of cases.
subtype_term:
preferred_term: Rubinstein-Taybi syndrome due to CREBBP mutations
term:
id: MONDO:0008393
label: Rubinstein-Taybi syndrome due to CREBBP mutations
evidence:
- reference: PMID:38471765
reference_title: "Diagnosis and management in Rubinstein-Taybi syndrome: first international consensus statement."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "clinical diagnostic criteria for types of RTS (RTS1: CREBBP; RTS2: EP300)"
explanation: >
The 2024 international consensus defines RTS1 as the CREBBP-related type of
Rubinstein-Taybi syndrome.
- name: RSTS2
display_name: Rubinstein-Taybi syndrome type 2 (EP300)
description: >
A less common form caused by heterozygous pathogenic variants in EP300,
accounting for approximately 5-10% of cases. EP300-related RSTS tends to be
associated with milder intellectual disability, and some individuals have
normal intellect.
subtype_term:
preferred_term: Rubinstein-Taybi syndrome due to EP300 haploinsufficiency
term:
id: MONDO:0013364
label: Rubinstein-Taybi syndrome due to EP300 haploinsufficiency
evidence:
- reference: PMID:38471765
reference_title: "Diagnosis and management in Rubinstein-Taybi syndrome: first international consensus statement."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "clinical diagnostic criteria for types of RTS (RTS1: CREBBP; RTS2: EP300)"
explanation: >
The 2024 international consensus defines RTS2 as the EP300-related type of
Rubinstein-Taybi syndrome.
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Some individuals with EP300-related RSTS have normal intellect."
explanation: >
GeneReviews notes that EP300-related RSTS can present with milder cognitive
involvement, distinguishing it from the CREBBP type.
inheritance:
- name: Autosomal dominant inheritance
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "RSTS is inherited in an autosomal dominant manner."
explanation: GeneReviews documents autosomal dominant inheritance for RSTS.
- reference: PMID:7630403
reference_title: "Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Because the patients are heterozygous for the mutations, we propose that the loss of one functional copy of the CBP gene underlies the developmental abnormalities in RTS"
explanation: >
Petrij et al. established that heterozygous loss-of-function of CBP
(haploinsufficiency) underlies RSTS, consistent with dominant inheritance.
pathophysiology:
- name: CBP/p300 Haploinsufficiency and Loss of Histone Acetyltransferase Activity
description: >
Heterozygous loss-of-function variants or deletions in CREBBP or EP300
reduce the dosage of the paralogous histone acetyltransferases CBP and p300.
Both proteins function as transcriptional coactivators that acetylate
histones and other substrates; a certain level of CBP is essential for
normal development. Loss of one functional allele lowers histone
acetyltransferase activity, producing aberrant chromatin regulation and
dysregulated transcription that drives the developmental phenotype.
biological_processes:
- preferred_term: Histone (peptidyl-lysine) acetylation
term:
id: GO:0018394
label: peptidyl-lysine acetylation
modifier: DECREASED
- preferred_term: CBP/p300-mediated transcriptional coactivation
term:
id: GO:0045944
label: positive regulation of transcription by RNA polymerase II
modifier: DECREASED
- preferred_term: Chromatin organization
term:
id: GO:0006325
label: chromatin organization
modifier: ABNORMAL
evidence:
- reference: PMID:15706485
reference_title: "Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "There is a direct link between loss of acetyl transferase activity and RSTS, which indicates that the disorder is caused by aberrant chromatin regulation."
explanation: >
Roelfsema et al. directly link loss of CBP/p300 histone acetyltransferase
activity to RSTS via aberrant chromatin regulation.
- reference: PMID:15706485
reference_title: "Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "CREB-binding protein and p300 function as transcriptional coactivators in the regulation of gene expression through various signal-transduction pathways. Both are potent histone acetyl transferases."
explanation: >
Establishes the molecular function of CBP and p300 as transcriptional
coactivators and histone acetyltransferases central to the mechanism.
- reference: PMID:7630403
reference_title: "Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "the gene for the human CREB binding protein (CBP), a nuclear protein participating as a co-activator in cyclic-AMP-regulated gene expression"
explanation: >
Identifies CBP as the transcriptional coactivator whose loss underlies
RSTS, supporting the coactivator/transcription mechanism.
- reference: PMID:38471765
reference_title: "Diagnosis and management in Rubinstein-Taybi syndrome: first international consensus statement."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "It is caused by variants in either of two genes (CREBBP, EP300) which encode for the proteins CBP and p300, which both have a function in transcription regulation and histone acetylation."
explanation: >
The 2024 international consensus statement confirms that RTS is caused by
CREBBP/EP300 variants affecting CBP/p300 transcription regulation and
histone acetylation.
downstream:
- target: Impaired Neurodevelopment and Intellectual Disability
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
description: >
Reduced CBP/p300-dependent histone acetylation impairs neurodevelopmental
transcriptional programs, contributing to intellectual disability.
evidence:
- reference: PMID:15706485
reference_title: "Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A certain level of CREB-binding protein is essential for normal development, since inactivation of one allele causes Rubinstein-Taybi syndrome (RSTS)."
explanation: >
Roelfsema et al. link insufficient CBP dosage to the developmental
(including neurodevelopmental) abnormalities of RSTS.
- target: Tumor Predisposition
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
description: >
Loss of one functional CBP/p300 copy, which have tumor-suppressor roles,
is proposed to underlie the increased incidence of certain benign tumors
in RSTS.
evidence:
- reference: PMID:7630403
reference_title: "Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "the loss of one functional copy of the CBP gene underlies the developmental abnormalities in RTS and possibly the propensity for malignancy"
explanation: >
Petrij et al. proposed that CBP haploinsufficiency may underlie the
tumor predisposition; classified PARTIAL because the malignancy link was
proposed rather than demonstrated.
- name: Impaired Neurodevelopment and Intellectual Disability
description: >
CBP/p300-dependent histone acetylation is required for the transcriptional
programs underlying neurogenesis, neuronal differentiation, and
activity-dependent gene expression involved in learning and memory.
Reduced acetyltransferase activity impairs these programs, contributing to
the global developmental delay and moderate-to-severe intellectual
disability characteristic of RSTS.
cell_types:
- preferred_term: Neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: Nervous system development
term:
id: GO:0007399
label: nervous system development
modifier: ABNORMAL
- preferred_term: Regulation of neurogenesis
term:
id: GO:0050767
label: regulation of neurogenesis
modifier: ABNORMAL
- preferred_term: Learning or memory
term:
id: GO:0007611
label: learning or memory
modifier: ABNORMAL
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Rubinstein-Taybi syndrome (RSTS) is characterized by distinctive facial features, broad and often angulated thumbs and halluces, short stature, and moderate-to-severe intellectual disability."
explanation: >
GeneReviews documents moderate-to-severe intellectual disability as a core
feature, the clinical correlate of impaired CBP/p300-dependent
neurodevelopment.
downstream:
- target: Broad thumb
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Broad hallux
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Intellectual disability
causal_link_type: DIRECT
- target: Neuropsychiatric and behavioral challenges
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Distinctive facial features
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: High palate
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Talon cusps
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Short stature
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Postnatal microcephaly
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Obesity
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Congenital heart defects
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Cryptorchidism
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Recurrent infections
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Primary immunodeficiency with antibody defect
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Severe constipation
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Hearing loss
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Ocular abnormalities
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Renal abnormalities
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Respiratory difficulties
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Feeding problems
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- name: Tumor Predisposition
description: >
CBP and p300 have tumor-suppressor roles, and loss of one functional copy
is associated with an increased incidence of certain benign tumors,
particularly meningiomas and pilomatricomas. Petrij et al. proposed that
loss of one functional CBP allele may underlie the propensity for
malignancy; population-based data confirm a significantly elevated incidence
of meningiomas and pilomatricomas in RSTS.
cell_types:
- preferred_term: Arachnoid (meningothelial) cell
term:
id: CL:4023097
label: arachnoid barrier cell
biological_processes:
- preferred_term: Cell population proliferation
term:
id: GO:0008283
label: cell population proliferation
modifier: INCREASED
evidence:
- reference: PMID:29359884
reference_title: "Benign and malignant tumors in Rubinstein-Taybi syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Meningiomas and pilomatricomas were the most frequent benign tumors and their incidence was significantly elevated in comparison to the general Dutch population."
explanation: >
Population-based Dutch registry data demonstrate significantly elevated
incidence of meningiomas and pilomatricomas in RSTS.
- reference: PMID:7630403
reference_title: "Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "the loss of one functional copy of the CBP gene underlies the developmental abnormalities in RTS and possibly the propensity for malignancy"
explanation: >
Petrij et al. proposed that CBP haploinsufficiency may underlie the tumor
predisposition; classified PARTIAL because the malignancy link was
proposed rather than demonstrated.
downstream:
- target: Meningioma
causal_link_type: DIRECT
- target: Pilomatricoma
causal_link_type: DIRECT
phenotypes:
- category: Skeletal
name: Broad thumb
description: >
Broad and often angulated thumbs are a hallmark feature of RSTS.
phenotype_term:
preferred_term: Broad thumb
term:
id: HP:0011304
label: Broad thumb
evidence:
- reference: PMID:7630403
reference_title: "Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "a well-defined syndrome with facial abnormalities, broad thumbs, broad big toes and mental retardation as the main clinical features"
explanation: Petrij et al. list broad thumbs among the main clinical features of RSTS.
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "broad and often angulated thumbs and halluces"
explanation: GeneReviews documents broad, often angulated thumbs as a hallmark.
- category: Skeletal
name: Broad hallux
description: >
Broad great toes (halluces), often angulated, are a hallmark feature.
phenotype_term:
preferred_term: Broad hallux
term:
id: HP:0010055
label: Broad hallux
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "broad and often angulated thumbs and halluces"
explanation: GeneReviews documents broad, often angulated halluces (great toes).
- reference: PMID:7630403
reference_title: "Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "broad thumbs, broad big toes"
explanation: Petrij et al. list broad big toes among the main clinical features.
- category: Neurologic
name: Intellectual disability
description: >
Moderate-to-severe intellectual disability is characteristic, with average
IQ between 35 and 50, though developmental outcome varies; some individuals
with EP300-related RSTS have normal intellect.
phenotype_term:
preferred_term: Intellectual disability
term:
id: HP:0001249
label: Intellectual disability
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Average IQ ranges between 35 and 50; however, developmental outcome varies considerably. Some individuals with EP300-related RSTS have normal intellect."
explanation: >
GeneReviews documents moderate-to-severe intellectual disability with
variable outcome and milder EP300-related presentations.
- category: Behavioral
name: Neuropsychiatric and behavioral challenges
description: >
Individuals with RSTS show a high prevalence of neuropsychiatric and
behavioral challenges across the lifespan, including OCD-like symptoms,
anxiety, and challenging behaviors. RSTS2 (EP300) individuals tend to have
better adaptive behavior but higher social phobia than RSTS1 (CREBBP).
phenotype_term:
preferred_term: Neuropsychiatric and behavioral challenges
term:
id: HP:0000708
label: Atypical behavior
evidence:
- reference: PMID:37415602
reference_title: "Behavioral and neuropsychiatric challenges across the lifespan in individuals with Rubinstein-Taybi syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Results revealed a high prevalence of neuropsychiatric and behavioral challenges across ages."
explanation: >
A caregiver-survey cohort documents a high prevalence of neuropsychiatric
and behavioral challenges in RSTS.
- reference: PMID:37415602
reference_title: "Behavioral and neuropsychiatric challenges across the lifespan in individuals with Rubinstein-Taybi syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "individuals with RSTS2 had better adaptive behavior and living skills and less stereotypic behaviors but higher social phobia than individuals with RSTS1"
explanation: >
Documents type-specific behavioral differences between RSTS2 (EP300) and
RSTS1 (CREBBP).
- category: Craniofacial
name: Distinctive facial features
description: >
Characteristic craniofacial features include downslanted palpebral
fissures, low-hanging columella, high palate, grimacing smile, and talon
cusps.
phenotype_term:
preferred_term: Downslanted palpebral fissures
term:
id: HP:0000494
label: Downslanted palpebral fissures
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Characteristic craniofacial features include downslanted palpebral fissures, low-hanging columella, high palate, grimacing smile, and talon cusps."
explanation: GeneReviews lists downslanted palpebral fissures among characteristic facial features.
- category: Craniofacial
name: High palate
description: >
A high-arched palate is a characteristic craniofacial feature.
phenotype_term:
preferred_term: High palate
term:
id: HP:0000218
label: High palate
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Characteristic craniofacial features include downslanted palpebral fissures, low-hanging columella, high palate, grimacing smile, and talon cusps."
explanation: GeneReviews lists high palate among characteristic craniofacial features.
