Ritscher-Schinzel Syndrome
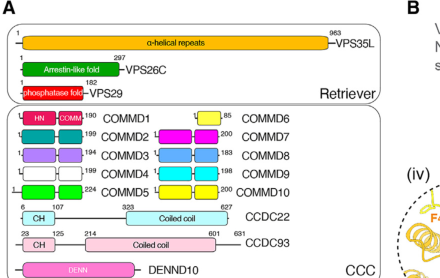
Ritscher-Schinzel syndrome (RSS), also called 3C (cranio-cerebello-cardiac) syndrome, is a rare multiple congenital anomalies syndrome defined by a triad of craniofacial dysmorphism, cerebellar malformations (notably Dandy-Walker malformation and cerebellar vermis hypoplasia), and congenital heart defects, with variable developmental delay/intellectual disability. RSS is genetically heterogeneous and is now understood as an endosomal "recyclinopathy": it is caused by biallelic or X-linked variants in genes encoding subunits of the Commander multiprotein assembly and the functionally coupled WASH complex (WASHC5, CCDC22, VPS35L, and the CCC-complex subunits COMMD4, COMMD9, CCDC93), which together drive SNX17-dependent endosomal recycling of integral membrane cargo proteins required for tissue development.
Ask OpenScientist
Ask a research question about Ritscher-Schinzel Syndrome. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Subtypes
4Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Pathophysiology
4Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Pathograph
Phenotypes
17Blood 1
Show evidence (1 reference)
Cardiovascular 2
Show evidence (1 reference)
Show evidence (1 reference)
Eye 1
Show evidence (1 reference)
Genitourinary 1
Show evidence (1 reference)
Head and Neck 3
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Metabolism 1
Show evidence (1 reference)
Nervous System 4
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Growth 1
Show evidence (1 reference)
Other 3
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
6Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Medical Actions
4Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Source YAML
click to showname: Ritscher-Schinzel Syndrome
creation_date: "2026-06-05T12:00:00Z"
category: Mendelian
description: >-
Ritscher-Schinzel syndrome (RSS), also called 3C (cranio-cerebello-cardiac)
syndrome, is a rare multiple congenital anomalies syndrome defined by a triad
of craniofacial dysmorphism, cerebellar malformations (notably Dandy-Walker
malformation and cerebellar vermis hypoplasia), and congenital heart defects,
with variable developmental delay/intellectual disability. RSS is genetically
heterogeneous and is now understood as an endosomal "recyclinopathy": it is
caused by biallelic or X-linked variants in genes encoding subunits of the
Commander multiprotein assembly and the functionally coupled WASH complex
(WASHC5, CCDC22, VPS35L, and the CCC-complex subunits COMMD4, COMMD9, CCDC93),
which together drive SNX17-dependent endosomal recycling of integral membrane
cargo proteins required for tissue development.
disease_term:
preferred_term: Ritscher-Schinzel Syndrome
term:
id: MONDO:0019078
label: Ritscher-Schinzel syndrome
parents:
- Multiple congenital anomalies syndrome
- Endosomal recycling disorder
references:
- reference: PMID:31971710
title: "Ritscher-Schinzel Syndrome."
tags:
- GeneReviews
has_subtypes:
- name: RSS1
display_name: RSS1 (WASHC5-related)
subtype_term:
preferred_term: Ritscher-Schinzel syndrome 1
term:
id: MONDO:0009073
label: Ritscher-Schinzel syndrome 1
description: >-
Classic autosomal recessive form caused by biallelic loss-of-function
variants in WASHC5 (formerly KIAA0196), which encodes the WASH complex
subunit strumpellin. The first identified molecular cause of RSS, presenting
with the full 3C triad plus developmental delay.
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
genes:
- preferred_term: WASHC5
term:
id: hgnc:28984
label: WASHC5
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
the identification of biallelic pathogenic variants in WASHC5 in a male or
female or a hemizygous pathogenic variant in CCDC22 in a male by molecular
genetic testing
explanation: >-
Biallelic WASHC5 variants define the autosomal recessive WASHC5-related
subtype of RSS.
- name: RSS2
display_name: RSS2 (CCDC22-related)
subtype_term:
preferred_term: Ritscher-Schinzel syndrome 2
term:
id: MONDO:0010499
label: Ritscher-Schinzel syndrome 2
description: >-
X-linked form caused by hemizygous variants in CCDC22, a CCC-complex subunit
of Commander. Overlaps with X-linked intellectual disability; some missense
alleles that impair COMMD binding cause an attenuated 3C phenotype without
major cardiac or neuroanatomical abnormalities.
inheritance:
- name: X-linked
inheritance_term:
preferred_term: X-linked inheritance
term:
id: HP:0001417
label: X-linked inheritance
genes:
- preferred_term: CCDC22
term:
id: hgnc:28909
label: CCDC22
evidence:
- reference: PMID:40448120
reference_title: "CCDC22 mutations that impair COMMD binding cause attenuated 3C/Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Here, we report a new CCDC22 missense mutation, p.E208K, that results in
attenuated 3 C syndrome, without cardiac or neuroanatomical abnormalities.
explanation: >-
CCDC22 variants define the X-linked subtype, with some alleles producing
an attenuated 3C phenotype.
- name: RSS3
display_name: RSS3 (VPS35L-related)
subtype_term:
preferred_term: Ritscher-Schinzel syndrome 3
term:
id: MONDO:0030864
label: Ritscher-Schinzel syndrome 3
description: >-
Autosomal recessive form caused by biallelic VPS35L variants. VPS35L is a
Retriever subunit and the third identified RSS gene after WASHC5 and CCDC22.
Associated with a distinct, often more severe spectrum including
hypercholesterolemia, hypogammaglobulinemia, intestinal lymphangiectasia,
and proteinuria.
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
genes:
- preferred_term: VPS35L
term:
id: hgnc:24641
label: VPS35L
evidence:
- reference: PMID:36113987
reference_title: "Clinical diversity and molecular mechanism of VPS35L-associated Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The Retriever subunit VPS35L is the third responsible gene for
Ritscher-Schinzel syndrome (RSS) after WASHC5 and CCDC22.
explanation: >-
VPS35L variants define the third (autosomal recessive) molecular subtype
of RSS.
- name: CCC-related
display_name: CCC-complex-related (COMMD4/COMMD9/CCDC93)
description: >-
Autosomal recessive forms caused by biallelic variants in additional
CCC-complex subunits (COMMD4, COMMD9, CCDC93), identified as Commander
pathway RSS genes. Severity varies with residual Commander activity; severe
biallelic COMMD4 genotypes have been associated with early childhood death.
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
genes:
- preferred_term: COMMD4
term:
id: hgnc:26027
label: COMMD4
- preferred_term: COMMD9
term:
id: hgnc:25014
label: COMMD9
- preferred_term: CCDC93
term:
id: hgnc:25611
label: CCDC93
evidence:
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
to identify causative genes in the copper metabolic murr1
domain-containing (COMMD) proteins COMMD4, COMMD9, and coiled-coil domain
containing 93 (CCDC93) subunits of the Commander complex
explanation: >-
COMMD4, COMMD9, and CCDC93 variants define additional CCC-complex subtypes
of RSS identified in newly recognized patient cohorts.
pathophysiology:
- name: Commander/WASH complex deficiency
description: >-
Pathogenic variants in Commander and WASH complex genes (WASHC5, CCDC22,
VPS35L, COMMD4, COMMD9, CCDC93) destabilize or disrupt assembly of the
16-subunit Commander assembly (Retriever: VPS35L/VPS26C/VPS29 plus the CCC
complex: COMMD1-10 with CCDC22 and CCDC93) and the functionally coupled WASH
complex. Most variants are loss-of-function or complex-destabilizing,
reducing endosomal recycling capacity.
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: endocytic recycling
term:
id: GO:0032456
label: endocytic recycling
modifier: DECREASED
evidence:
- reference: PMID:37172566
reference_title: "Structure of the endosomal Commander complex linked to Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
The Commander complex is required for endosomal recycling of diverse
transmembrane cargos and is mutated in Ritscher-Schinzel syndrome. It
comprises two sub-assemblies: Retriever composed of VPS35L, VPS26C, and
VPS29; and the CCC complex which contains twelve subunits: COMMD1-COMMD10
and the coiled-coil domain-containing (CCDC) proteins CCDC22 and CCDC93.
explanation: >-
The structural study establishes that Commander, mutated in RSS, is
composed of the Retriever and CCC sub-assemblies whose genes are the
causative RSS genes.