- category: Other
name: Talon cusps
description: >
Talon cusps (accessory cusp-like structures on teeth) are a characteristic
dental finding in RSTS.
phenotype_term:
preferred_term: Talon cusp
term:
id: HP:0011087
label: Talon cusp
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "downslanted palpebral fissures, low-hanging columella, high palate, grimacing smile, and talon cusps"
explanation: GeneReviews documents talon cusps as a characteristic dental feature.
- category: Growth
name: Short stature
description: >
Prenatal growth is often normal, then height, weight, and head
circumference percentiles rapidly drop in the first months of life; short
stature is typical in adulthood.
phenotype_term:
preferred_term: Short stature
term:
id: HP:0004322
label: Short stature
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Prenatal growth is often normal, then height, weight, and head circumference percentiles rapidly drop in the first few months of life. Short stature is typical in adulthood."
explanation: GeneReviews documents postnatal growth retardation and short stature.
- category: Growth
name: Postnatal microcephaly
description: >
Head circumference is often normal at birth, then drops in percentile over
the first months of life, resulting in postnatal microcephaly.
phenotype_term:
preferred_term: Microcephaly
term:
id: HP:0000252
label: Microcephaly
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "height, weight, and head circumference percentiles rapidly drop in the first few months of life"
explanation: >
GeneReviews documents a postnatal drop in head circumference percentiles,
the basis of postnatal microcephaly in RSTS.
- category: Metabolic
name: Obesity
description: >
Obesity may develop in childhood or adolescence.
phenotype_term:
preferred_term: Obesity
term:
id: HP:0001513
label: Obesity
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Obesity may develop in childhood or adolescence."
explanation: GeneReviews documents childhood/adolescent obesity in RSTS.
- category: Cardiovascular
name: Congenital heart defects
description: >
Congenital heart defects are among the additional features of RSTS.
phenotype_term:
preferred_term: Congenital heart defect
term:
id: HP:0001627
label: Abnormal heart morphology
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Additional features include ocular abnormalities, hearing loss, respiratory difficulties, congenital heart defects, renal abnormalities, cryptorchidism, feeding problems, recurrent infections, and severe constipation."
explanation: GeneReviews lists congenital heart defects among additional features.
- category: Genitourinary
name: Cryptorchidism
description: >
Cryptorchidism (undescended testes) is reported among additional features.
phenotype_term:
preferred_term: Cryptorchidism
term:
id: HP:0000028
label: Cryptorchidism
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "cryptorchidism, feeding problems, recurrent infections, and severe constipation"
explanation: GeneReviews lists cryptorchidism among additional features.
- category: Immunologic
name: Recurrent infections
description: >
Recurrent infections are reported among additional features of RSTS.
phenotype_term:
preferred_term: Recurrent infections
term:
id: HP:0002719
label: Recurrent infections
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "cryptorchidism, feeding problems, recurrent infections, and severe constipation"
explanation: GeneReviews lists recurrent infections among additional features.
- reference: PMID:32594341
reference_title: "Prevalence of Immunological Defects in a Cohort of 97 Rubinstein-Taybi Syndrome Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Recurrent or severe infections, autoimmune/autoinflammatory complications, and lymphoproliferation were observed in 72.1%, 12.3%, and 8.2% of patients."
explanation: >
An international cohort of 97 RSTS patients quantifies recurrent/severe
infections at 72.1%, supporting this as a frequent manifestation.
- category: Immunologic
name: Primary immunodeficiency with antibody defect
description: >
Manifestations of immune dysfunction, affecting mostly B cells, are more
common than previously recognized in RSTS. Antibody defects (including
hypogammaglobulinemia with low B cell counts and reduced switched memory B
cells) occur in a subset of patients, and syndromic immunodeficiency is
frequently diagnosed.
phenotype_term:
preferred_term: Antibody defect (abnormal immunoglobulin level)
term:
id: HP:0010701
label: Abnormal circulating immunoglobulin concentration
evidence:
- reference: PMID:32594341
reference_title: "Prevalence of Immunological Defects in a Cohort of 97 Rubinstein-Taybi Syndrome Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "antibody defects were observed in 11.3% of subjects. In particular, these patients presented hypogammaglobulinemia associated with low B cell counts and reduction of switched memory B cell numbers."
explanation: >
Documents B-cell-predominant antibody defects (hypogammaglobulinemia, low
switched memory B cells) in RSTS.
- reference: PMID:32594341
reference_title: "Prevalence of Immunological Defects in a Cohort of 97 Rubinstein-Taybi Syndrome Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Syndromic immunodeficiency was diagnosed in 46.4% of individuals."
explanation: >
Syndromic immunodeficiency was diagnosed in nearly half of the cohort,
supporting immunodeficiency as a feature of RSTS.
- category: Gastrointestinal
name: Severe constipation
description: >
Severe constipation is reported among additional features.
phenotype_term:
preferred_term: Constipation
term:
id: HP:0002019
label: Constipation
severity: SEVERE
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "cryptorchidism, feeding problems, recurrent infections, and severe constipation"
explanation: GeneReviews lists severe constipation among additional features.
- category: Other
name: Hearing loss
description: >
Hearing loss is reported among additional features of RSTS.
phenotype_term:
preferred_term: Hearing impairment
term:
id: HP:0000365
label: Hearing impairment
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Additional features include ocular abnormalities, hearing loss, respiratory difficulties"
explanation: GeneReviews lists hearing loss among additional features.
- category: Ophthalmologic
name: Ocular abnormalities
description: >
Ocular abnormalities are reported among additional features of RSTS, and
annual eye evaluations are recommended in surveillance.
phenotype_term:
preferred_term: Ocular abnormalities
term:
id: HP:0000478
label: Abnormality of the eye
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Additional features include ocular abnormalities, hearing loss, respiratory difficulties"
explanation: GeneReviews lists ocular abnormalities among additional features.
- category: Genitourinary
name: Renal abnormalities
description: >
Renal abnormalities are reported among additional features of RSTS, with
routine monitoring for renal anomalies recommended.
phenotype_term:
preferred_term: Renal abnormalities
term:
id: HP:0000077
label: Abnormality of the kidney
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "congenital heart defects, renal abnormalities, cryptorchidism"
explanation: GeneReviews lists renal abnormalities among additional features.
- category: Respiratory
name: Respiratory difficulties
description: >
Respiratory difficulties are reported among additional features of RSTS.
phenotype_term:
preferred_term: Respiratory difficulties
term:
id: HP:0002795
label: Abnormal respiratory system physiology
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Additional features include ocular abnormalities, hearing loss, respiratory difficulties"
explanation: GeneReviews lists respiratory difficulties among additional features.
- category: Gastrointestinal
name: Feeding problems
description: >
Feeding problems are reported among additional features of RSTS, with
monitoring of feeding recommended especially in the first year of life.
phenotype_term:
preferred_term: Feeding problems
term:
id: HP:0011968
label: Feeding difficulties
onset:
onset_category: INFANTILE
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "cryptorchidism, feeding problems, recurrent infections, and severe constipation"
explanation: GeneReviews lists feeding problems among additional features.
- category: Neoplasm
name: Meningioma
description: >
Meningiomas are the most frequent benign tumor in RSTS, with significantly
elevated incidence compared with the general population.
phenotype_term:
preferred_term: Meningioma
term:
id: HP:0002858
label: Meningioma
evidence:
- reference: PMID:29359884
reference_title: "Benign and malignant tumors in Rubinstein-Taybi syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Meningiomas and pilomatricomas were the most frequent benign tumors and their incidence was significantly elevated in comparison to the general Dutch population."
explanation: Population-based registry data document significantly elevated meningioma incidence in RSTS.
- category: Neoplasm
name: Pilomatricoma
description: >
Pilomatricomas (benign skin adnexal tumors of hair matrix origin) are,
alongside meningiomas, the most frequent benign tumor in RSTS, with
significantly elevated incidence compared with the general population.
phenotype_term:
preferred_term: Pilomatricoma
term:
id: HP:0008069
label: Neoplasm of the skin
evidence:
- reference: PMID:29359884
reference_title: "Benign and malignant tumors in Rubinstein-Taybi syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Meningiomas and pilomatricomas were the most frequent benign tumors and their incidence was significantly elevated in comparison to the general Dutch population."
explanation: Population-based registry data document significantly elevated pilomatricoma incidence in RSTS.
genetic:
- name: CREBBP pathogenic variants
gene_term:
preferred_term: CREBBP
term:
id: hgnc:2348
label: CREBBP
association: Causative
notes: >
CREBBP (16p13.3) encodes CREB-binding protein (CBP), a histone
acetyltransferase and transcriptional coactivator. Heterozygous
loss-of-function point mutations, small lesions, microdeletions, and
intragenic duplications cause RSTS type 1 (~70% of cases). DNA sequencing
detects CREBBP mutations in the majority of patients with unequivocal RSTS.
evidence:
- reference: PMID:7630403
reference_title: "Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "RTS results not only from gross chromosomal rearrangements of chromosome 16p, but also from point mutations in the CBP gene itself."
explanation: >
Petrij et al. established CREBBP (CBP) point mutations and 16p
rearrangements as the cause of RSTS.
- reference: PMID:16021471
reference_title: "DNA sequencing of CREBBP demonstrates mutations in 56% of patients with Rubinstein-Taybi syndrome (RSTS) and in another patient with incomplete RSTS."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "17 mutations in 30 patients with unequivocal RSTS (detection rate 56.6%)"
explanation: >
Bartsch et al. report a 56.6% CREBBP point-mutation detection rate in
unequivocal RSTS, establishing CREBBP as the major causative gene.
- name: EP300 pathogenic variants
gene_term:
preferred_term: EP300
term:
id: hgnc:3373
label: EP300
association: Causative
notes: >
EP300 (22q13.2) encodes p300, a paralog of CBP that is also a histone
acetyltransferase and transcriptional coactivator. Heterozygous EP300
mutations cause RSTS type 2 (~5-10% of cases); these were the first
mutations identified in EP300 for a congenital disorder.
evidence:
- reference: PMID:15706485
reference_title: "Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We extended the search for mutations to the EP300 gene and showed that mutations in EP300 also cause this disorder. These are the first mutations identified in EP300 for a congenital disorder."
explanation: >
Roelfsema et al. established EP300 as a second causative gene for RSTS,
demonstrating genetic heterogeneity.
- reference: PMID:38471765
reference_title: "Diagnosis and management in Rubinstein-Taybi syndrome: first international consensus statement."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "clinical diagnostic criteria for types of RTS (RTS1: CREBBP; RTS2: EP300)"
explanation: >
The 2024 international consensus formalizes the two molecular types of RTS,
RTS1 (CREBBP) and RTS2 (EP300).
clinical_trials:
- name: NCT01619644
phase: PHASE_II
status: COMPLETED
description: >
Exploratory phase 2 trial of sodium valproate (a histone deacetylase
inhibitor) versus placebo in children with genetically confirmed RSTS,
intended to test whether restoring histone acetylation balance improves
learning, memory, and fine motor skills. Rationale: CBP and p300 act through
neuronal and synaptic plasticity, so HDAC inhibition is hypothesized to
counteract the histone-acetylation deficit.
target_phenotypes:
- preferred_term: Intellectual disability
term:
id: HP:0001249
label: Intellectual disability
evidence:
- reference: clinicaltrials:NCT01619644
reference_title: "Rubinstein-Taybi Syndrome: Functional Imaging and Therapeutic Trial"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "An exploratory phase 2 therapeutic trial in children from 6 to 21, RTS carriers, randomized to be treated either with sodium valproate with the usual pediatric dosage (30 mg/kg/j), or by placebo for one year."
explanation: >
ClinicalTrials.gov documents a phase 2 randomized trial of sodium
valproate (HDAC inhibitor) in RSTS targeting the histone-acetylation
mechanism.
- reference: clinicaltrials:NCT01619644
reference_title: "Rubinstein-Taybi Syndrome: Functional Imaging and Therapeutic Trial"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "CBP and EP300 take effect through neuronal and synaptic plasticity"
explanation: >
The trial rationale links CBP/EP300 function to neuronal and synaptic
plasticity, the basis for the HDAC-inhibitor therapeutic hypothesis.
treatments:
- name: Surgical repair of thumb and hallux anomalies
description: >
Surgical repair of significantly angulated thumbs or duplicated halluces is
used to address the characteristic digital anomalies and improve function.
treatment_term:
preferred_term: surgical procedure
term:
id: MAXO:0000004
label: surgical procedure
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "surgical repair of significantly angulated thumbs or duplicated halluces"
explanation: GeneReviews lists surgical repair of angulated thumbs/duplicated halluces as a management approach.
- name: Genetic counseling
description: >
Genetic counseling addresses the autosomal dominant inheritance, the high
proportion of de novo variants, the 50% recurrence risk for offspring of an
affected individual, and the availability of prenatal and preimplantation
genetic testing once the familial variant is known.
treatment_term:
preferred_term: Genetic Counseling
term:
id: NCIT:C15240
label: Genetic Counseling
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Once the RSTS-related pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing for RSTS are possible."
explanation: GeneReviews describes genetic counseling content, including prenatal/preimplantation testing options.
- name: Developmental and educational support
description: >
Early intervention programs, special education, vocational training, and
referral to behavioral specialists / psychologists address the
developmental disabilities and behavioral characteristics of RSTS.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Early intervention programs, special education, vocational training to address developmental disabilities, referral to behavioral specialists / psychologists, and support groups / resources for family members"
explanation: GeneReviews lists developmental and educational support as a management approach.
- name: Surveillance
description: >
Recommended surveillance includes monitoring of growth and feeding
(especially in the first year of life), annual eye and hearing evaluations,
and routine monitoring for cardiac, renal, and dental anomalies.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
evidence:
- reference: PMID:20301699
reference_title: "Rubinstein-Taybi Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Monitoring of growth and feeding, especially in the first year of life; annual eye and hearing evaluations; routine monitoring for cardiac, renal, and dental anomalies."
explanation: GeneReviews describes the recommended surveillance schedule for RSTS.
References & Deep Research
References
2Deep Research
1Rubinstein–Taybi Syndrome (RSTS) — Disease Characteristics Research Report
Executive summary
Rubinstein–Taybi syndrome (RSTS; also written RTS) is a rare, typically de novo, autosomal dominant neurodevelopmental disorder characterized by intellectual disability/developmental delay, distinctive facial features, distal limb anomalies (classically broad thumbs and halluces), and atypical growth. It is primarily caused by heterozygous pathogenic variants in the lysine acetyltransferase coactivators CREBBP (RSTS1) and EP300 (RSTS2), placing it among “chromatinopathies”/epigenetic disorders due to impaired chromatin regulation and histone acetylation. (lacombe2024diagnosisandmanagement pages 1-2, gils2021rubinsteintaybisyndromea pages 1-2, lacombe2024diagnosisandmanagement pages 3-4)
Recent (2023–2024) developments include (i) the first international consensus statement for diagnosis and management (2024) and (ii) human iPSC neuronal differentiation multi-omics (transcriptome + acetylome) mapping that identifies a vulnerable neurodevelopmental transition with concentrated transcriptional dysregulation (2024). (lacombe2024diagnosisandmanagement pages 1-2, gils2024transcriptomeandacetylome pages 1-2, gils2024transcriptomeandacetylome pages 3-4)
Target disease
- Disease name: Rubinstein–Taybi syndrome (RSTS/RTS)
- Category: Mendelian
- MONDO ID: Not retrieved in the available tool outputs; should be added from MONDO/OBO Foundry in downstream curation.
1. Disease information
1.1 Definition / concise overview
The 2024 international consensus describes Rubinstein–Taybi syndrome as an “archetypical genetic syndrome” characterized by “intellectual disability, well-defined facial features, distal limb anomalies and atypical growth,” among other multisystem findings, and caused by variants in CREBBP or EP300, encoding CBP and p300 with roles in transcription regulation and histone acetylation. (lacombe2024diagnosisandmanagement pages 1-2)
1.2 Key identifiers
- OMIM/MIM: RSTS1 (CREBBP): MIM 180849; RSTS2 (EP300): MIM 613684. (gils2021rubinsteintaybisyndromea pages 1-2, marchetti2024thephenotypebasedapproach pages 1-2)
- Additional identifiers (Orphanet, ICD-10/11, MeSH, MONDO) were not retrievable using the current tool calls and should be added from those databases during curation.
1.3 Synonyms / alternative names
- “Rubinstein–Taybi syndrome” / “Rubinstein-Taybi syndrome” / “RTS” / “RSTS”. (lacombe2024diagnosisandmanagement pages 1-2, gils2021rubinsteintaybisyndromea pages 1-2)
- Historical synonym noted in a review: “thumb syndrome and hallux larges” (former terminology). (gils2021rubinsteintaybisyndromea pages 1-2)
1.4 Evidence source type
The evidence summarized here comes from: - Aggregated guideline/review resources (international consensus; review articles). (lacombe2024diagnosisandmanagement pages 1-2, gils2021rubinsteintaybisyndromea pages 1-2) - Human cohorts/case series (molecular diagnostic cohorts; immunology cohort; caregiver behavioral cohort). (cross2020screeningofa pages 1-2, saettini2020prevalenceofimmunological pages 1-2, qu’d2023behavioralandneuropsychiatric pages 1-2) - Mechanistic human cellular/organoid models (patient-derived iPSCs/iNPCs/organoids; omics profiling). (thonel2022cbphsf2structuraland pages 1-2, gils2024transcriptomeandacetylome pages 1-2) - ClinicalTrials.gov registry entries. (NCT01619644 chunk 1, NCT04122742 chunk 1)
2. Etiology
2.1 Disease causal factors
Primary cause: Germline heterozygous pathogenic variants in CREBBP or EP300 (autosomal dominant), typically leading to haploinsufficiency and impaired lysine acetyltransferase (KAT/HAT) activity and transcriptional coactivation. (lacombe2024diagnosisandmanagement pages 3-4, gils2021rubinsteintaybisyndromea pages 1-2)
2.2 Risk factors
- Genetic: Pathogenic variants in CREBBP or EP300. Detection rates in clinically suspected RSTS are approximately ~55–75% for CREBBP and ~8–11% for EP300, with ~2–3% complete gene deletions and ~5–20% lacking a detectable molecular anomaly in current testing pipelines (consensus statement). (lacombe2024diagnosisandmanagement pages 3-4, lacombe2024diagnosisandmanagement pages 4-5)
- De novo predominance: The review notes that the vast majority are sporadic/de novo (reported as ~99% in the 2021 review). (gils2021rubinsteintaybisyndromea pages 1-2)
- Mosaicism: Mosaic CREBBP variants can occur and may underlie milder phenotypes; high-depth sequencing can help detect mosaic cases. (marchetti2024thephenotypebasedapproach pages 1-2, lacombe2024diagnosisandmanagement pages 5-6)
2.3 Protective factors
No protective genetic or environmental factors were identified in the retrieved evidence.
2.4 Gene–environment interactions
No specific gene–environment interactions were identified in the retrieved evidence.
3. Phenotypes
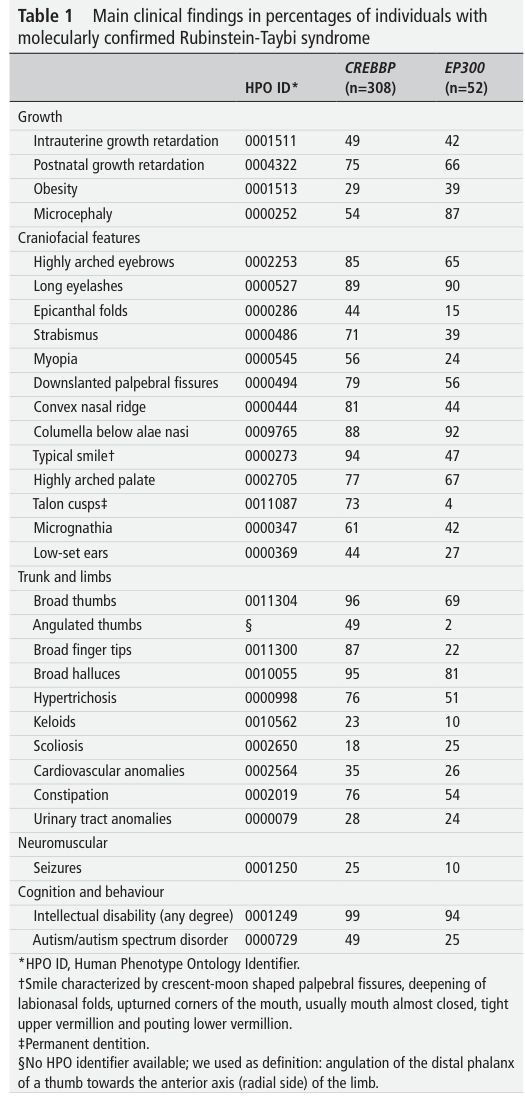
3.1 Core phenotype spectrum (with frequencies)
The 2024 consensus statement provides feature frequencies in large molecularly confirmed cohorts (CREBBP n=308; EP300 n=52) and forms the most authoritative quantitative phenotype baseline retrieved here. (lacombe2024diagnosisandmanagement pages 1-2)
Growth/development (examples): - Postnatal growth retardation: 75% (CREBBP) vs 66% (EP300). (lacombe2024diagnosisandmanagement pages 1-2) - Microcephaly: 54% vs 87%. (lacombe2024diagnosisandmanagement pages 1-2) - Intellectual disability (any degree): 99% vs 94%. (lacombe2024diagnosisandmanagement pages 2-3)
Craniofacial (examples): - Highly arched eyebrows: 85% vs 65%. (lacombe2024diagnosisandmanagement pages 1-2) - Downslanted palpebral fissures: 79% vs 56%. (lacombe2024diagnosisandmanagement pages 1-2) - Columella below alae nasi: 88% vs 92%. (lacombe2024diagnosisandmanagement pages 1-2) - Typical smile: 94% vs 47%. (lacombe2024diagnosisandmanagement pages 1-2)
Distal limbs / skeletal (examples): - Broad thumbs: 96% vs 69%. (lacombe2024diagnosisandmanagement pages 1-2) - Broad halluces: 95% vs 81%. (lacombe2024diagnosisandmanagement pages 1-2) - Angulated thumbs: 49% vs 2%. (lacombe2024diagnosisandmanagement pages 1-2)
Multisystem features (examples): - Cardiovascular anomalies: 35% vs 26%. (lacombe2024diagnosisandmanagement pages 1-2) - Urinary tract anomalies: 28% vs 24%. (lacombe2024diagnosisandmanagement pages 2-3) - Seizures: 25% vs 10%. (lacombe2024diagnosisandmanagement pages 2-3) - Autism/autism spectrum disorder: 49% vs 25%. (lacombe2024diagnosisandmanagement pages 2-3)
Visual evidence: The consensus Table 1 and Table 2 (diagnostic criteria) are captured in the extracted figure/table crops. (lacombe2024diagnosisandmanagement media d0762497, lacombe2024diagnosisandmanagement media cac74578)
3.2 Behavioral and neuropsychiatric phenotype (recent quantitative cohort)
A 2023 caregiver survey of 71 individuals aged 1–61 years reported high prevalence of behavioral and neuropsychiatric issues. (qu’d2023behavioralandneuropsychiatric pages 1-2, qu’d2023behavioralandneuropsychiatric pages 3-4)
Key statistics from the study include: - “Behavioral issues” endorsed for 88% of the sample. (qu’d2023behavioralandneuropsychiatric pages 11-13) - OCD-like symptomatology: 82% had mild-to-severe OCD-like symptoms; 21% reported an OCD diagnosis. (qu’d2023behavioralandneuropsychiatric pages 11-13) - Anxiety: 34% reported an anxiety diagnosis (despite elevated symptom measures). (qu’d2023behavioralandneuropsychiatric pages 11-13) - Genotype/type differences: RSTS2 tended to have better adaptive functioning and less stereotypic behavior, but higher social phobia than RSTS1. (qu’d2023behavioralandneuropsychiatric pages 1-2, qu’d2023behavioralandneuropsychiatric pages 8-10)
3.3 Immunologic phenotype (human cohort)
In a 2020 cohort of 97 RSTS patients, immune dysfunction and infection susceptibility were common: - Recurrent/severe infections: 72.1%. - Autoimmune/autoinflammatory complications: 12.3%. - Lymphoproliferation: 8.2%. - “Syndromic immunodeficiency”: 46.4%. - Antibody defects: 11.3%. - Interventions used in practice included immunoglobulin replacement (16.4%) and antibiotic prophylaxis (8.2%). (saettini2020prevalenceofimmunological pages 1-2)
3.4 Phenotype characteristics (age of onset; progression)
- Onset/presentation: The consensus statement notes early recognition; 86% present within the first month of life. (lacombe2024diagnosisandmanagement pages 6-7)
- Course: No robust longitudinal natural-history statistics (e.g., survival curves) were retrievable from the current evidence set; the caregiver cohort indicates adaptive skill gaps widen with age, implying increasing functional divergence over time. (qu’d2023behavioralandneuropsychiatric pages 1-2)
3.5 Quality-of-life impact
Behavioral challenges are described as a primary factor impacting quality of life in RSTS in the 2023 behavioral cohort; however, standardized QoL instruments (e.g., SF-36/EQ-5D) were not retrieved. (qu’d2023behavioralandneuropsychiatric pages 1-2)
3.6 Suggested HPO terms (non-exhaustive; based on consensus tables)
Examples directly supported by the consensus tables include: - Postnatal growth retardation HP:0004322 (lacombe2024diagnosisandmanagement pages 1-2) - Microcephaly HP:0000252 (lacombe2024diagnosisandmanagement pages 1-2) - Highly arched eyebrows HP:0002253 (lacombe2024diagnosisandmanagement pages 1-2) - Downslanted palpebral fissures HP:0000494 (lacombe2024diagnosisandmanagement pages 1-2) - Broad thumbs HP:0011304 (lacombe2024diagnosisandmanagement pages 1-2) - Broad halluces HP:0010055 (lacombe2024diagnosisandmanagement pages 2-3) - Cardiovascular anomalies HP:0002564 (lacombe2024diagnosisandmanagement pages 2-3) - Urinary tract anomalies HP:0000079 (lacombe2024diagnosisandmanagement pages 2-3) - Seizures HP:0001250 (lacombe2024diagnosisandmanagement pages 2-3) - Autism/autism spectrum disorder HP:0000729 (lacombe2024diagnosisandmanagement pages 2-3)
4. Genetic / molecular information
4.1 Causal genes
- CREBBP (CBP; located at 16p13.3) — major causal gene; RSTS1. (marchetti2024thephenotypebasedapproach pages 1-2, lacombe2024diagnosisandmanagement pages 3-4)
- EP300 (p300; located at 22q13) — RSTS2, often described as clinically milder on average. (lacombe2024diagnosisandmanagement pages 3-4, qu’d2023behavioralandneuropsychiatric pages 1-2)
4.2 Variant classes and functional consequences
- The disorder is typically attributed to heterozygous pathogenic variants or rearrangements leading to haploinsufficiency of CREBBP/EP300. (lacombe2024diagnosisandmanagement pages 3-4)
- Missense pathogenicity evidence: in a diagnostic cohort, 17/19 likely pathogenic CREBBP missense variants localized to the HAT (histone acetyltransferase) domain, and the authors concluded that missense variants in this domain can serve as “moderate evidence of pathogenicity” in variant interpretation. (cross2020screeningofa pages 1-2)
- Copy-number changes and mosaicism can contribute; mosaic CREBBP truncating variants are reported, emphasizing high-depth sequencing and multi-tissue assessment. (marchetti2024thephenotypebasedapproach pages 1-2, lacombe2024diagnosisandmanagement pages 5-6)
4.3 Modifier genes
No modifier genes were identified in the retrieved evidence.