- reference: PMID:40448120
reference_title: "CCDC22 mutations that impair COMMD binding cause attenuated 3C/Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
Mutations in genes encoding subunits of these three complexes, CCDC22,
VPS35L, and WASHC5, have been linked with a developmental syndrome known
as 3 C (cranio-cerebello-cardiac) or Ritscher-Schinzel syndrome.
explanation: >-
Confirms that CCDC22, VPS35L, and WASHC5 variants underlie RSS through
defects in the CCC/Retriever/WASH recycling machinery.
downstream:
- target: Impaired SNX17-dependent endosomal recycling
description: >-
Disruption of Commander assembly impairs the SNX17-dependent retrieval and
recycling of integral membrane cargo proteins from endosomes to the cell
surface.
evidence:
- reference: PMID:40448120
reference_title: "CCDC22 mutations that impair COMMD binding cause attenuated 3C/Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
The CCC complex, composed of CCDC22, CCDC93, and ten proteins of the
COMMD family, coordinates several critical steps required to recycle
internalized plasma membrane proteins from endosomes to the cell surface.
explanation: >-
Loss of CCC/Commander function impairs endosome-to-surface recycling of
internalized membrane proteins.
- name: Impaired SNX17-dependent endosomal recycling
description: >-
Commander organizes the sorting nexin-17 (SNX17)-dependent recycling of
hundreds of integral membrane proteins through the endosomal network.
Commander dysfunction reduces cell-surface presentation of cargos bearing
SNX17-recognized ΦxNPxY/F or ΦxNxxY/F sorting motifs, including integrins and
lipoprotein receptors, in a tissue-specific manner.
cell_types:
- preferred_term: hepatocyte
term:
id: CL:0000182
label: hepatocyte
- preferred_term: epithelial cell of proximal tubule
term:
id: CL:0002306
label: epithelial cell of proximal tubule
biological_processes:
- preferred_term: endosome to plasma membrane protein transport
term:
id: GO:0099638
label: endosome to plasma membrane protein transport
modifier: DECREASED
- preferred_term: receptor recycling
term:
id: GO:0001881
label: receptor recycling
modifier: DECREASED
evidence:
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Commander organizes the sorting nexin-17 (SNX17)-dependent recycling of
hundreds of integral membrane proteins through the endosomal network.
explanation: >-
Establishes the SNX17-Commander recycling pathway as the molecular hub
whose dysfunction underlies RSS.
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
these integral proteins contained ΦxNPxY/F or ΦxNxxY/F sorting motifs in
their cytoplasmic-facing domains (where Φ is a hydrophobic residue and x
is any residue) that are recognized by SNX17 to drive their
Commander-dependent endosomal recycling
explanation: >-
Defines the cargo sorting motifs recognized by SNX17 for
Commander-dependent recycling, the process impaired in RSS.
downstream:
- target: Reduced cell-surface receptor density
description: >-
Impaired recycling reduces tissue-specific presentation of cell-surface
integral membrane proteins essential for kidney, bone, and brain
development, and reduces surface lipoprotein receptors.
evidence:
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
through cell surface proteomics, that this reduces tissue-specific
presentation of cell surface integral membrane proteins essential for
kidney, bone, and brain development
explanation: >-
Disrupted recycling lowers cell-surface presentation of developmentally
essential cargo proteins.
- name: Reduced cell-surface receptor density
description: >-
Defective recycling lowers steady-state cell-surface levels of receptors and
adhesion molecules. In VPS35L-associated RSS, ablation decreases surface
LRP1 and LDLR, reducing LDL uptake and providing a mechanism for
hypercholesterolemia. Reduced tissue-specific surface cargo presentation
drives the multisystem (cerebellar, cardiac, craniofacial, renal, skeletal)
developmental defects.
cell_types:
- preferred_term: hepatocyte
term:
id: CL:0000182
label: hepatocyte
biological_processes:
- preferred_term: receptor-mediated endocytosis
term:
id: GO:0006898
label: receptor-mediated endocytosis
modifier: DECREASED
evidence:
- reference: PMID:36113987
reference_title: "Clinical diversity and molecular mechanism of VPS35L-associated Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
Cellular analysis found VPS35L ablation decreased the cell surface level
of lipoprotein receptor-related protein 1 and low-density lipoprotein
receptor, resulting in reduced low-density lipoprotein cellular uptake.
explanation: >-
Demonstrates that loss of the Retriever subunit VPS35L reduces surface
lipoprotein receptors and LDL uptake, the proposed mechanism for
hypercholesterolemia in RSS.
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
through cell surface proteomics, that this reduces tissue-specific
presentation of cell surface integral membrane proteins essential for
kidney, bone, and brain development
explanation: >-
Links reduced surface cargo presentation to the multisystem developmental
phenotypes (kidney, bone, brain) of RSS.
downstream:
- target: Multisystem developmental malformation
description: >-
Tissue-specific loss of surface cargo during organogenesis produces
cerebellar, cardiac, craniofacial, renal, and skeletal malformations.
evidence:
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
through generation of mouse models of RSS, we show replication of
RSS-associated clinical phenotypes including proteinuria, skeletal
malformation, and neurological impairment
explanation: >-
Mouse models confirm that Commander dysfunction produces the multisystem
malformation phenotypes of RSS.
- target: Hypercholesterolemia
causal_link_type: DIRECT
- name: Multisystem developmental malformation
description: >-
The convergent consequence of impaired Commander-dependent recycling is a
congenital multiple-organ malformation syndrome affecting the cerebellum
(Dandy-Walker malformation, vermis hypoplasia), heart (septal and
atrioventricular canal defects, tetralogy of Fallot), craniofacial
structures, kidney (proteinuria), liver, and skeleton, together with
neurodevelopmental impairment.
cell_types:
- preferred_term: Purkinje cell
term:
id: CL:0000121
label: Purkinje cell
- preferred_term: migratory cranial neural crest cell
term:
id: CL:0000008
label: migratory cranial neural crest cell
evidence:
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Ritscher-Schinzel syndrome (RSS) is a congenital malformation syndrome
characterized by cerebellar, cardiac, and craniofacial malformations and
phenotypes associated with liver, skeletal, and kidney dysfunction.
explanation: >-
Summarizes the multisystem malformation phenotype that results from the
recycling defect.
downstream:
- target: Dandy-Walker malformation
causal_link_type: DIRECT
- target: Cerebellar vermis hypoplasia
causal_link_type: DIRECT
- target: Congenital heart defect
causal_link_type: DIRECT
- target: Atrial septal defect
causal_link_type: DIRECT
- target: Craniofacial dysmorphism
causal_link_type: DIRECT
- target: Hypertelorism
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Downslanted palpebral fissures
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Cleft palate
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Intellectual disability
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Global developmental delay
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Hypogammaglobulinemia
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Intestinal lymphangiectasia
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Proteinuria
causal_link_type: DIRECT
- target: Eye anomalies
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Obesity
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Obstructive sleep apnea
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
phenotypes:
- category: Phenotypic
name: Dandy-Walker malformation
description: >-
Cerebellar malformation characteristic of the RSS triad, part of the
spectrum of cerebellar/posterior fossa anomalies.
phenotype_term:
preferred_term: Dandy-Walker malformation
term:
id: HP:0001305
label: Dandy-Walker malformation
evidence:
- reference: PMID:24916641
reference_title: "Missense variant in CCDC22 causes X-linked recessive intellectual disability with features of Ritscher-Schinzel/3C syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Ritscher-Schinzel syndrome (RSS)/3C (cranio-cerebro-cardiac) syndrome
(OMIM#220210) is a rare and clinically heterogeneous developmental
disorder characterized by intellectual disability, cerebellar brain
malformations, congenital heart defects, and craniofacial abnormalities.
explanation: >-
Cerebellar brain malformations, including Dandy-Walker malformation, are a
cardinal feature of RSS.
- category: Phenotypic
name: Cerebellar vermis hypoplasia
description: >-
Hypoplasia of the cerebellar vermis is part of the posterior fossa anomaly
spectrum in RSS.
phenotype_term:
preferred_term: Cerebellar vermis hypoplasia
term:
id: HP:0001320
label: Cerebellar vermis hypoplasia
evidence:
- reference: PMID:24916641
reference_title: "Missense variant in CCDC22 causes X-linked recessive intellectual disability with features of Ritscher-Schinzel/3C syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
characterized by intellectual disability, cerebellar brain malformations,
congenital heart defects, and craniofacial abnormalities
explanation: >-
Cerebellar malformations including vermis hypoplasia are a defining
cerebellar component of the RSS triad.
- category: Phenotypic
name: Congenital heart defect
description: >-
Congenital cardiac malformations, including atrial and ventricular septal
defects, atrioventricular canal defects, and tetralogy of Fallot, are the
cardiac component of the 3C triad and are reported in the majority of cases.
phenotype_term:
preferred_term: Congenital heart defect
term:
id: HP:0001627
label: Abnormal heart morphology
frequency: VERY_FREQUENT
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Ritscher-Schinzel syndrome (RSS) is a clinically recognizable condition
that includes the cardinal findings of craniofacial features, cerebellar
defects, and cardiovascular malformations resulting in the alternate
diagnostic name of 3C syndrome.
explanation: >-
Cardiovascular malformations are one of the three cardinal findings of
RSS/3C syndrome.