4.4 Epigenetic information
RSTS is framed as an epigenetic disorder/chromatinopathy because CREBBP/EP300 encode lysine acetyltransferases affecting chromatin remodeling and transcriptional regulation. (gils2021rubinsteintaybisyndromea pages 1-2, lacombe2024diagnosisandmanagement pages 1-2)
5. Environmental information
No specific environmental, lifestyle, or infectious causal contributors were identified in the retrieved evidence; RSTS is primarily genetic. (lacombe2024diagnosisandmanagement pages 1-2, gils2021rubinsteintaybisyndromea pages 1-2)
6. Mechanism / pathophysiology
6.1 Core molecular mechanism (current understanding)
CBP/CREBBP and p300/EP300 are transcriptional coactivators with catalytic lysine acetyltransferase activity; loss of their function causes a deficit in acetylation (notably histone acetylation), with downstream transcriptional dysregulation during development—particularly neurodevelopment. (lacombe2024diagnosisandmanagement pages 1-2, gils2024transcriptomeandacetylome pages 1-2)
6.2 Recent omics and mechanistic advances (2024 emphasized)
(A) Transcriptome + acetylome profiling during neuronal differentiation (2024)
A 2024 Communications Biology study differentiated patient-derived iPSCs (with a recurrent CREBBP KAT-inactivating mutation) into cortical/pyramidal neurons and profiled acetylome + transcriptome across time. Major quantitative findings: - 25 specific acetylated histone residues were altered in RSTS. (gils2024transcriptomeandacetylome pages 1-2) - 2,973 differentially expressed genes (DEGs) overall, with 2,454 at day 20 (D20); D20 contained ~82.5% of all DEGs and ~75% were unique to that progenitor→immature neuron transition, identifying a critical developmental window. (gils2024transcriptomeandacetylome pages 3-4) - Specific residues frequently highlighted include H2B and H3 sites (e.g., H2BK5, H2BK43/46/108; H3K18/K23/K56/K79/K122) and others (H2AK95, H4K77). (gils2024transcriptomeandacetylome pages 6-7, gils2024transcriptomeandacetylome pages 3-4)
Suggested ontology mapping (examples): - GO biological process: neuron differentiation; neural progenitor cell differentiation; regulation of transcription, DNA-templated (supported broadly by mechanism and iPSC neuronal differentiation design). (gils2024transcriptomeandacetylome pages 1-2, gils2024transcriptomeandacetylome pages 3-4) - Cell types (CL): neural progenitor cell; cortical neuron (iPSC-derived cortical/pyramidal neurons). (gils2024transcriptomeandacetylome pages 1-2) - Anatomy (UBERON): cerebral cortex (modeled via cortical neuron differentiation/organoids). (gils2024transcriptomeandacetylome pages 1-2)
(B) CBP/EP300–HSF2–chaperone–N-cadherin cascade (2022; strong mechanistic)
A 2022 Nature Communications paper provides a mechanistic chain connecting CBP/EP300 dysfunction to neurodevelopmental phenotypes: - CBP/EP300 acetylate HSF2 (key lysines reported include K128/K135/K197) and acetylation stabilizes HSF2 by limiting proteasomal degradation. (thonel2022cbphsf2structuraland pages 2-3, thonel2022cbphsf2structuraland pages 11-11) - RSTS patient cells show reduced HSF2 and altered expression of HSF2-dependent molecular chaperones and stress response, and decreased N-cadherin–linked adhesion, which is recapitulated in patient-derived neural progenitors and cortical organoids. (thonel2022cbphsf2structuraland pages 7-9, thonel2022cbphsf2structuraland pages 15-16, thonel2022cbphsf2structuraland pages 1-2) - Rescue experiment: low, subthreshold doses of bortezomib (5–10 nM) restored HSF2 and rescued HSP110 and N-cadherin expression in cellular models, supporting causality in the pathway. (thonel2022cbphsf2structuraland pages 7-9)
Suggested ontology mapping (examples): - GO biological process: regulation of protein stability; response to heat; cell–cell adhesion. (thonel2022cbphsf2structuraland pages 7-9, thonel2022cbphsf2structuraland pages 15-16) - Cell types (CL): neural progenitor cell (iNPC); neuron (organoid neuronal layers). (thonel2022cbphsf2structuraland pages 15-16, thonel2022cbphsf2structuraland pages 3-4)
6.3 Epigenetic biomarkers: DNA methylation episignatures (clinical diagnostics)
The 2024 consensus notes that a genome-wide methylation signature can assist when molecular findings are absent. (lacombe2024diagnosisandmanagement pages 5-6)
A 2024 Human Genetics and Genomics Advances paper describes clinical deployment of EpiSign v3 (Illumina MethylationEPIC array) for NDDs. The assay compares to a large reference classifier (57 DNAm profiles representing 65 syndromes) and reports an SVM-derived “methylation variant pathogenicity (MVP)” score with secondary review above a threshold (MVP >0.01). The included table explicitly lists CREBBP and EP300 among genes with established episignatures and shows RSTS-related calls (e.g., RSTS1) alongside ACMG/AMP classifications, illustrating integration into variant interpretation workflows. (trajkova2024dnamethylationanalysis pages 2-3, trajkova2024dnamethylationanalysis pages 3-4)
7. Anatomical structures affected
RSTS is multisystem; major implicated systems include: - Nervous system / brain: neurodevelopmental delay/intellectual disability; modeled mechanisms involve cortical neuronal differentiation and neuroepithelial integrity in cortical organoids. (lacombe2024diagnosisandmanagement pages 1-2, gils2024transcriptomeandacetylome pages 1-2, thonel2022cbphsf2structuraland pages 1-2) - Craniofacial: characteristic facial gestalt (see phenotype frequencies). (lacombe2024diagnosisandmanagement pages 1-2) - Limbs: distal limb anomalies—broad/angulated thumbs and halluces. (lacombe2024diagnosisandmanagement pages 1-2) - Cardiovascular: congenital anomalies ~26–35%. (lacombe2024diagnosisandmanagement pages 2-3) - Genitourinary/urinary tract: urinary tract anomalies ~24–28%. (lacombe2024diagnosisandmanagement pages 2-3)
Suggested UBERON terms (non-exhaustive): cerebral cortex; heart; urinary system; limb. (Supported generally by organ/system-level phenotypes in consensus and organoid/cortical modeling.) (lacombe2024diagnosisandmanagement pages 2-3, gils2024transcriptomeandacetylome pages 1-2)
8. Temporal development
- Typical onset: congenital/early infancy with most presenting very early: 86% within the first month (consensus). (lacombe2024diagnosisandmanagement pages 6-7)
- Course: In the 2023 behavioral cohort, adaptive functioning deficits persist across the lifespan and the gap relative to typical peers may become more apparent at older ages. (qu’d2023behavioralandneuropsychiatric pages 1-2)
9. Inheritance and population
9.1 Epidemiology
- Incidence commonly cited as ~1/100,000–1/125,000 births in reviews and cohorts. (gils2021rubinsteintaybisyndromea pages 1-2, cross2020screeningofa pages 1-2)
9.2 Inheritance pattern
- Autosomal dominant, usually de novo. (gils2021rubinsteintaybisyndromea pages 1-2, lacombe2024diagnosisandmanagement pages 3-4)
- Recurrence risk: empirical recurrence risk for unaffected parents with one affected child ~0.5–1% (due to possible germline mosaicism); if a parent is affected, 50% transmission risk. (lacombe2024diagnosisandmanagement pages 6-7)
9.3 Penetrance/expressivity
The consensus and reviews emphasize clinical heterogeneity and incomplete molecular confirmation in some clinically typical cases; no quantitative penetrance estimate was retrieved. (lacombe2024diagnosisandmanagement pages 1-1, lacombe2024diagnosisandmanagement pages 3-4)
10. Diagnostics
10.1 Clinical criteria (2024 international consensus)
The consensus defines a weighted clinical diagnostic scoring system based on craniofacial, skeletal, growth, and development domains, with a “cardinal score” requiring ≥2 groups positive including craniofacial or skeletal. Diagnostic thresholds: - Definitive: score ≥12 + positive cardinal score. - Likely: 8–11 + positive cardinal score (warrants molecular confirmation). - Possible: 5–7 + negative cardinal score; “warrants molecular analyses of CREBBP and EP300.” (lacombe2024diagnosisandmanagement pages 1-2, lacombe2024diagnosisandmanagement pages 2-3)
10.2 Genetic testing strategy (consensus)
For individuals with suspected RSTS: - First-line targeted testing of CREBBP and EP300 via Sanger sequencing + MLPA, or high-throughput approaches (aCGH; WES/WGS depending on scenario). Variant interpretation should follow ACMG guidelines; RNA studies can clarify splicing; mosaicism can require multi-tissue testing. (lacombe2024diagnosisandmanagement pages 5-6) - A genome-wide methylation signature may support diagnosis when molecular findings are absent. (lacombe2024diagnosisandmanagement pages 5-6)
10.3 Omics-based diagnostics (episignatures)
EpiSign methylation profiling can support variant interpretation in CREBBP/EP300 cases and provide syndrome-level classification signals (e.g., RSTS1) integrated with ACMG/AMP classification. (trajkova2024dnamethylationanalysis pages 3-4)
10.4 Differential diagnosis
The consensus notes overlap with related chromatinopathies and that careful differential diagnosis is needed when specificity is reduced (e.g., overlap with Wiedemann–Steiner syndrome is mentioned). (lacombe2024diagnosisandmanagement pages 3-4)
11. Outcome / prognosis
Robust survival and life expectancy statistics were not retrievable from the tool evidence set. However, morbidity can be significant due to congenital anomalies, neurodevelopmental impairment, infections/immunodeficiency, and behavioral challenges. (saettini2020prevalenceofimmunological pages 1-2, qu’d2023behavioralandneuropsychiatric pages 1-2)
12. Treatment
12.1 Current management (evidence available)
The 2024 international consensus exists to standardize diagnostic and care practices, but the retrieved excerpts did not include the detailed baseline evaluations/surveillance schedules (noted as potentially in supplemental materials). (lacombe2024diagnosisandmanagement pages 1-2, lacombe2024diagnosisandmanagement pages 6-7)
Immunologic management in practice (cohort evidence): Immunoglobulin replacement and antibiotic prophylaxis were used in subsets of patients in the immunology cohort. (saettini2020prevalenceofimmunological pages 1-2)
12.2 Experimental / clinical trials
(A) Sodium valproate (HDAC inhibitor) trial — completed - NCT01619644 (RUBIVAL); sponsor: University Hospital Bordeaux; start year in registry entry: 2012. - Design: randomized, double-blind phase 2, sodium valproate 30 mg/kg/day vs placebo for 1 year in genetically confirmed RSTS age 6–21. - Primary endpoints: long-term memory “point location” (CMS) and “image recognition” (RBMT); responder defined as ≥1-point improvement on at least one subtest. (NCT01619644 chunk 1)
(B) Acetylome biomarker / functional assay study — ongoing - NCT04122742 (GENEPI); University Hospital Bordeaux; registry year: 2019; recruiting with estimated completion Oct 2025. - Objective: identify CBP/p300-dependent acetylation markers and regulated genes during neuronal differentiation of iPSC-derived neurons; methods include LC–MS/MS acetylome profiling, ChIP-seq, RNA-seq, and CRISPR correction of CREBBP mutations to generate isogenic controls. (NCT04122742 chunk 1, NCT04122742 chunk 2)
Suggested MAXO terms (non-exhaustive): - Histone deacetylase inhibitor therapy (sodium valproate trial) (NCT01619644 chunk 1) - Immunoglobulin replacement therapy; antibiotic prophylaxis (saettini2020prevalenceofimmunological pages 1-2)
13. Prevention
Primary prevention is generally not applicable because most cases are de novo.
Genetic counseling / reproductive options (consensus): - Prenatal testing is primarily recommended when there is a previously affected child or known familial CREBBP/EP300 pathogenic variant; invasive sampling (CVS/amniocentesis or embryonic cells with IVF) enables reliable molecular prenatal diagnosis. - Non-invasive cfDNA screening is not advocated without a known familial variant. (lacombe2024diagnosisandmanagement pages 6-7)
14. Other species / natural disease
No naturally occurring veterinary disease analogs were retrieved in the tool evidence.
15. Model organisms
15.1 Human cellular models (most directly supported)
- Patient-derived iPSCs differentiated into cortical/pyramidal neurons with paired acetylome/transcriptome profiling identified stage-specific vulnerabilities (D20 transition). (gils2024transcriptomeandacetylome pages 1-2, gils2024transcriptomeandacetylome pages 3-4)
- Patient-derived iNPCs and human cortical organoids demonstrated cell–cell adhesion and neuroepithelial integrity abnormalities linked to CBP/EP300–HSF2 cascade; rescue of molecular deficits via proteasome inhibition supports causality. (thonel2022cbphsf2structuraland pages 15-16, thonel2022cbphsf2structuraland pages 1-2)
15.2 Mouse developmental context (limited)
HSF2 acetylation and co-localization with CBP/EP300 is described in developing mouse cortex, supporting conservation of the axis in mammalian neurodevelopment, but no full mouse disease model characterization was retrieved. (thonel2022cbphsf2structuraland pages 3-4)
Knowledge-base curation table
| Category | Key items | High-value statistics/data | Key sources |
|---|---|---|---|
| Disease identifiers | Rubinstein–Taybi syndrome (RSTS/RTS); OMIM #180849 (RSTS1, CREBBP-related), OMIM #613684 (RSTS2, EP300-related); named for Rubinstein and Taybi; chromatinopathy / epigenetic disorder | >800 publications noted by 2024 consensus; incidence generally cited as ~1:100,000–1:125,000 births | (lacombe2024diagnosisandmanagement pages 1-2, gils2021rubinsteintaybisyndromea pages 1-2) |
| Causal genes and inheritance | Autosomal dominant disorder caused mainly by heterozygous pathogenic variants in CREBBP and EP300; most cases are de novo; rare familial transmission and mosaicism reported | CREBBP explains ~55–75% of cases; EP300 ~8–11%; complete gene deletions ~2–3%; ~5–20% to ~30% remain without a molecular diagnosis depending on cohort/series; empirical recurrence risk for unaffected parents with one affected child ~0.5–1%; affected parent transmission risk 50% | (lacombe2024diagnosisandmanagement pages 3-4, gils2021rubinsteintaybisyndromea pages 1-2, lacombe2024diagnosisandmanagement pages 6-7, marchetti2024thephenotypebasedapproach pages 1-2) |
| Variant spectrum | Loss-of-function predominates; SNVs, indels, splice variants, CNVs, whole-gene deletions, rare mosaic variants; CREBBP missense clustering in HAT domain supports domain-critical pathogenicity | In a 395-referral cohort: 129 CREBBP P/LP and 16 EP300 P/LP variants; 145 molecular diagnoses (37%); 103/133 variants novel; 17/19 likely pathogenic CREBBP missense variants were in the HAT domain | (cross2020screeningofa pages 1-2, marchetti2024thephenotypebasedapproach pages 1-2, lacombe2024diagnosisandmanagement pages 5-6) |
| Core phenotype: growth and craniofacial | Characteristic face, growth disturbance, developmental delay/intellectual disability, distal limb anomalies | Molecularly confirmed cohorts (CREBBP vs EP300): postnatal growth retardation 75% vs 66%; microcephaly 54% vs 87%; highly arched eyebrows 85% vs 65%; downslanted palpebral fissures 79% vs 56%; convex nasal ridge 81% vs 44%; columella below alae nasi 88% vs 92%; typical smile 94% vs 47%; highly arched palate 77% vs 67% | (lacombe2024diagnosisandmanagement pages 1-2) |
| Core phenotype: limbs and multisystem involvement | Broad/angulated thumbs and halluces; hypertrichosis; cardiac, urinary, neurologic, GI, behavioral involvement | Broad thumbs 96% vs 69%; angulated thumbs 49% vs 2%; broad halluces 95% vs 81%; broad fingertips 87% vs 22%; hypertrichosis 76% vs 51%; cardiovascular anomalies 35% vs 26%; urinary tract anomalies 28% vs 24%; constipation 76% vs 54%; seizures 25% vs 10%; intellectual disability 99% vs 94%; autism/ASD 49% vs 25% | (lacombe2024diagnosisandmanagement pages 1-2) |
| Immunologic phenotype | Recurrent infections, antibody defects, syndromic immunodeficiency in a substantial subset; B-cell abnormalities highlighted | In 97 patients: recurrent/severe infections 72.1%; autoimmune/autoinflammatory complications 12.3%; lymphoproliferation 8.2%; syndromic immunodeficiency 46.4%; antibody defects 11.3%; immunoglobulin replacement 16.4%; antibiotic prophylaxis 8.2%; immunosuppressive therapy 9.8% | (saettini2020prevalenceofimmunological pages 1-2) |
| Behavioral / neuropsychiatric features | Anxiety, OCD-like symptoms, hyperactivity/inattention, stereotypies, challenging behavior, adaptive-function impairment across lifespan; RSTS2 tends to have milder adaptive impairment but more social phobia | Caregiver study: n=71, ages 1–61; behavioral issues 88%; OCD diagnosis 21%; anxiety diagnosis 34%; 82% had mild-to-severe OCD-like symptoms; 90% of RSTS1 vs 73% of RSTS2 had elevated OCD-like symptoms; RSTS2 had higher social phobia and better adaptive scores; school-age group showed peak challenging behaviors | (qu’d2023behavioralandneuropsychiatric pages 1-2, qu’d2023behavioralandneuropsychiatric pages 11-13, qu’d2023behavioralandneuropsychiatric pages 8-10, qu’d2023behavioralandneuropsychiatric pages 3-4) |
| Clinical diagnostic criteria | 2024 international consensus uses weighted craniofacial, skeletal, growth, and development features; cardinal score requires 2 of 4 groups positive, including skeletal or craniofacial | Definitive clinical diagnosis: score ≥12 + positive cardinal score; likely: 8–11 + positive cardinal score; possible: 5–7 + negative cardinal score; both likely and possible categories warrant CREBBP/EP300 testing | (lacombe2024diagnosisandmanagement pages 1-2, lacombe2024diagnosisandmanagement pages 3-4, lacombe2024diagnosisandmanagement pages 2-3) |
| Recommended molecular testing | If RSTS clinically suspected: first-line targeted CREBBP/EP300 testing by Sanger + MLPA or high-throughput methods; if not specifically suspected but ID/malformations present: aCGH/WES/WGS first-tier; RNA studies for splice uncertainty; mosaicism assessment across tissues; methylation/episignature can aid unresolved cases | Consensus specifically recommends CREBBP/EP300 molecular analysis for likely/possible clinical diagnoses; prenatal molecular testing mainly when familial variant/previously affected child is known | (lacombe2024diagnosisandmanagement pages 5-6, lacombe2024diagnosisandmanagement pages 6-7, lacombe2024diagnosisandmanagement pages 1-2) |
| DNA methylation / episignature diagnostics | Genome-wide methylation signatures (EpiSign) are clinically useful adjuncts for variant interpretation and unresolved neurodevelopmental cases; CREBBP and EP300 are included among genes with established episignatures | EpiSign v3 compares against 57 DNAm profiles spanning 65 syndromes; SVM-based MVP score >0.01 triggers secondary review; study table includes CREBBP/EP300 with RSTS-related diagnostic calls | (trajkova2024dnamethylationanalysis pages 2-3, trajkova2024dnamethylationanalysis pages 3-4, lacombe2024diagnosisandmanagement pages 5-6) |
| Molecular mechanism: core disease biology | CBP/CREBBP and p300/EP300 are lysine acetyltransferase (KAT/HAT) transcriptional coactivators governing histone acetylation, chromatin remodeling, and transcriptional regulation; RSTS is a chromatinopathy | Reported pathogenic variant counts in review: ~500 CREBBP and 118 EP300; HAT-domain missense enrichment is strong evidence for pathogenicity | (gils2021rubinsteintaybisyndromea pages 1-2, cross2020screeningofa pages 1-2, lacombe2024diagnosisandmanagement pages 4-5) |
| Omics mechanism: neuronal differentiation defect | iPSC-derived neuron multi-omics identified altered histone acetylation and delayed maturation during neural progenitor → immature neuron transition | 25 altered acetylated histone residues; 2,973 DEGs overall; 2,454 DEGs at D20; ~82.5% of all DEGs concentrated at D20; ~75% of all DEGs unique to D20; residues repeatedly implicated include H2BK5, H2BK11/43/46/108, H3K18, H3K23, H3K27, H3K56, H3K79, H3K122, H2AK95, H4K77 | (gils2024transcriptomeandacetylome pages 1-2, gils2024transcriptomeandacetylome pages 3-4, gils2024transcriptomeandacetylome pages 7-8, gils2024transcriptomeandacetylome pages 9-10, gils2024transcriptomeandacetylome pages 6-7) |
| Mechanistic pathway: HSF2 / stress / adhesion | CBP/EP300 acetylate and stabilize HSF2; RSTS-associated CBP/p300 dysfunction lowers HSF2, impairing chaperone/stress responses and N-cadherin–dependent neuroepithelial integrity | Key acetylated HSF2 lysines include K128, K135, K197; reduced HSF2 lowers HSP70/HSP90/HSP110-related responses and N-cadherin; low-dose bortezomib (5–10 nM) restored HSF2 and rescued HSP110/N-cadherin expression in cell models; defects reproduced in iNPCs and cortical organoids | (thonel2022cbphsf2structuraland pages 2-3, thonel2022cbphsf2structuraland pages 7-9, thonel2022cbphsf2structuraland pages 15-16, thonel2022cbphsf2structuraland pages 11-11, thonel2022cbphsf2structuraland pages 1-2) |
| Natural history / demographics | Usually congenital/early-childhood onset; many cases recognized neonatally; EP300-related disease can be milder/less typical | Consensus notes 86% present within the first month of life; equal sex distribution reported in immunology cohort summary; adults survive into later life, though morbidity depends on associated anomalies | (lacombe2024diagnosisandmanagement pages 6-7, saettini2020prevalenceofimmunological pages 1-2, wang2023geneticscornera pages 4-5) |
| Key trial / translational study: sodium valproate | NCT01619644 (RUBIVAL) phase 2 randomized, double-blind trial of oral sodium valproate vs placebo in genetically confirmed RSTS | Completed; enrolled 41; ages 6–21; sodium valproate 30 mg/kg/day for 1 year; primary endpoint was ≥1-point improvement in at least one long-term memory subtest; included imaging and histone-acetylation biomarker outcomes | (NCT01619644 chunk 1) |
| Key trial / translational study: acetylome biomarkers | NCT04122742 (GENEPI) observational Bordeaux study using patient-derived cells to define acetylation profiles as epigenetic markers for CREBBP/EP300 variant causality | Recruiting/ongoing in registry; target enrollment 154; start 2019-10-08; estimated completion Oct 2025; uses blood/skin biopsy, iPSC neuronal differentiation, LC-MS/MS acetylome profiling, ChIP-seq, RNA-seq, and CRISPR-corrected isogenic lines | (NCT04122742 chunk 1, NCT04122742 chunk 2) |
Table: This table condenses the highest-yield disease, genotype, phenotype, diagnostic, mechanistic, and clinical-trial facts for Rubinstein–Taybi syndrome using only the gathered evidence. It is designed to support rapid knowledge-base curation with direct traceability to the cited context IDs.