- category: Phenotypic
name: Atrial septal defect
description: >-
Atrial septal defect is among the recurrent congenital heart defects in RSS.
phenotype_term:
preferred_term: Atrial septal defect
term:
id: HP:0001631
label: Atrial septal defect
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
Standard treatment for obesity, obstructive sleep apnea, cleft palate,
congenital heart defects, hypercholesterolemia, renal anomalies,
immunodeficiency, and developmental delay / intellectual disability.
explanation: >-
The cited source documents congenital heart defects in RSS generally;
atrial septal defect is a recognized component of the RSS cardiac spectrum
(Dandy-Walker-like malformation with atrioventricular/atrial septal
defect), though this snippet supports CHD generically rather than ASD
specifically.
- category: Phenotypic
name: Craniofacial dysmorphism
description: >-
Characteristic craniofacial features include brachycephaly, hypotonic face
with protruding tongue, short midface, widely spaced eyes, downslanted
palpebral fissures, low-set ears, smooth or short philtrum, and high or
cleft palate.
phenotype_term:
preferred_term: Craniofacial dysmorphism
term:
id: HP:0001999
label: Abnormal facial shape
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Dysmorphic facial features may include brachycephaly, hypotonic face with
protruding tongue, flat appearance of the face on profile view, short
midface, widely spaced eyes, downslanted palpebral fissures, low-set ears
with overfolding of the upper helix, smooth or short philtrum, and high or
cleft palate.
explanation: >-
GeneReviews details the characteristic craniofacial dysmorphism of RSS.
- category: Phenotypic
name: Hypertelorism
description: >-
Widely spaced eyes (hypertelorism) is a recurrent craniofacial feature of
RSS.
phenotype_term:
preferred_term: Hypertelorism
term:
id: HP:0000316
label: Hypertelorism
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
short midface, widely spaced eyes, downslanted palpebral fissures, low-set
ears with overfolding of the upper helix
explanation: >-
Widely spaced eyes (hypertelorism) is part of the RSS facial gestalt.
- category: Phenotypic
name: Downslanted palpebral fissures
description: >-
Downslanted palpebral fissures are part of the characteristic facial gestalt
of RSS.
phenotype_term:
preferred_term: Downslanted palpebral fissures
term:
id: HP:0000494
label: Downslanted palpebral fissures
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
widely spaced eyes, downslanted palpebral fissures, low-set ears with
overfolding of the upper helix
explanation: >-
Downslanted palpebral fissures are an explicit RSS facial feature.
- category: Phenotypic
name: Cleft palate
description: >-
High-arched or cleft palate is a recurrent craniofacial feature in RSS.
phenotype_term:
preferred_term: Cleft palate
term:
id: HP:0000175
label: Cleft palate
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
smooth or short philtrum, and high or cleft palate
explanation: >-
Cleft (or high-arched) palate is part of the RSS craniofacial spectrum.
- category: Phenotypic
name: Intellectual disability
description: >-
RSS is associated with variable degrees of developmental delay and
intellectual disability.
phenotype_term:
preferred_term: Intellectual disability
term:
id: HP:0001249
label: Intellectual disability
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
RSS is associated with variable degrees of developmental delay and
intellectual disability.
explanation: >-
GeneReviews documents variable developmental delay and intellectual
disability as characteristic of RSS.
- category: Phenotypic
name: Global developmental delay
description: >-
Developmental delay is a near-universal feature; in a WASHC5-associated
cohort it was reported in all assessed cases.
phenotype_term:
preferred_term: Global developmental delay
term:
id: HP:0001263
label: Global developmental delay
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
RSS is associated with variable degrees of developmental delay and
intellectual disability.
explanation: >-
Developmental delay is a recognized characteristic of RSS.
- category: Phenotypic
name: Hypercholesterolemia
description: >-
Hypercholesterolemia may be variably present and is a confirmed complication
of VPS35L-associated RSS, mechanistically linked to reduced surface
lipoprotein receptors.
phenotype_term:
preferred_term: Hypercholesterolemia
term:
id: HP:0003124
label: Hypercholesterolemia
subtype: RSS3
evidence:
- reference: PMID:36113987
reference_title: "Clinical diversity and molecular mechanism of VPS35L-associated Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
In addition to typical features of RSS, we confirmed hypercholesterolaemia,
hypogammaglobulinaemia and intestinal lymphangiectasia as novel
complications of VPS35L-associated RSS.
explanation: >-
Hypercholesterolemia is a confirmed complication of VPS35L-associated RSS.
- category: Phenotypic
name: Hypogammaglobulinemia
description: >-
Decreased circulating immunoglobulin (hypogammaglobulinemia) is a novel
complication confirmed in VPS35L-associated RSS.
phenotype_term:
preferred_term: Hypogammaglobulinemia
term:
id: HP:0004313
label: Decreased circulating immunoglobulin concentration
subtype: RSS3
evidence:
- reference: PMID:36113987
reference_title: "Clinical diversity and molecular mechanism of VPS35L-associated Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
In addition to typical features of RSS, we confirmed hypercholesterolaemia,
hypogammaglobulinaemia and intestinal lymphangiectasia as novel
complications of VPS35L-associated RSS.
explanation: >-
Hypogammaglobulinemia is a confirmed immunologic complication of
VPS35L-associated RSS.
- category: Phenotypic
name: Intestinal lymphangiectasia
description: >-
Intestinal lymphangiectasia is a novel complication confirmed in
VPS35L-associated RSS.
phenotype_term:
preferred_term: Intestinal lymphangiectasia
term:
id: HP:0002593
label: Intestinal lymphangiectasia
subtype: RSS3
evidence:
- reference: PMID:36113987
reference_title: "Clinical diversity and molecular mechanism of VPS35L-associated Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
In addition to typical features of RSS, we confirmed hypercholesterolaemia,
hypogammaglobulinaemia and intestinal lymphangiectasia as novel
complications of VPS35L-associated RSS.
explanation: >-
Intestinal lymphangiectasia is a confirmed gastrointestinal complication
of VPS35L-associated RSS.
- category: Phenotypic
name: Proteinuria
description: >-
Proteinuria reflects renal involvement in RSS and is recapitulated in mouse
models of the disease.
phenotype_term:
preferred_term: Proteinuria
term:
id: HP:0000093
label: Proteinuria
evidence:
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
through generation of mouse models of RSS, we show replication of
RSS-associated clinical phenotypes including proteinuria, skeletal
malformation, and neurological impairment
explanation: >-
Proteinuria is an RSS-associated clinical phenotype recapitulated in mouse
models.
- category: Phenotypic
name: Eye anomalies
description: >-
Ocular anomalies (including coloboma per Orphanet) may be variably present
in RSS.
phenotype_term:
preferred_term: Eye anomalies
term:
id: HP:0000478
label: Abnormality of the eye
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Eye anomalies and hypercholesterolemia may be variably present.
explanation: >-
GeneReviews documents variably present eye anomalies as a feature of RSS.
- category: Phenotypic
name: Obesity
description: >-
Obesity is a recognized manifestation of RSS addressed in clinical
management per GeneReviews.
phenotype_term:
preferred_term: Obesity
term:
id: HP:0001513
label: Obesity
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Standard treatment for obesity, obstructive sleep apnea, cleft palate,
congenital heart defects, hypercholesterolemia, renal anomalies,
immunodeficiency, and developmental delay / intellectual disability.
explanation: >-
GeneReviews lists obesity among the manifestations of RSS requiring
standard management, establishing it as a recognized clinical feature.
- category: Phenotypic
name: Obstructive sleep apnea
description: >-
Obstructive sleep apnea is a recognized manifestation of RSS addressed in
clinical management per GeneReviews.
phenotype_term:
preferred_term: Obstructive sleep apnea
term:
id: HP:0002870
label: Obstructive sleep apnea
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Standard treatment for obesity, obstructive sleep apnea, cleft palate,
congenital heart defects, hypercholesterolemia, renal anomalies,
immunodeficiency, and developmental delay / intellectual disability.
explanation: >-
GeneReviews lists obstructive sleep apnea among the manifestations of RSS
requiring standard management, establishing it as a recognized clinical
feature.
genetic:
- name: WASHC5
gene_term:
preferred_term: WASHC5
term:
id: hgnc:28984
label: WASHC5
association: CAUSATIVE
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
WASHC5-related RSS is inherited in an autosomal recessive manner;
CCDC22-related RSS is inherited in an X-linked manner.
explanation: >-
GeneReviews specifies autosomal recessive inheritance for WASHC5-related
RSS.