Notes on evidence gaps vs requested template
- MONDO, Orphanet, ICD-10/11, MeSH identifiers were not retrievable via the tool calls used in this run; they should be added from those resources during curation.
- The 2024 consensus statement includes long-term management recommendations, but in the retrieved excerpts, detailed surveillance/baseline evaluation schedules were referenced as being in supplemental materials not captured here.
- Non-human model organisms (e.g., zebrafish/Drosophila disease models) were not retrieved in the current evidence set, so this section is intentionally conservative.
Key recent/authoritative sources (with dates and URLs)
- Lacombe D, et al. “Diagnosis and management in Rubinstein–Taybi syndrome: first international consensus statement.” J Med Genet. Mar 2024. https://doi.org/10.1136/jmg-2023-109438 (lacombe2024diagnosisandmanagement pages 1-2)
- Van Gils J, et al. “Transcriptome and acetylome profiling identify crucial steps of neuronal differentiation in Rubinstein-Taybi syndrome.” Communications Biology. Oct 2024. https://doi.org/10.1038/s42003-024-06939-3 (gils2024transcriptomeandacetylome pages 1-2)
- Trajkova S, et al. “DNA methylation analysis in patients with neurodevelopmental disorders improves variant interpretation and reveals complexity.” Hum Genet Genomics Adv. Jul 2024. https://doi.org/10.1016/j.xhgg.2024.100309 (trajkova2024dnamethylationanalysis pages 2-3)
- Qu’d D, et al. “Behavioral and neuropsychiatric challenges across the lifespan in individuals with Rubinstein-Taybi syndrome.” Front Genet. Jun 2023. https://doi.org/10.3389/fgene.2023.1116919 (qu’d2023behavioralandneuropsychiatric pages 1-2)
- de Thonel A, et al. “CBP-HSF2 structural and functional interplay in Rubinstein-Taybi neurodevelopmental disorder.” Nat Commun. Nov 2022. https://doi.org/10.1038/s41467-022-34476-2 (thonel2022cbphsf2structuraland pages 1-2)
- Saettini F, et al. “Prevalence of Immunological Defects in a Cohort of 97 Rubinstein–Taybi Syndrome Patients.” J Clin Immunol. Jun 2020. https://doi.org/10.1007/s10875-020-00808-4 (saettini2020prevalenceofimmunological pages 1-2)
- Cross E, et al. “Screening of a large Rubinstein–Taybi cohort identified many novel variants and emphasizes the importance of the CREBBP histone acetyltransferase domain.” Am J Med Genet A. Aug 2020. https://doi.org/10.1002/ajmg.a.61813 (cross2020screeningofa pages 1-2)
- ClinicalTrials.gov: NCT01619644 (RUBIVAL; posted 2012). (NCT01619644 chunk 1)
- ClinicalTrials.gov: NCT04122742 (GENEPI; posted 2019). (NCT04122742 chunk 1)
References
-
(lacombe2024diagnosisandmanagement pages 1-2): Didier Lacombe, Agnès Bloch-Zupan, Cecilie Bredrup, Edward B Cooper, Sofia Douzgou Houge, Sixto García-Miñaúr, Hülya Kayserili, Lidia Larizza, Vanesa Lopez Gonzalez, Leonie A Menke, Donatella Milani, Francesco Saettini, Cathy A Stevens, Lloyd Tooke, Jill A Van der Zee, Maria M Van Genderen, Julien Van-Gils, Jane Waite, Jean-Louis Adrien, Oliver Bartsch, Pierre Bitoun, Antonia H M Bouts, Anna M Cueto-González, Elena Dominguez-Garrido, Floor A Duijkers, Patricia Fergelot, Elizabeth Halstead, Sylvia A Huisman, Camilla Meossi, Jo Mullins, Sarah M Nikkel, Chris Oliver, Elisabetta Prada, Alessandra Rei, Ilka Riddle, Cristina Rodriguez-Fonseca, Rebecca Rodríguez Pena, Janet Russell, Alicia Saba, Fernando Santos-Simarro, Brittany N Simpson, David F Smith, Markus F Stevens, Katalin Szakszon, Emmanuelle Taupiac, Nadia Totaro, Irene Valenzuena Palafoll, Daniëlle C M Van Der Kaay, Michiel P Van Wijk, Klea Vyshka, Susan Wiley, and Raoul C Hennekam. Diagnosis and management in rubinstein-taybi syndrome: first international consensus statement. Journal of Medical Genetics, 61:503-519, Mar 2024. URL: https://doi.org/10.1136/jmg-2023-109438, doi:10.1136/jmg-2023-109438. This article has 46 citations and is from a domain leading peer-reviewed journal.
-
(gils2021rubinsteintaybisyndromea pages 1-2): Julien Van Gils, Frederique Magdinier, Patricia Fergelot, and Didier Lacombe. Rubinstein-taybi syndrome: a model of epigenetic disorder. Genes, 12:968, Jun 2021. URL: https://doi.org/10.3390/genes12070968, doi:10.3390/genes12070968. This article has 111 citations.
-
(lacombe2024diagnosisandmanagement pages 3-4): Didier Lacombe, Agnès Bloch-Zupan, Cecilie Bredrup, Edward B Cooper, Sofia Douzgou Houge, Sixto García-Miñaúr, Hülya Kayserili, Lidia Larizza, Vanesa Lopez Gonzalez, Leonie A Menke, Donatella Milani, Francesco Saettini, Cathy A Stevens, Lloyd Tooke, Jill A Van der Zee, Maria M Van Genderen, Julien Van-Gils, Jane Waite, Jean-Louis Adrien, Oliver Bartsch, Pierre Bitoun, Antonia H M Bouts, Anna M Cueto-González, Elena Dominguez-Garrido, Floor A Duijkers, Patricia Fergelot, Elizabeth Halstead, Sylvia A Huisman, Camilla Meossi, Jo Mullins, Sarah M Nikkel, Chris Oliver, Elisabetta Prada, Alessandra Rei, Ilka Riddle, Cristina Rodriguez-Fonseca, Rebecca Rodríguez Pena, Janet Russell, Alicia Saba, Fernando Santos-Simarro, Brittany N Simpson, David F Smith, Markus F Stevens, Katalin Szakszon, Emmanuelle Taupiac, Nadia Totaro, Irene Valenzuena Palafoll, Daniëlle C M Van Der Kaay, Michiel P Van Wijk, Klea Vyshka, Susan Wiley, and Raoul C Hennekam. Diagnosis and management in rubinstein-taybi syndrome: first international consensus statement. Journal of Medical Genetics, 61:503-519, Mar 2024. URL: https://doi.org/10.1136/jmg-2023-109438, doi:10.1136/jmg-2023-109438. This article has 46 citations and is from a domain leading peer-reviewed journal.
-
(gils2024transcriptomeandacetylome pages 1-2): Julien Van Gils, Slim Karkar, Aurélien Barre, Seyta Ley-Ngardigal, Sophie Nothof, Stéphane Claverol, Caroline Tokarski, Jean-Philippe Trani, Raphael Chevalier, Natacha Broucqsault, Claire El Yazidi, Didier Lacombe, Patricia Fergelot, and Frédérique Magdinier. Transcriptome and acetylome profiling identify crucial steps of neuronal differentiation in rubinstein-taybi syndrome. Communications Biology, Oct 2024. URL: https://doi.org/10.1038/s42003-024-06939-3, doi:10.1038/s42003-024-06939-3. This article has 3 citations and is from a peer-reviewed journal.
-
(gils2024transcriptomeandacetylome pages 3-4): Julien Van Gils, Slim Karkar, Aurélien Barre, Seyta Ley-Ngardigal, Sophie Nothof, Stéphane Claverol, Caroline Tokarski, Jean-Philippe Trani, Raphael Chevalier, Natacha Broucqsault, Claire El Yazidi, Didier Lacombe, Patricia Fergelot, and Frédérique Magdinier. Transcriptome and acetylome profiling identify crucial steps of neuronal differentiation in rubinstein-taybi syndrome. Communications Biology, Oct 2024. URL: https://doi.org/10.1038/s42003-024-06939-3, doi:10.1038/s42003-024-06939-3. This article has 3 citations and is from a peer-reviewed journal.
-
(marchetti2024thephenotypebasedapproach pages 1-2): Giulia Bruna Marchetti, Donatella Milani, Livia Pisciotta, Laura Pezzoli, Paola Marchisio, Berardo Rinaldi, and Maria Iascone. The phenotype-based approach can solve cold cases: the paradigm of mosaic mutations of the crebbp gene. Genes, 15:654, May 2024. URL: https://doi.org/10.3390/genes15060654, doi:10.3390/genes15060654. This article has 0 citations.
-
(cross2020screeningofa pages 1-2): Esther Cross, Philippa J. Duncan‐Flavell, Rachel J. Howarth, James I. Hobbs, Nicholas Simon Thomas, and David J. Bunyan. Screening of a large rubinstein–taybi cohort identified many novel variants and emphasizes the importance of the crebbp histone acetyltransferase domain. American Journal of Medical Genetics Part A, 182:2508-2520, Aug 2020. URL: https://doi.org/10.1002/ajmg.a.61813, doi:10.1002/ajmg.a.61813. This article has 24 citations.
-
(saettini2020prevalenceofimmunological pages 1-2): Francesco Saettini, Richard Herriot, Elisabetta Prada, Mathilde Nizon, Daniele Zama, Antonio Marzollo, Igor Romaniouk, Vassilios Lougaris, Manuela Cortesi, Alessia Morreale, Rika Kosaki, Fabio Cardinale, Silvia Ricci, Elena Domínguez-Garrido, Davide Montin, Marie Vincent, Donatella Milani, Andrea Biondi, Cristina Gervasini, and Raffaele Badolato. Prevalence of immunological defects in a cohort of 97 rubinstein–taybi syndrome patients. Journal of Clinical Immunology, 40:851-860, Jun 2020. URL: https://doi.org/10.1007/s10875-020-00808-4, doi:10.1007/s10875-020-00808-4. This article has 40 citations and is from a domain leading peer-reviewed journal.
-
(qu’d2023behavioralandneuropsychiatric pages 1-2): Dima Qu’d, Lauren M. Schmitt, Amber Leston, Jacqueline R. Harris, Anne Slavotinek, Ilka Riddle, Diana S. Brightman, and Brittany N. Simpson. Behavioral and neuropsychiatric challenges across the lifespan in individuals with rubinstein-taybi syndrome. Frontiers in Genetics, Jun 2023. URL: https://doi.org/10.3389/fgene.2023.1116919, doi:10.3389/fgene.2023.1116919. This article has 6 citations and is from a peer-reviewed journal.
-
(thonel2022cbphsf2structuraland pages 1-2): Aurélie de Thonel, Johanna K. Ahlskog, Kevin Daupin, Véronique Dubreuil, Jérémy Berthelet, Carole Chaput, Geoffrey Pires, Camille Leonetti, Ryma Abane, Lluís Cordón Barris, Isabelle Leray, Anna L. Aalto, Sarah Naceri, Marine Cordonnier, Carène Benasolo, Matthieu Sanial, Agathe Duchateau, Anniina Vihervaara, Mikael C. Puustinen, Federico Miozzo, Patricia Fergelot, Élise Lebigot, Alain Verloes, Pierre Gressens, Didier Lacombe, Jessica Gobbo, Carmen Garrido, Sandy D. Westerheide, Laurent David, Michel Petitjean, Olivier Taboureau, Fernando Rodrigues-Lima, Sandrine Passemard, Délara Sabéran-Djoneidi, Laurent Nguyen, Madeline Lancaster, Lea Sistonen, and Valérie Mezger. Cbp-hsf2 structural and functional interplay in rubinstein-taybi neurodevelopmental disorder. Nature Communications, Nov 2022. URL: https://doi.org/10.1038/s41467-022-34476-2, doi:10.1038/s41467-022-34476-2. This article has 27 citations and is from a highest quality peer-reviewed journal.