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
the identification of biallelic pathogenic variants in WASHC5 in a male or
female or a hemizygous pathogenic variant in CCDC22 in a male by molecular
genetic testing
explanation: >-
Biallelic WASHC5 variants establish the diagnosis of RSS.
- name: CCDC22
gene_term:
preferred_term: CCDC22
term:
id: hgnc:28909
label: CCDC22
association: CAUSATIVE
inheritance:
- name: X-linked
inheritance_term:
preferred_term: X-linked inheritance
term:
id: HP:0001417
label: X-linked inheritance
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
WASHC5-related RSS is inherited in an autosomal recessive manner;
CCDC22-related RSS is inherited in an X-linked manner.
explanation: >-
GeneReviews specifies X-linked inheritance for CCDC22-related RSS.
evidence:
- reference: PMID:24916641
reference_title: "Missense variant in CCDC22 causes X-linked recessive intellectual disability with features of Ritscher-Schinzel/3C syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Exome sequencing detected a missense variant (c.1670A>G; p.(Tyr557Cys)) in
exon 15 of the CCDC22 gene, which maps to chromosome Xp11.23.
explanation: >-
A CCDC22 missense variant on the X chromosome causes an X-linked phenotype
with features of RSS/3C syndrome.
- name: VPS35L
gene_term:
preferred_term: VPS35L
term:
id: hgnc:24641
label: VPS35L
association: CAUSATIVE
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:36113987
reference_title: "Clinical diversity and molecular mechanism of VPS35L-associated Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We report three new patients with biallelic VPS35L variants.
explanation: >-
Biallelic VPS35L variants indicate autosomal recessive inheritance of
VPS35L-associated RSS.
evidence:
- reference: PMID:36113987
reference_title: "Clinical diversity and molecular mechanism of VPS35L-associated Ritscher-Schinzel syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The Retriever subunit VPS35L is the third responsible gene for
Ritscher-Schinzel syndrome (RSS) after WASHC5 and CCDC22.
explanation: >-
VPS35L is established as the third causative RSS gene.
- name: COMMD4
gene_term:
preferred_term: COMMD4
term:
id: hgnc:26027
label: COMMD4
association: CAUSATIVE
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
to identify causative genes in the copper metabolic murr1
domain-containing (COMMD) proteins COMMD4, COMMD9, and coiled-coil domain
containing 93 (CCDC93) subunits of the Commander complex
explanation: >-
COMMD4 is identified as a causative Commander-complex gene for RSS.
- name: COMMD9
gene_term:
preferred_term: COMMD9
term:
id: hgnc:25014
label: COMMD9
association: CAUSATIVE
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
to identify causative genes in the copper metabolic murr1
domain-containing (COMMD) proteins COMMD4, COMMD9, and coiled-coil domain
containing 93 (CCDC93) subunits of the Commander complex
explanation: >-
COMMD9 is identified as a causative Commander-complex gene for RSS.
- name: CCDC93
gene_term:
preferred_term: CCDC93
term:
id: hgnc:25611
label: CCDC93
association: CAUSATIVE
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:40601774
reference_title: "Ritscher-Schinzel syndrome can be characterized as an endosomal recyclinopathy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
to identify causative genes in the copper metabolic murr1
domain-containing (COMMD) proteins COMMD4, COMMD9, and coiled-coil domain
containing 93 (CCDC93) subunits of the Commander complex
explanation: >-
CCDC93 is identified as a causative Commander-complex gene for RSS.
treatments:
- name: Multidisciplinary Supportive Care
description: >-
No disease-modifying therapy is established; management is multidisciplinary
supportive care addressing congenital heart defects, cleft palate,
hypercholesterolemia, renal anomalies, immunodeficiency, and developmental
delay / intellectual disability.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Standard treatment for obesity, obstructive sleep apnea, cleft palate,
congenital heart defects, hypercholesterolemia, renal anomalies,
immunodeficiency, and developmental delay / intellectual disability.
explanation: >-
GeneReviews recommends standard supportive treatment across the
multisystem manifestations of RSS.
- name: Congenital Heart Defect Repair
description: >-
Surgical or interventional repair of congenital heart defects as indicated
by lesion severity.
treatment_term:
preferred_term: surgical procedure
term:
id: MAXO:0000004
label: surgical procedure
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Standard treatment for obesity, obstructive sleep apnea, cleft palate,
congenital heart defects, hypercholesterolemia, renal anomalies,
immunodeficiency, and developmental delay / intellectual disability.
explanation: >-
Standard treatment of congenital heart defects in RSS includes surgical
repair.
- name: Immunoglobulin Replacement Therapy
description: >-
Immunoglobulin replacement for patients with hypogammaglobulinemia /
immunodeficiency, particularly in VPS35L-associated RSS.
treatment_term:
preferred_term: immunoglobulin replacement therapy
term:
id: NCIT:C15986
label: Pharmacotherapy
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Standard treatment for obesity, obstructive sleep apnea, cleft palate,
congenital heart defects, hypercholesterolemia, renal anomalies,
immunodeficiency, and developmental delay / intellectual disability.
explanation: >-
Immunodeficiency in RSS is managed with standard treatment, which for
hypogammaglobulinemia includes immunoglobulin replacement.
- name: Genetic Counseling
description: >-
Genetic counseling for affected families, including carrier testing in
autosomal recessive families and X-linked counseling in CCDC22 families;
prenatal and preimplantation genetic testing are possible once the
causative variant(s) are identified.
treatment_term:
preferred_term: Genetic Counseling
term:
id: NCIT:C15240
label: Genetic Counseling
evidence:
- reference: PMID:31971710
reference_title: "Ritscher-Schinzel Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Once the causative pathogenic variant(s) have been identified in an
affected family member, prenatal and preimplantation genetic testing are
possible.
explanation: >-
Genetic counseling and reproductive genetic testing are core management
components for RSS families.
datasets: []
References & Deep Research
References
1Deep Research
11. Disease Information
1.1 Concise overview (current understanding)
Ritscher–Schinzel syndrome is a multisystem developmental disorder classically defined by a triad of craniofacial features, cerebellar anomalies, and congenital heart defects, hence “3C syndrome.” (otsuji2023clinicaldiversityand pages 1-2). In a recent mechanistic reframing, RSS is proposed to be an endosomal recycling disorder (“endosomal recyclinopathy”) arising from dysfunction of the Commander endosomal recycling pathway (kato2024thecongenitalmultiple pages 1-4).
1.2 Key identifiers and synonyms
A subset of identifiers could be extracted directly from retrieved sources (OMIM only); other identifier systems (Orphanet/ICD/MeSH/MONDO) were not present in the retrieved full texts and should be populated via the authoritative databases.
| Identifier system | Code/ID | Label | Notes | URL |
|---|---|---|---|---|
| OMIM | MIM:220210 | Ritscher-Schinzel syndrome / 3C syndrome | Retrieved evidence links RSS/3C syndrome to OMIM 220210; classic disease label/synonym supported by 2015 and 2023 literature summaries and a syndrome list noting “3 C-syndrom, cranio-cerebello-cardiale Dysplasie.” (hirschsprungUnknownyearsyndrome pages 7-7, otsuji2023clinicaldiversityand pages 1-2) | https://omim.org/entry/220210 |
| OMIM | MIM:300963 | CCDC22-associated Ritscher-Schinzel syndrome | Otsuji 2023 notes OMIM 300963 in connection with RSS via CCDC22, reflecting the X-linked form/gene-specific entry rather than the aggregate syndrome label. (otsuji2023clinicaldiversityand pages 1-2) | https://omim.org/entry/300963 |
| OMIM | MIM:619135 | VPS35L-associated Ritscher-Schinzel syndrome | Otsuji 2023 identifies VPS35L as the “third responsible gene” for RSS and cites MIM 619135 for this gene-associated form. (otsuji2023clinicaldiversityand pages 1-2) | https://omim.org/entry/619135 |
| Synonym | RSS | Ritscher-Schinzel syndrome | Common abbreviation used in recent peer-reviewed and preprint literature. (otsuji2023clinicaldiversityand pages 1-2, kato2024thecongenitalmultiple pages 1-4) | N/A |
| Synonym | 3C syndrome | Cranio-cerebello-cardiac syndrome / cranio-cerebello-cardiac dysplasia | Widely used alternative name reflecting the core triad of craniofacial, cerebellar, and cardiac abnormalities. (hirschsprungUnknownyearsyndrome pages 7-7, otsuji2023clinicaldiversityand pages 1-2) | N/A |
| Synonym | 3 C syndrome | Ritscher-Schinzel/3 C syndrome | Variant spacing/formatting appears in the literature, especially in gene-specific CCDC22 reports. (singla2025ccdc22mutationsthat pages 10-10) | N/A |
| Disease concept | N/A | Multi-system developmental disorder | Recent sources describe RSS as a congenital multiple-organ malformation syndrome characterized by craniofacial, cerebellar, and cardiac defects; newer mechanistic framing is an “endosomal recyclinopathy.” (otsuji2023clinicaldiversityand pages 1-2, kato2024thecongenitalmultiple pages 1-4) | N/A |
| Orphanet | Not found in retrieved evidence | To be filled from external database | No ORPHA identifier was present in the retrieved evidence; verify directly in Orphanet before KB ingestion. (hirschsprungUnknownyearsyndrome pages 7-7, otsuji2023clinicaldiversityand pages 1-2) | https://www.orpha.net |
| ICD-10 / ICD-11 | Not found in retrieved evidence | To be filled from external database | No ICD code was present in the retrieved evidence; confirm from WHO/clinical coding resources. (hirschsprungUnknownyearsyndrome pages 7-7, otsuji2023clinicaldiversityand pages 1-2) | https://icd.who.int/ |
| MeSH | Not found in retrieved evidence | To be filled from external database | No MeSH term/ID was present in the retrieved evidence; confirm in MeSH Browser. (hirschsprungUnknownyearsyndrome pages 7-7, otsuji2023clinicaldiversityand pages 1-2) | https://meshb.nlm.nih.gov/ |
| MONDO | Not found in retrieved evidence | To be filled from external database | No MONDO identifier was present in the retrieved evidence; confirm in Mondo/OBO resources. (hirschsprungUnknownyearsyndrome pages 7-7, otsuji2023clinicaldiversityand pages 1-2) | https://monarchinitiative.org/ |
| Evidence provenance | Aggregated disease-level literature and syndrome catalogs | Not EHR-derived in retrieved evidence | Available evidence comes from peer-reviewed case series/reviews and syndrome listings rather than individual EHR datasets; examples include Otsuji 2023 J Med Genet and a syndrome list containing the 220210 identifier. (hirschsprungUnknownyearsyndrome pages 7-7, otsuji2023clinicaldiversityand pages 1-2) | N/A |
Table: This table summarizes the disease identifiers and naming conventions for Ritscher-Schinzel syndrome based strictly on retrieved evidence. It highlights confirmed OMIM entries and synonyms while flagging ORPHA, ICD, MeSH, and MONDO as requiring direct verification from external databases.
Evidence source types represented in this report: aggregated disease-level literature (peer-reviewed research and case series; preprint cohort analyses), not EHR-derived datasets (otsuji2023clinicaldiversityand pages 1-2, kato2024thecongenitalmultiple pages 1-4).
2. Etiology
2.1 Disease causal factors
Primary cause: pathogenic variants affecting genes encoding subunits of the endosomal recycling machinery—especially the Commander pathway (Retriever + CCC complex, functionally coupled to WASH complex). This has been linked to RSS by structural biology (Commander complex structure) and by patient genetic and functional studies (healy2023structureofthe pages 1-3, otsuji2023clinicaldiversityand pages 1-2).
2.2 Risk factors
- Genetic risk factors (causal): rare pathogenic germline variants in Commander/WASH pathway genes, with inheritance depending on the gene (biallelic vs X-linked) (otsuji2023clinicaldiversityand pages 1-1, otsuji2023clinicaldiversityand pages 1-2).
- Environmental risk factors: none established in retrieved evidence (not typically expected for a congenital Mendelian malformation syndrome).
2.3 Protective factors / gene–environment interactions
No protective factors or gene–environment interactions were described in the retrieved evidence.
3. Phenotypes
3.1 Core phenotypic triad and spectrum
RSS/3C is defined by the triad: - Craniofacial anomalies (craniofacial dysmorphism/abnormal craniofacial features) (healy2023structureofthe pages 1-3, otsuji2023clinicaldiversityand pages 1-2) - Cerebellar anomalies (often described as cerebellar hypoplasia) (healy2023structureofthe pages 1-3, kato2024thecongenitalmultiple pages 66-68) - Cardiac defects (stunted cardiovascular development / congenital heart defects) (healy2023structureofthe pages 1-3, otsuji2023clinicaldiversityand pages 1-2)
Expanded multisystem involvement reported in recent sources includes renal, skeletal, hepatic, gastrointestinal, immunologic, and lipid phenotypes (kato2024thecongenitalmultiple pages 1-4, otsuji2023clinicaldiversityand pages 1-1).
3.2 Frequencies/statistics from recent/available evidence
- Congenital heart defects are reported in ~80% of RSS/3C cases in a literature summary (kolanczyk2015missensevariantin pages 1-2).
- In a 2024 cohort synthesis table spanning Commander/WASH-related genes, WASHC5-associated cases included:
- Developmental delay: 11/11
- Cardiac abnormalities: 7/11
- Cerebellar hypoplasia: reported as 6/7 in a comparison row (table excerpt) (kato2024thecongenitalmultiple pages 66-68).
- VPS35L-associated cases in that same 2024 synthesis table showed proteinuria and dyslipidemia reported as 3/3 (kato2024thecongenitalmultiple pages 66-68).
3.3 Age of onset, severity, progression (general)
- Onset: congenital/early-life (implied by malformations; prenatal/infant presentations are common) (kato2024thecongenitalmultiple pages 1-4).
- Severity/expressivity: variable across genes and even within the same gene; VPS35L-associated RSS shows diverse severity, and milder phenotypes correlated with relatively higher VPS35L protein levels in patient-derived cells (otsuji2023clinicaldiversityand pages 1-1).
- Course: chronic/lifelong multisystem disease; early mortality occurs in severe forms (e.g., biallelic COMMD4-L41R family with deaths ages 0–5 in 2024 preprint cohort) (kato2024thecongenitalmultiple pages 16-19).
3.4 Suggested HPO terms (examples; not exhaustive)
(These are ontology suggestions based on the phenotypes explicitly described in retrieved sources.) - Abnormal craniofacial morphology (e.g., Abnormal facial shape; Craniofacial dysmorphism) (healy2023structureofthe pages 1-3) - Cerebellar hypoplasia (healy2023structureofthe pages 1-3) - Congenital heart defect (otsuji2023clinicaldiversityand pages 1-2) - Global developmental delay / Intellectual disability (kolanczyk2015missensevariantin pages 1-2) - Proteinuria (otsuji2023clinicaldiversityand pages 1-1) - Hypercholesterolemia (otsuji2023clinicaldiversityand pages 1-1) - Hypogammaglobulinemia (otsuji2023clinicaldiversityand pages 1-1) - Intestinal lymphangiectasia (otsuji2023clinicaldiversityand pages 1-1)
3.5 Quality-of-life impact
Direct patient-reported QoL instruments (EQ-5D/SF-36/PROMIS) were not described in retrieved evidence; however, neurodevelopmental impairment and multi-organ morbidity (cardiac, renal, GI, immunologic) are expected to substantially affect daily function (kato2024thecongenitalmultiple pages 66-68).
4. Genetic / Molecular Information
4.1 Causal genes and inheritance patterns (current)
Recent literature supports RSS as a disorder of Commander/WASH pathway genes. Key genes with disease association in retrieved evidence: - WASHC5 (WASH complex; RSS/3C association with biallelic loss-of-function summarized in 2024 synthesis) (kato2024thecongenitalmultiple pages 66-68) - CCDC22 (CCC complex; X-linked/hemizygous form; overlaps with XLID and RSS features) (otsuji2023clinicaldiversityand pages 1-2) - VPS35L (Retriever complex; biallelic; “third responsible gene” for RSS after WASHC5 and CCDC22) (otsuji2023clinicaldiversityand pages 1-1)
A 2024 cohort/mechanistic preprint proposed additional candidate/causal genes within the Commander pathway: - COMMD4, COMMD9, CCDC93 (CCC complex components; biallelic) (kato2024thecongenitalmultiple pages 6-9).