-
(NCT01619644 chunk 1): Rubinstein-Taybi Syndrome: Functional Imaging and Therapeutic Trial. University Hospital, Bordeaux. 2012. ClinicalTrials.gov Identifier: NCT01619644
-
(NCT04122742 chunk 1): Diagnosis of RSTS: Identification of the Acetylation Profiles as Epigenetic Markers for Assessing Causality of CREBBP and EP300 Variants.. University Hospital, Bordeaux. 2019. ClinicalTrials.gov Identifier: NCT04122742
-
(lacombe2024diagnosisandmanagement pages 4-5): Didier Lacombe, Agnès Bloch-Zupan, Cecilie Bredrup, Edward B Cooper, Sofia Douzgou Houge, Sixto García-Miñaúr, Hülya Kayserili, Lidia Larizza, Vanesa Lopez Gonzalez, Leonie A Menke, Donatella Milani, Francesco Saettini, Cathy A Stevens, Lloyd Tooke, Jill A Van der Zee, Maria M Van Genderen, Julien Van-Gils, Jane Waite, Jean-Louis Adrien, Oliver Bartsch, Pierre Bitoun, Antonia H M Bouts, Anna M Cueto-González, Elena Dominguez-Garrido, Floor A Duijkers, Patricia Fergelot, Elizabeth Halstead, Sylvia A Huisman, Camilla Meossi, Jo Mullins, Sarah M Nikkel, Chris Oliver, Elisabetta Prada, Alessandra Rei, Ilka Riddle, Cristina Rodriguez-Fonseca, Rebecca Rodríguez Pena, Janet Russell, Alicia Saba, Fernando Santos-Simarro, Brittany N Simpson, David F Smith, Markus F Stevens, Katalin Szakszon, Emmanuelle Taupiac, Nadia Totaro, Irene Valenzuena Palafoll, Daniëlle C M Van Der Kaay, Michiel P Van Wijk, Klea Vyshka, Susan Wiley, and Raoul C Hennekam. Diagnosis and management in rubinstein-taybi syndrome: first international consensus statement. Journal of Medical Genetics, 61:503-519, Mar 2024. URL: https://doi.org/10.1136/jmg-2023-109438, doi:10.1136/jmg-2023-109438. This article has 46 citations and is from a domain leading peer-reviewed journal.
-
(lacombe2024diagnosisandmanagement pages 5-6): Didier Lacombe, Agnès Bloch-Zupan, Cecilie Bredrup, Edward B Cooper, Sofia Douzgou Houge, Sixto García-Miñaúr, Hülya Kayserili, Lidia Larizza, Vanesa Lopez Gonzalez, Leonie A Menke, Donatella Milani, Francesco Saettini, Cathy A Stevens, Lloyd Tooke, Jill A Van der Zee, Maria M Van Genderen, Julien Van-Gils, Jane Waite, Jean-Louis Adrien, Oliver Bartsch, Pierre Bitoun, Antonia H M Bouts, Anna M Cueto-González, Elena Dominguez-Garrido, Floor A Duijkers, Patricia Fergelot, Elizabeth Halstead, Sylvia A Huisman, Camilla Meossi, Jo Mullins, Sarah M Nikkel, Chris Oliver, Elisabetta Prada, Alessandra Rei, Ilka Riddle, Cristina Rodriguez-Fonseca, Rebecca Rodríguez Pena, Janet Russell, Alicia Saba, Fernando Santos-Simarro, Brittany N Simpson, David F Smith, Markus F Stevens, Katalin Szakszon, Emmanuelle Taupiac, Nadia Totaro, Irene Valenzuena Palafoll, Daniëlle C M Van Der Kaay, Michiel P Van Wijk, Klea Vyshka, Susan Wiley, and Raoul C Hennekam. Diagnosis and management in rubinstein-taybi syndrome: first international consensus statement. Journal of Medical Genetics, 61:503-519, Mar 2024. URL: https://doi.org/10.1136/jmg-2023-109438, doi:10.1136/jmg-2023-109438. This article has 46 citations and is from a domain leading peer-reviewed journal.
-
(lacombe2024diagnosisandmanagement pages 2-3): Didier Lacombe, Agnès Bloch-Zupan, Cecilie Bredrup, Edward B Cooper, Sofia Douzgou Houge, Sixto García-Miñaúr, Hülya Kayserili, Lidia Larizza, Vanesa Lopez Gonzalez, Leonie A Menke, Donatella Milani, Francesco Saettini, Cathy A Stevens, Lloyd Tooke, Jill A Van der Zee, Maria M Van Genderen, Julien Van-Gils, Jane Waite, Jean-Louis Adrien, Oliver Bartsch, Pierre Bitoun, Antonia H M Bouts, Anna M Cueto-González, Elena Dominguez-Garrido, Floor A Duijkers, Patricia Fergelot, Elizabeth Halstead, Sylvia A Huisman, Camilla Meossi, Jo Mullins, Sarah M Nikkel, Chris Oliver, Elisabetta Prada, Alessandra Rei, Ilka Riddle, Cristina Rodriguez-Fonseca, Rebecca Rodríguez Pena, Janet Russell, Alicia Saba, Fernando Santos-Simarro, Brittany N Simpson, David F Smith, Markus F Stevens, Katalin Szakszon, Emmanuelle Taupiac, Nadia Totaro, Irene Valenzuena Palafoll, Daniëlle C M Van Der Kaay, Michiel P Van Wijk, Klea Vyshka, Susan Wiley, and Raoul C Hennekam. Diagnosis and management in rubinstein-taybi syndrome: first international consensus statement. Journal of Medical Genetics, 61:503-519, Mar 2024. URL: https://doi.org/10.1136/jmg-2023-109438, doi:10.1136/jmg-2023-109438. This article has 46 citations and is from a domain leading peer-reviewed journal.
-
(lacombe2024diagnosisandmanagement media d0762497): Didier Lacombe, Agnès Bloch-Zupan, Cecilie Bredrup, Edward B Cooper, Sofia Douzgou Houge, Sixto García-Miñaúr, Hülya Kayserili, Lidia Larizza, Vanesa Lopez Gonzalez, Leonie A Menke, Donatella Milani, Francesco Saettini, Cathy A Stevens, Lloyd Tooke, Jill A Van der Zee, Maria M Van Genderen, Julien Van-Gils, Jane Waite, Jean-Louis Adrien, Oliver Bartsch, Pierre Bitoun, Antonia H M Bouts, Anna M Cueto-González, Elena Dominguez-Garrido, Floor A Duijkers, Patricia Fergelot, Elizabeth Halstead, Sylvia A Huisman, Camilla Meossi, Jo Mullins, Sarah M Nikkel, Chris Oliver, Elisabetta Prada, Alessandra Rei, Ilka Riddle, Cristina Rodriguez-Fonseca, Rebecca Rodríguez Pena, Janet Russell, Alicia Saba, Fernando Santos-Simarro, Brittany N Simpson, David F Smith, Markus F Stevens, Katalin Szakszon, Emmanuelle Taupiac, Nadia Totaro, Irene Valenzuena Palafoll, Daniëlle C M Van Der Kaay, Michiel P Van Wijk, Klea Vyshka, Susan Wiley, and Raoul C Hennekam. Diagnosis and management in rubinstein-taybi syndrome: first international consensus statement. Journal of Medical Genetics, 61:503-519, Mar 2024. URL: https://doi.org/10.1136/jmg-2023-109438, doi:10.1136/jmg-2023-109438. This article has 46 citations and is from a domain leading peer-reviewed journal.
-
(lacombe2024diagnosisandmanagement media cac74578): Didier Lacombe, Agnès Bloch-Zupan, Cecilie Bredrup, Edward B Cooper, Sofia Douzgou Houge, Sixto García-Miñaúr, Hülya Kayserili, Lidia Larizza, Vanesa Lopez Gonzalez, Leonie A Menke, Donatella Milani, Francesco Saettini, Cathy A Stevens, Lloyd Tooke, Jill A Van der Zee, Maria M Van Genderen, Julien Van-Gils, Jane Waite, Jean-Louis Adrien, Oliver Bartsch, Pierre Bitoun, Antonia H M Bouts, Anna M Cueto-González, Elena Dominguez-Garrido, Floor A Duijkers, Patricia Fergelot, Elizabeth Halstead, Sylvia A Huisman, Camilla Meossi, Jo Mullins, Sarah M Nikkel, Chris Oliver, Elisabetta Prada, Alessandra Rei, Ilka Riddle, Cristina Rodriguez-Fonseca, Rebecca Rodríguez Pena, Janet Russell, Alicia Saba, Fernando Santos-Simarro, Brittany N Simpson, David F Smith, Markus F Stevens, Katalin Szakszon, Emmanuelle Taupiac, Nadia Totaro, Irene Valenzuena Palafoll, Daniëlle C M Van Der Kaay, Michiel P Van Wijk, Klea Vyshka, Susan Wiley, and Raoul C Hennekam. Diagnosis and management in rubinstein-taybi syndrome: first international consensus statement. Journal of Medical Genetics, 61:503-519, Mar 2024. URL: https://doi.org/10.1136/jmg-2023-109438, doi:10.1136/jmg-2023-109438. This article has 46 citations and is from a domain leading peer-reviewed journal.
-
(qu’d2023behavioralandneuropsychiatric pages 3-4): Dima Qu’d, Lauren M. Schmitt, Amber Leston, Jacqueline R. Harris, Anne Slavotinek, Ilka Riddle, Diana S. Brightman, and Brittany N. Simpson. Behavioral and neuropsychiatric challenges across the lifespan in individuals with rubinstein-taybi syndrome. Frontiers in Genetics, Jun 2023. URL: https://doi.org/10.3389/fgene.2023.1116919, doi:10.3389/fgene.2023.1116919. This article has 6 citations and is from a peer-reviewed journal.
-
(qu’d2023behavioralandneuropsychiatric pages 11-13): Dima Qu’d, Lauren M. Schmitt, Amber Leston, Jacqueline R. Harris, Anne Slavotinek, Ilka Riddle, Diana S. Brightman, and Brittany N. Simpson. Behavioral and neuropsychiatric challenges across the lifespan in individuals with rubinstein-taybi syndrome. Frontiers in Genetics, Jun 2023. URL: https://doi.org/10.3389/fgene.2023.1116919, doi:10.3389/fgene.2023.1116919. This article has 6 citations and is from a peer-reviewed journal.
-
(qu’d2023behavioralandneuropsychiatric pages 8-10): Dima Qu’d, Lauren M. Schmitt, Amber Leston, Jacqueline R. Harris, Anne Slavotinek, Ilka Riddle, Diana S. Brightman, and Brittany N. Simpson. Behavioral and neuropsychiatric challenges across the lifespan in individuals with rubinstein-taybi syndrome. Frontiers in Genetics, Jun 2023. URL: https://doi.org/10.3389/fgene.2023.1116919, doi:10.3389/fgene.2023.1116919. This article has 6 citations and is from a peer-reviewed journal.
-
(lacombe2024diagnosisandmanagement pages 6-7): Didier Lacombe, Agnès Bloch-Zupan, Cecilie Bredrup, Edward B Cooper, Sofia Douzgou Houge, Sixto García-Miñaúr, Hülya Kayserili, Lidia Larizza, Vanesa Lopez Gonzalez, Leonie A Menke, Donatella Milani, Francesco Saettini, Cathy A Stevens, Lloyd Tooke, Jill A Van der Zee, Maria M Van Genderen, Julien Van-Gils, Jane Waite, Jean-Louis Adrien, Oliver Bartsch, Pierre Bitoun, Antonia H M Bouts, Anna M Cueto-González, Elena Dominguez-Garrido, Floor A Duijkers, Patricia Fergelot, Elizabeth Halstead, Sylvia A Huisman, Camilla Meossi, Jo Mullins, Sarah M Nikkel, Chris Oliver, Elisabetta Prada, Alessandra Rei, Ilka Riddle, Cristina Rodriguez-Fonseca, Rebecca Rodríguez Pena, Janet Russell, Alicia Saba, Fernando Santos-Simarro, Brittany N Simpson, David F Smith, Markus F Stevens, Katalin Szakszon, Emmanuelle Taupiac, Nadia Totaro, Irene Valenzuena Palafoll, Daniëlle C M Van Der Kaay, Michiel P Van Wijk, Klea Vyshka, Susan Wiley, and Raoul C Hennekam. Diagnosis and management in rubinstein-taybi syndrome: first international consensus statement. Journal of Medical Genetics, 61:503-519, Mar 2024. URL: https://doi.org/10.1136/jmg-2023-109438, doi:10.1136/jmg-2023-109438. This article has 46 citations and is from a domain leading peer-reviewed journal.
-
(gils2024transcriptomeandacetylome pages 6-7): Julien Van Gils, Slim Karkar, Aurélien Barre, Seyta Ley-Ngardigal, Sophie Nothof, Stéphane Claverol, Caroline Tokarski, Jean-Philippe Trani, Raphael Chevalier, Natacha Broucqsault, Claire El Yazidi, Didier Lacombe, Patricia Fergelot, and Frédérique Magdinier. Transcriptome and acetylome profiling identify crucial steps of neuronal differentiation in rubinstein-taybi syndrome. Communications Biology, Oct 2024. URL: https://doi.org/10.1038/s42003-024-06939-3, doi:10.1038/s42003-024-06939-3. This article has 3 citations and is from a peer-reviewed journal.