| Gene (HGNC symbol) | Protein/complex | Inheritance pattern reported | Variant types (general) | Key clinical notes/complications | Key supporting recent sources with publication year and URL |
|---|---|---|---|---|---|
| WASHC5 | Strumpellin; core WASH complex subunit functionally linked to Commander-mediated recycling | Autosomal recessive for RSS/3C in retrieved evidence; biallelic loss-of-function reported | General loss-of-function; splice/disruptive variants reported in RSS literature summaries | Classic RSS/3C phenotype with developmental delay, cerebellar hypoplasia, cardiac abnormalities; 2024 summary table notes developmental delay in 11/11 and cardiac abnormalities in 7/11 WASHC5-associated cases; mechanism tied to reduced recycling of surface cargo proteins (kato2024thecongenitalmultiple pages 66-68, kato2024thecongenitalmultiple pages 14-16, otsuji2023clinicaldiversityand pages 1-2) | Kato et al., 2024, medRxiv, https://doi.org/10.1101/2024.08.17.24311658; Otsuji et al., 2023, J Med Genet, https://doi.org/10.1136/jmg-2022-108602 (kato2024thecongenitalmultiple pages 66-68, kato2024thecongenitalmultiple pages 14-16, otsuji2023clinicaldiversityand pages 1-2) |
| CCDC22 | CCC complex subunit within Commander | X-linked / hemizygous form reported; gene-specific RSS/3C overlap with XLID | Missense and other variants that disrupt CCC assembly/COMMD binding; loss-of-function/functional impairment reported | RSS/3C with craniofacial, cerebellar, cardiac, and neurodevelopmental involvement; some attenuated phenotypes may lack major cardiac/neuroanatomical abnormalities; CCDC22 dysfunction perturbs CCC assembly and Commander function (otsuji2023clinicaldiversityand pages 1-2, singla2025ccdc22mutationsthat pages 10-10, singla2025ccdc22mutationsthat pages 1-2) | Healy et al., 2023, Cell, https://doi.org/10.1016/j.cell.2023.04.003; Singla et al., 2025, BMC Med Genomics, https://doi.org/10.1186/s12920-025-02168-7; Otsuji et al., 2023, J Med Genet, https://doi.org/10.1136/jmg-2022-108602 (otsuji2023clinicaldiversityand pages 1-2, singla2025ccdc22mutationsthat pages 10-10, singla2025ccdc22mutationsthat pages 1-2) |
| VPS35L | Retriever subunit (with VPS26C and VPS29) within Commander | Autosomal recessive / biallelic | Biallelic pathogenic variants including truncating, splice-altering, in-frame deletion, and missense alleles with reduced protein stability/function | Distinct VPS35L-associated RSS spectrum with variable severity; novel 2023 complications include hypercholesterolaemia, hypogammaglobulinaemia, intestinal lymphangiectasia, and proteinuria; mechanism includes reduced cell-surface LRP1/LDLR and reduced LDL uptake (otsuji2023clinicaldiversityand pages 1-1, otsuji2023clinicaldiversityand pages 5-6, otsuji2023clinicaldiversityand pages 8-8) | Otsuji et al., 2023, J Med Genet, https://doi.org/10.1136/jmg-2022-108602; Healy et al., 2023, Cell, https://doi.org/10.1016/j.cell.2023.04.003 (otsuji2023clinicaldiversityand pages 1-1, otsuji2023clinicaldiversityand pages 5-6, healy2023structureofthe pages 1-3, otsuji2023clinicaldiversityand pages 8-8) |
| COMMD4 | CCC complex subunit; Commander-associated | Autosomal recessive / biallelic in 2024 preprint cohort | Biallelic pathogenic variants; severe COMMD4-L41R genotype highlighted | Newly proposed RSS gene; associated with severe multisystem disease and early childhood death (ages 0–5) in reported family; functional studies suggest major Commander cargo-recycling defects (kato2024thecongenitalmultiple pages 16-19, kato2024thecongenitalmultiple pages 6-9) | Kato et al., 2024, medRxiv, https://doi.org/10.1101/2024.08.17.24311658 (kato2024thecongenitalmultiple pages 16-19, kato2024thecongenitalmultiple pages 6-9) |
| COMMD9 | CCC complex subunit; Commander-associated | Autosomal recessive / biallelic in 2024 preprint cohort | Biallelic pathogenic/truncating variants reported in candidate-gene expansion study | Newly proposed RSS gene; functional data indicate milder cargo-trafficking defects than COMMD4 or CCDC93 loss, suggesting residual pathway activity may moderate severity (kato2024thecongenitalmultiple pages 16-19, kato2024thecongenitalmultiple pages 6-9) | Kato et al., 2024, medRxiv, https://doi.org/10.1101/2024.08.17.24311658 (kato2024thecongenitalmultiple pages 16-19, kato2024thecongenitalmultiple pages 6-9) |
| CCDC93 | CCC complex scaffold subunit within Commander | Autosomal recessive / biallelic in 2024 preprint cohort | Biallelic pathogenic variants causing loss of CCC/Commander function | Newly proposed RSS gene; linked to dysgenic corpus callosum, cerebellar abnormalities, limb/nail anomalies, and broader multisystem RSS manifestations; knockout/cell studies support defective endosomal recycling (kato2024thecongenitalmultiple pages 16-19, kato2024thecongenitalmultiple pages 6-9) | Kato et al., 2024, medRxiv, https://doi.org/10.1101/2024.08.17.24311658 (kato2024thecongenitalmultiple pages 16-19, kato2024thecongenitalmultiple pages 6-9) |
| Pathway-level note | Commander = Retriever + CCC, acting with the WASH complex in SNX17-dependent endosomal recycling | Not applicable | Not applicable | Retrieved evidence supports RSS as an endosomal recyclinopathy caused by impaired retrieval/recycling of membrane cargoes including integrins and lipoprotein receptors; this provides a unifying mechanism across WASHC5, CCDC22, VPS35L, and newly proposed CCC-gene cases (kato2024thecongenitalmultiple pages 14-16, healy2023structureofthe pages 1-3, kato2024thecongenitalmultiple pages 4-6, kato2024thecongenitalmultiple pages 6-9) | Healy et al., 2023, Cell, https://doi.org/10.1016/j.cell.2023.04.003; Kato et al., 2024, medRxiv, https://doi.org/10.1101/2024.08.17.24311658; Otsuji et al., 2023, J Med Genet, https://doi.org/10.1136/jmg-2022-108602 (kato2024thecongenitalmultiple pages 14-16, healy2023structureofthe pages 1-3, kato2024thecongenitalmultiple pages 4-6, kato2024thecongenitalmultiple pages 6-9) |
Table: This table summarizes the currently supported and newly proposed genetic causes of Ritscher-Schinzel syndrome/3C syndrome, emphasizing the shared Commander-Retriever-CCC-WASH endosomal recycling mechanism. It is useful for quickly comparing inheritance, variant classes, and distinctive complications across genes.
4.2 Pathogenic variant classes and functional consequences
Across genes, reported variants are primarily loss-of-function or complex-destabilizing alleles (frameshift/truncating, splice-altering, in-frame deletions, or missense variants that impair complex assembly/interactions) resulting in reduced endosomal recycling capacity and decreased cell-surface expression of key cargos (otsuji2023clinicaldiversityand pages 5-6, kato2024thecongenitalmultiple pages 6-9).
4.3 Population allele frequencies
Population allele frequency data (e.g., gnomAD) were not provided in the retrieved sources.
4.4 Modifier genes / epigenetic information / chromosomal abnormalities
No specific modifier genes or epigenetic signatures were described in the retrieved evidence. Chromosomal microarray (array-CGH) was used diagnostically in at least one CCDC22-related study, but this is a testing modality rather than a recurrent chromosomal cause in the retrieved evidence (kolanczyk2015missensevariantin pages 1-2).
5. Environmental Information
No non-genetic environmental contributors, lifestyle associations, or infectious triggers were described in the retrieved evidence, consistent with a congenital Mendelian malformation syndrome.
6. Mechanism / Pathophysiology
6.1 Key concept: RSS as an “endosomal recyclinopathy”
A 2024 cohort/mechanistic study explicitly frames RSS as an endosomal recycling disorder: it “establishes RSS as a 'recyclinopathy' that arises from a dysfunction in the Commander endosomal recycling pathway” (kato2024thecongenitalmultiple pages 1-4). Commander is required for endosomal recycling of diverse transmembrane cargos and is mutated in RSS (healy2023structureofthe pages 1-3).
6.2 Molecular pathway description (Commander / SNX17–Retriever–CCC–WASH)
- Commander architecture: a 16-subunit assembly comprising two subassemblies—Retriever (VPS35L, VPS26C, VPS29) and CCC complex (COMMD proteins with CCDC22 and CCDC93) (healy2023structureofthe pages 1-3).
- Functional role: Commander regulates retromer-independent retrieval and recycling of many proteins, including integrins and lipoprotein receptors (healy2023structureofthe pages 1-3).
- Disease mechanism: impairment of this recycling pathway reduces cell-surface expression of integral membrane proteins, providing a mechanistic basis for multisystem developmental defects (otsuji2023clinicaldiversityand pages 1-2).
6.3 Mechanistic links to key complications
- Hypercholesterolemia: VPS35L ablation decreases surface LRP1 and LDLR, reducing LDL uptake; authors propose this as a molecular mechanism for hypercholesterolemia in VPS35L-associated RSS (otsuji2023clinicaldiversityand pages 1-1). Structural work also notes Commander mutations can lead to hypercholesterolemia via reduced trafficking of LDL receptors (healy2023structureofthe pages 1-3).
- Proteinuria / renal phenotype: Commander pathway perturbation is linked to altered recycling of kidney-relevant receptors (e.g., LRP2) and proteinuria as a clinical phenotype (kato2024thecongenitalmultiple pages 6-9).
6.4 Suggested GO biological process terms (examples)
Ontology suggestions aligned with the mechanistic evidence: - Endosomal transport / endosome-to-plasma membrane recycling (Commander-dependent recycling) (healy2023structureofthe pages 1-3) - Receptor-mediated endocytosis / receptor recycling (LRP1/LDLR trafficking) (otsuji2023clinicaldiversityand pages 1-1) - Actin filament organization (endosomal branched actin) (WASH complex functional coupling to recycling) (otsuji2023clinicaldiversityand pages 1-2)
6.5 Suggested Cell Ontology (CL) cell types (examples)
Based on tissues/organs implicated by cargo and phenotype: - Hepatocyte (lipoprotein receptor recycling and cholesterol phenotype) (otsuji2023clinicaldiversityand pages 1-1) - Renal proximal tubule epithelial cell (proteinuria/LRP2-related proximal tubular reabsorption context) (kato2024thecongenitalmultiple pages 6-9) - Neurons (synaptic cargo defects and neurodevelopmental impairment) (kato2024thecongenitalmultiple pages 14-16)
7. Anatomical Structures Affected
7.1 Organ/system level
- Central nervous system: cerebellum (cerebellar hypoplasia), other brain malformations (healy2023structureofthe pages 1-3, kato2024thecongenitalmultiple pages 66-68)
- Cardiovascular system: congenital cardiac defects and cardiovascular development abnormalities (healy2023structureofthe pages 1-3, otsuji2023clinicaldiversityand pages 1-2)
- Craniofacial structures: craniofacial dysmorphism (healy2023structureofthe pages 1-3)
- Kidney: proteinuria and renal involvement (otsuji2023clinicaldiversityand pages 1-1, kato2024thecongenitalmultiple pages 6-9)
- Immune system / lymphatics: hypogammaglobulinemia; intestinal lymphangiectasia (VPS35L-associated) (otsuji2023clinicaldiversityand pages 1-1)
- Gastrointestinal tract and liver: NEC, protein-losing enteropathy, cholestasis/hepatic dysfunction reported in 2024 cohort summary (kato2024thecongenitalmultiple pages 66-68)
7.2 Suggested UBERON terms (examples)
- Cerebellum; heart; kidney; liver; intestine (supported by described malformations and complications) (kato2024thecongenitalmultiple pages 66-68, healy2023structureofthe pages 1-3).
8. Temporal Development
- Onset: congenital / infancy (malformation syndrome) (kato2024thecongenitalmultiple pages 1-4).
- Critical periods: embryonic development/organogenesis implied by congenital structural anomalies.
- Progression: variable; some complications (e.g., lipid abnormalities, immunodeficiency, proteinuria) emerge with postnatal physiology and require longitudinal monitoring (otsuji2023clinicaldiversityand pages 1-1).
9. Inheritance and Population
9.1 Inheritance
- Autosomal recessive (biallelic): VPS35L-associated RSS (otsuji2023clinicaldiversityand pages 1-1); WASHC5-associated RSS (summarized as biallelic LoF in 2024 synthesis) (kato2024thecongenitalmultiple pages 66-68).
- X-linked: CCDC22-associated RSS/3C overlap and XLID phenotypes (kolanczyk2015missensevariantin pages 1-2, otsuji2023clinicaldiversityand pages 1-2).
9.2 Epidemiology
Prevalence/incidence, geographic distribution, and sex ratio were not present in the retrieved evidence and should be obtained from Orphanet and registry-based sources.
10. Diagnostics
10.1 Clinical recognition and imaging
Diagnosis is typically initiated by recognizing the 3C triad (craniofacial, cerebellar, cardiac) (otsuji2023clinicaldiversityand pages 1-2), followed by: - Brain MRI focused on posterior fossa/cerebellar anomalies (singla2025ccdc22mutationsthat pages 4-5) - Echocardiography/cardiac evaluation for congenital heart disease (singla2025ccdc22mutationsthat pages 1-2)
10.2 Genetic testing (real-world implementations)
A representative diagnostic workflow from a CCDC22-associated report included: - Chromosomal microarray (array-CGH) - Whole-exome sequencing (WES) with standard filtering against population databases and internal controls, followed by confirmatory and functional studies (e.g., western blot) (kolanczyk2015missensevariantin pages 1-2).
Given the expanding gene set, contemporary practice is well aligned with exome/genome-first testing or multigene panels targeting Commander/WASH pathway genes (WASHC5, CCDC22, VPS35L, and potentially CCC subunits as evidence matures) (kato2024thecongenitalmultiple pages 6-9).
10.3 Differential diagnosis
Not exhaustively enumerated in retrieved evidence. In practice, major differentials include other syndromic congenital heart + posterior fossa malformation disorders, and other endosomal trafficking disorders; gene-centric testing reduces diagnostic ambiguity (otsuji2023clinicaldiversityand pages 1-2).
11. Outcome / Prognosis
Outcomes are variable and depend on gene and severity of organ involvement. - Early mortality: reported in severe biallelic CCC-gene cases (e.g., COMMD4-L41R family with deaths ages 0–5) (kato2024thecongenitalmultiple pages 16-19). - Ongoing morbidity: neurodevelopmental impairment, congenital heart disease, renal proteinuria, lipid abnormalities, and immunologic complications may require chronic follow-up (otsuji2023clinicaldiversityand pages 1-1).
No formal survival curves or life expectancy estimates were present in retrieved evidence.
12. Treatment
No disease-modifying therapy is established in the retrieved evidence; management is multidisciplinary supportive care.
12.1 Organ-directed management (examples documented)
- Seizure management: anti-epileptic medications used in affected individuals (singla2025ccdc22mutationsthat pages 1-2).
- Immunologic support: immunoglobulin replacement is suggested by clinical practice in cases with hypogammaglobulinemia (reported as a complication in VPS35L-associated RSS) (kato2024thecongenitalmultiple pages 66-68, otsuji2023clinicaldiversityand pages 1-1).
- Gastroesophageal reflux / feeding: severe GERD managed with Nissen fundoplication in one reported case within the 2024 cohort summary (kato2024thecongenitalmultiple pages 66-68).
- Congenital heart disease: standard cardiology evaluation and surgical/interventional repair as indicated by lesion severity (supported by high CHD frequency and clinical practice; specific operative series not present in retrieved evidence) (kolanczyk2015missensevariantin pages 1-2).
12.2 Suggested MAXO terms (examples)
- Cardiac surgical repair; developmental therapy/rehabilitation; antiseizure pharmacotherapy; immunoglobulin replacement therapy; nutritional support/feeding therapy (supported as care domains by clinical features and documented interventions) (kato2024thecongenitalmultiple pages 66-68, singla2025ccdc22mutationsthat pages 1-2).
No clinical trials specific to RSS were identified in retrieved evidence.
13. Prevention
Primary prevention is not applicable in the usual environmental sense for a Mendelian disorder; prevention focuses on genetic counseling and reproductive options (carrier testing in autosomal recessive families; X-linked counseling in CCDC22 families) (kolanczyk2015missensevariantin pages 1-2, otsuji2023clinicaldiversityand pages 1-1).
14. Other Species / Natural Disease
No naturally occurring non-human disease analogs were identified in the retrieved evidence.
15. Model Organisms
A mouse model was used to probe pathophysiology: - Vps35l conditional knockout in Nestin-lineage (Vps35l-cKONestin) showed strong neurodevelopmental phenotypes; notably ~30% (23/72) had profound hydrocephalus, alongside growth impairment, behavioral problems, and high mortality (kato2024thecongenitalmultiple pages 14-16).
This supports causal links between Retriever/Commander dysfunction and neurodevelopmental structural phenotypes.
Visual evidence (figures)
Structural studies directly connect RSS/XLID mutations to the Commander complex architecture. - Commander/Retriever/CCC subunit schematic and mutation mapping onto the structural model are shown in extracted figures from Healy et al. (Cell 2023) (healy2023structureofthe media 9aee47de, healy2023structureofthe media 5bc4c3b6, healy2023structureofthe media 274803a4).
Expert synthesis and interpretation (authoritative-source analysis)
Recent authoritative work supports a unifying concept: RSS is best understood as a developmental “recyclinopathy” caused by impaired SNX17–Retriever–CCC–WASH endosomal recycling, leading to reduced cell-surface presentation of tissue-critical receptors and adhesion molecules (kato2024thecongenitalmultiple pages 1-4, otsuji2023clinicaldiversityand pages 1-2). This framework explains why seemingly disparate features (cardiac malformations, cerebellar hypoplasia, renal proteinuria, lipid abnormalities, and immunologic/GI complications) can co-occur, and it predicts that ongoing gene discovery will likely continue within the Commander pathway and its regulators/cargo adaptors (kato2024thecongenitalmultiple pages 6-9).