-
(thonel2022cbphsf2structuraland pages 2-3): Aurélie de Thonel, Johanna K. Ahlskog, Kevin Daupin, Véronique Dubreuil, Jérémy Berthelet, Carole Chaput, Geoffrey Pires, Camille Leonetti, Ryma Abane, Lluís Cordón Barris, Isabelle Leray, Anna L. Aalto, Sarah Naceri, Marine Cordonnier, Carène Benasolo, Matthieu Sanial, Agathe Duchateau, Anniina Vihervaara, Mikael C. Puustinen, Federico Miozzo, Patricia Fergelot, Élise Lebigot, Alain Verloes, Pierre Gressens, Didier Lacombe, Jessica Gobbo, Carmen Garrido, Sandy D. Westerheide, Laurent David, Michel Petitjean, Olivier Taboureau, Fernando Rodrigues-Lima, Sandrine Passemard, Délara Sabéran-Djoneidi, Laurent Nguyen, Madeline Lancaster, Lea Sistonen, and Valérie Mezger. Cbp-hsf2 structural and functional interplay in rubinstein-taybi neurodevelopmental disorder. Nature Communications, Nov 2022. URL: https://doi.org/10.1038/s41467-022-34476-2, doi:10.1038/s41467-022-34476-2. This article has 27 citations and is from a highest quality peer-reviewed journal.
-
(thonel2022cbphsf2structuraland pages 11-11): Aurélie de Thonel, Johanna K. Ahlskog, Kevin Daupin, Véronique Dubreuil, Jérémy Berthelet, Carole Chaput, Geoffrey Pires, Camille Leonetti, Ryma Abane, Lluís Cordón Barris, Isabelle Leray, Anna L. Aalto, Sarah Naceri, Marine Cordonnier, Carène Benasolo, Matthieu Sanial, Agathe Duchateau, Anniina Vihervaara, Mikael C. Puustinen, Federico Miozzo, Patricia Fergelot, Élise Lebigot, Alain Verloes, Pierre Gressens, Didier Lacombe, Jessica Gobbo, Carmen Garrido, Sandy D. Westerheide, Laurent David, Michel Petitjean, Olivier Taboureau, Fernando Rodrigues-Lima, Sandrine Passemard, Délara Sabéran-Djoneidi, Laurent Nguyen, Madeline Lancaster, Lea Sistonen, and Valérie Mezger. Cbp-hsf2 structural and functional interplay in rubinstein-taybi neurodevelopmental disorder. Nature Communications, Nov 2022. URL: https://doi.org/10.1038/s41467-022-34476-2, doi:10.1038/s41467-022-34476-2. This article has 27 citations and is from a highest quality peer-reviewed journal.
-
(thonel2022cbphsf2structuraland pages 7-9): Aurélie de Thonel, Johanna K. Ahlskog, Kevin Daupin, Véronique Dubreuil, Jérémy Berthelet, Carole Chaput, Geoffrey Pires, Camille Leonetti, Ryma Abane, Lluís Cordón Barris, Isabelle Leray, Anna L. Aalto, Sarah Naceri, Marine Cordonnier, Carène Benasolo, Matthieu Sanial, Agathe Duchateau, Anniina Vihervaara, Mikael C. Puustinen, Federico Miozzo, Patricia Fergelot, Élise Lebigot, Alain Verloes, Pierre Gressens, Didier Lacombe, Jessica Gobbo, Carmen Garrido, Sandy D. Westerheide, Laurent David, Michel Petitjean, Olivier Taboureau, Fernando Rodrigues-Lima, Sandrine Passemard, Délara Sabéran-Djoneidi, Laurent Nguyen, Madeline Lancaster, Lea Sistonen, and Valérie Mezger. Cbp-hsf2 structural and functional interplay in rubinstein-taybi neurodevelopmental disorder. Nature Communications, Nov 2022. URL: https://doi.org/10.1038/s41467-022-34476-2, doi:10.1038/s41467-022-34476-2. This article has 27 citations and is from a highest quality peer-reviewed journal.
-
(thonel2022cbphsf2structuraland pages 15-16): Aurélie de Thonel, Johanna K. Ahlskog, Kevin Daupin, Véronique Dubreuil, Jérémy Berthelet, Carole Chaput, Geoffrey Pires, Camille Leonetti, Ryma Abane, Lluís Cordón Barris, Isabelle Leray, Anna L. Aalto, Sarah Naceri, Marine Cordonnier, Carène Benasolo, Matthieu Sanial, Agathe Duchateau, Anniina Vihervaara, Mikael C. Puustinen, Federico Miozzo, Patricia Fergelot, Élise Lebigot, Alain Verloes, Pierre Gressens, Didier Lacombe, Jessica Gobbo, Carmen Garrido, Sandy D. Westerheide, Laurent David, Michel Petitjean, Olivier Taboureau, Fernando Rodrigues-Lima, Sandrine Passemard, Délara Sabéran-Djoneidi, Laurent Nguyen, Madeline Lancaster, Lea Sistonen, and Valérie Mezger. Cbp-hsf2 structural and functional interplay in rubinstein-taybi neurodevelopmental disorder. Nature Communications, Nov 2022. URL: https://doi.org/10.1038/s41467-022-34476-2, doi:10.1038/s41467-022-34476-2. This article has 27 citations and is from a highest quality peer-reviewed journal.
-
(thonel2022cbphsf2structuraland pages 3-4): Aurélie de Thonel, Johanna K. Ahlskog, Kevin Daupin, Véronique Dubreuil, Jérémy Berthelet, Carole Chaput, Geoffrey Pires, Camille Leonetti, Ryma Abane, Lluís Cordón Barris, Isabelle Leray, Anna L. Aalto, Sarah Naceri, Marine Cordonnier, Carène Benasolo, Matthieu Sanial, Agathe Duchateau, Anniina Vihervaara, Mikael C. Puustinen, Federico Miozzo, Patricia Fergelot, Élise Lebigot, Alain Verloes, Pierre Gressens, Didier Lacombe, Jessica Gobbo, Carmen Garrido, Sandy D. Westerheide, Laurent David, Michel Petitjean, Olivier Taboureau, Fernando Rodrigues-Lima, Sandrine Passemard, Délara Sabéran-Djoneidi, Laurent Nguyen, Madeline Lancaster, Lea Sistonen, and Valérie Mezger. Cbp-hsf2 structural and functional interplay in rubinstein-taybi neurodevelopmental disorder. Nature Communications, Nov 2022. URL: https://doi.org/10.1038/s41467-022-34476-2, doi:10.1038/s41467-022-34476-2. This article has 27 citations and is from a highest quality peer-reviewed journal.
-
(trajkova2024dnamethylationanalysis pages 2-3): Slavica Trajkova, Jennifer Kerkhof, Matteo Rossi Sebastiano, Lisa Pavinato, Enza Ferrero, Chiara Giovenino, Diana Carli, Eleonora Di Gregorio, Roberta Marinoni, Giorgia Mandrile, Flavia Palermo, Silvia Carestiato, Simona Cardaropoli, Verdiana Pullano, Antonina Rinninella, Elisa Giorgio, Tommaso Pippucci, Paola Dimartino, Jessica Rzasa, Kathleen Rooney, Haley McConkey, Aleksandar Petlichkovski, Barbara Pasini, Elena Sukarova-Angelovska, Christopher M. Campbell, Kay Metcalfe, Sarah Jenkinson, Siddharth Banka, Alessandro Mussa, Giovanni Battista Ferrero, Bekim Sadikovic, and Alfredo Brusco. Dna methylation analysis in patients with neurodevelopmental disorders improves variant interpretation and reveals complexity. Human Genetics and Genomics Advances, 5:100309, Jul 2024. URL: https://doi.org/10.1016/j.xhgg.2024.100309, doi:10.1016/j.xhgg.2024.100309. This article has 18 citations and is from a peer-reviewed journal.
-
(trajkova2024dnamethylationanalysis pages 3-4): Slavica Trajkova, Jennifer Kerkhof, Matteo Rossi Sebastiano, Lisa Pavinato, Enza Ferrero, Chiara Giovenino, Diana Carli, Eleonora Di Gregorio, Roberta Marinoni, Giorgia Mandrile, Flavia Palermo, Silvia Carestiato, Simona Cardaropoli, Verdiana Pullano, Antonina Rinninella, Elisa Giorgio, Tommaso Pippucci, Paola Dimartino, Jessica Rzasa, Kathleen Rooney, Haley McConkey, Aleksandar Petlichkovski, Barbara Pasini, Elena Sukarova-Angelovska, Christopher M. Campbell, Kay Metcalfe, Sarah Jenkinson, Siddharth Banka, Alessandro Mussa, Giovanni Battista Ferrero, Bekim Sadikovic, and Alfredo Brusco. Dna methylation analysis in patients with neurodevelopmental disorders improves variant interpretation and reveals complexity. Human Genetics and Genomics Advances, 5:100309, Jul 2024. URL: https://doi.org/10.1016/j.xhgg.2024.100309, doi:10.1016/j.xhgg.2024.100309. This article has 18 citations and is from a peer-reviewed journal.
-
(lacombe2024diagnosisandmanagement pages 1-1): Didier Lacombe, Agnès Bloch-Zupan, Cecilie Bredrup, Edward B Cooper, Sofia Douzgou Houge, Sixto García-Miñaúr, Hülya Kayserili, Lidia Larizza, Vanesa Lopez Gonzalez, Leonie A Menke, Donatella Milani, Francesco Saettini, Cathy A Stevens, Lloyd Tooke, Jill A Van der Zee, Maria M Van Genderen, Julien Van-Gils, Jane Waite, Jean-Louis Adrien, Oliver Bartsch, Pierre Bitoun, Antonia H M Bouts, Anna M Cueto-González, Elena Dominguez-Garrido, Floor A Duijkers, Patricia Fergelot, Elizabeth Halstead, Sylvia A Huisman, Camilla Meossi, Jo Mullins, Sarah M Nikkel, Chris Oliver, Elisabetta Prada, Alessandra Rei, Ilka Riddle, Cristina Rodriguez-Fonseca, Rebecca Rodríguez Pena, Janet Russell, Alicia Saba, Fernando Santos-Simarro, Brittany N Simpson, David F Smith, Markus F Stevens, Katalin Szakszon, Emmanuelle Taupiac, Nadia Totaro, Irene Valenzuena Palafoll, Daniëlle C M Van Der Kaay, Michiel P Van Wijk, Klea Vyshka, Susan Wiley, and Raoul C Hennekam. Diagnosis and management in rubinstein-taybi syndrome: first international consensus statement. Journal of Medical Genetics, 61:503-519, Mar 2024. URL: https://doi.org/10.1136/jmg-2023-109438, doi:10.1136/jmg-2023-109438. This article has 46 citations and is from a domain leading peer-reviewed journal.
-
(NCT04122742 chunk 2): Diagnosis of RSTS: Identification of the Acetylation Profiles as Epigenetic Markers for Assessing Causality of CREBBP and EP300 Variants.. University Hospital, Bordeaux. 2019. ClinicalTrials.gov Identifier: NCT04122742
-
(gils2024transcriptomeandacetylome pages 7-8): Julien Van Gils, Slim Karkar, Aurélien Barre, Seyta Ley-Ngardigal, Sophie Nothof, Stéphane Claverol, Caroline Tokarski, Jean-Philippe Trani, Raphael Chevalier, Natacha Broucqsault, Claire El Yazidi, Didier Lacombe, Patricia Fergelot, and Frédérique Magdinier. Transcriptome and acetylome profiling identify crucial steps of neuronal differentiation in rubinstein-taybi syndrome. Communications Biology, Oct 2024. URL: https://doi.org/10.1038/s42003-024-06939-3, doi:10.1038/s42003-024-06939-3. This article has 3 citations and is from a peer-reviewed journal.
-
(gils2024transcriptomeandacetylome pages 9-10): Julien Van Gils, Slim Karkar, Aurélien Barre, Seyta Ley-Ngardigal, Sophie Nothof, Stéphane Claverol, Caroline Tokarski, Jean-Philippe Trani, Raphael Chevalier, Natacha Broucqsault, Claire El Yazidi, Didier Lacombe, Patricia Fergelot, and Frédérique Magdinier. Transcriptome and acetylome profiling identify crucial steps of neuronal differentiation in rubinstein-taybi syndrome. Communications Biology, Oct 2024. URL: https://doi.org/10.1038/s42003-024-06939-3, doi:10.1038/s42003-024-06939-3. This article has 3 citations and is from a peer-reviewed journal.
-
(wang2023geneticscornera pages 4-5): Hua Wang and L. Ann. Genetics corner: a new case of rubinstein-taybi syndrome with a novel variant in the crebbp gene detected through whole exome sequencing. Neonatology Today, pages 165, Dec 2023. URL: https://doi.org/10.51362/neonatology.today/20231812-165170, doi:10.51362/neonatology.today/20231812-165170. This article has 0 citations.