Notes on citation requirements and limitations
- PMIDs: Not available from the retrieved full-text evidence snippets for the key 2023–2024 sources used here (Cell 2023; J Med Genet 2023; medRxiv 2024). The report therefore cites DOIs and journal/preprint URLs.
- Orphanet/ICD/MeSH/MONDO identifiers and epidemiology: Not found in retrieved full texts and should be filled via direct database queries.
Key references (recent prioritized)
- Healy MD et al. “Structure of the endosomal Commander complex linked to Ritscher-Schinzel syndrome.” Cell (May 2023). https://doi.org/10.1016/j.cell.2023.04.003 (healy2023structureofthe pages 1-3)
- Otsuji S et al. “Clinical diversity and molecular mechanism of VPS35L-associated Ritscher-Schinzel syndrome.” Journal of Medical Genetics (Sep 2023). https://doi.org/10.1136/jmg-2022-108602 (otsuji2023clinicaldiversityand pages 1-1)
- Kato K et al. “The congenital multiple organ malformation syndrome, Ritscher-Schinzel syndrome is an endosomal recyclinopathy.” medRxiv (Aug 2024). https://doi.org/10.1101/2024.08.17.24311658 (kato2024thecongenitalmultiple pages 1-4)
References
-
(otsuji2023clinicaldiversityand pages 1-2): Shiomi Otsuji, Yosuke Nishio, Maki Tsujita, Marlene Rio, Céline Huber, Carlos Antón-Plágaro, Seiji Mizuno, Yoshihiko Kawano, Satoko Miyatake, Marleen Simon, Ellen van Binsbergen, Richard H van Jaarsveld, Naomichi Matsumoto, Valerie Cormier-Daire, Peter J.Cullen, Shinji Saitoh, and Kohji Kato. Clinical diversity and molecular mechanism of vps35l-associated ritscher-schinzel syndrome. Journal of Medical Genetics, 60:359-367, Sep 2023. URL: https://doi.org/10.1136/jmg-2022-108602, doi:10.1136/jmg-2022-108602. This article has 29 citations and is from a domain leading peer-reviewed journal.
-
(hirschsprungUnknownyearsyndrome pages 7-7): M Hirschsprung. Syndrome. Unknown journal, Unknown year.
-
(kato2024thecongenitalmultiple pages 1-4): Kohji Kato, Yosuke Nishio, Kirsty J McMillan, Aljazi Al-Maraghi, Hester Y Kroes, Mohamed S Abdel-Hamid, Emma Jones, Shrestha Shaw, Aya Yoshida, Shiomi Otsuji, Yuka Murofushi, Waleed H. O. Aamer, Ajaz A Bhat, Jehan AlRayahi, Ammira Al-Shabeeb Akil, Ellen van Binsbergen, Etienne J Janssen, Hisashi Oishi, Ryosuke Kobayashi, Takuro Horii, Izuho Hatada, Akihiko Saito, Mitsuharu Hattori, Yoshihiko Kawano, Philip A Lewis, Kate J Heesom, Takeshi Takarada, Kazunobu Sawamoto, Masaki Matsushita, Tomoo Ogi, Rebeka Butkovic, Chris Danson, Kevin A Wilkinson, Khalid A Fakhro, Maha S Zaki, Shinji Saitoh, and Peter J Cullen. The congenital multiple organ malformation syndrome, ritscher-schinzel syndrome is an endosomal recyclinopathy. MedRxiv, Aug 2024. URL: https://doi.org/10.1101/2024.08.17.24311658, doi:10.1101/2024.08.17.24311658. This article has 2 citations.
-
(singla2025ccdc22mutationsthat pages 10-10): Amika Singla, Carolyn Rogers, Mary-Joe Touma, Yassin El-Najjar, Alison Colley, Daniel J. Boesch, Daniel D. Billadeau, Jozef Gecz, Baoyu Chen, and Ezra Burstein. Ccdc22 mutations that impair commd binding cause attenuated 3c/ritscher-schinzel syndrome. BMC Medical Genomics, May 2025. URL: https://doi.org/10.1186/s12920-025-02168-7, doi:10.1186/s12920-025-02168-7. This article has 1 citations and is from a peer-reviewed journal.
-
(healy2023structureofthe pages 1-3): Michael D. Healy, Kerrie E. McNally, Rebeka Butkovič, Molly Chilton, Kohji Kato, Joanna Sacharz, Calum McConville, Edmund R.R. Moody, Shrestha Shaw, Vicente J. Planelles-Herrero, Sathish K.N. Yadav, Jennifer Ross, Ufuk Borucu, Catherine S. Palmer, Kai-En Chen, Tristan I. Croll, Ryan J. Hall, Nikeisha J. Caruana, Rajesh Ghai, Thi H.D. Nguyen, Kate J. Heesom, Shinji Saitoh, Imre Berger, Christiane Schaffitzel, Tom A. Williams, David A. Stroud, Emmanuel Derivery, Brett M. Collins, and Peter J. Cullen. Structure of the endosomal commander complex linked to ritscher-schinzel syndrome. Cell, 186:2219-2237.e29, May 2023. URL: https://doi.org/10.1016/j.cell.2023.04.003, doi:10.1016/j.cell.2023.04.003. This article has 88 citations and is from a highest quality peer-reviewed journal.
-
(otsuji2023clinicaldiversityand pages 1-1): Shiomi Otsuji, Yosuke Nishio, Maki Tsujita, Marlene Rio, Céline Huber, Carlos Antón-Plágaro, Seiji Mizuno, Yoshihiko Kawano, Satoko Miyatake, Marleen Simon, Ellen van Binsbergen, Richard H van Jaarsveld, Naomichi Matsumoto, Valerie Cormier-Daire, Peter J.Cullen, Shinji Saitoh, and Kohji Kato. Clinical diversity and molecular mechanism of vps35l-associated ritscher-schinzel syndrome. Journal of Medical Genetics, 60:359-367, Sep 2023. URL: https://doi.org/10.1136/jmg-2022-108602, doi:10.1136/jmg-2022-108602. This article has 29 citations and is from a domain leading peer-reviewed journal.
-
(kato2024thecongenitalmultiple pages 66-68): Kohji Kato, Yosuke Nishio, Kirsty J McMillan, Aljazi Al-Maraghi, Hester Y Kroes, Mohamed S Abdel-Hamid, Emma Jones, Shrestha Shaw, Aya Yoshida, Shiomi Otsuji, Yuka Murofushi, Waleed H. O. Aamer, Ajaz A Bhat, Jehan AlRayahi, Ammira Al-Shabeeb Akil, Ellen van Binsbergen, Etienne J Janssen, Hisashi Oishi, Ryosuke Kobayashi, Takuro Horii, Izuho Hatada, Akihiko Saito, Mitsuharu Hattori, Yoshihiko Kawano, Philip A Lewis, Kate J Heesom, Takeshi Takarada, Kazunobu Sawamoto, Masaki Matsushita, Tomoo Ogi, Rebeka Butkovic, Chris Danson, Kevin A Wilkinson, Khalid A Fakhro, Maha S Zaki, Shinji Saitoh, and Peter J Cullen. The congenital multiple organ malformation syndrome, ritscher-schinzel syndrome is an endosomal recyclinopathy. MedRxiv, Aug 2024. URL: https://doi.org/10.1101/2024.08.17.24311658, doi:10.1101/2024.08.17.24311658. This article has 2 citations.
-
(kolanczyk2015missensevariantin pages 1-2): Mateusz Kolanczyk, Peter Krawitz, Jochen Hecht, Anna Hupalowska, Marta Miaczynska, Katrin Marschner, Claire Schlack, Denise Emmerich, Karolina Kobus, Uwe Kornak, Peter N Robinson, Barbara Plecko, Gernot Grangl, Sabine Uhrig, Stefan Mundlos, and Denise Horn. Missense variant in ccdc22 causes x-linked recessive intellectual disability with features of ritscher-schinzel/3c syndrome. European Journal of Human Genetics, 23:633-638, Jun 2015. URL: https://doi.org/10.1038/ejhg.2014.109, doi:10.1038/ejhg.2014.109. This article has 88 citations and is from a domain leading peer-reviewed journal.
-
(kato2024thecongenitalmultiple pages 16-19): Kohji Kato, Yosuke Nishio, Kirsty J McMillan, Aljazi Al-Maraghi, Hester Y Kroes, Mohamed S Abdel-Hamid, Emma Jones, Shrestha Shaw, Aya Yoshida, Shiomi Otsuji, Yuka Murofushi, Waleed H. O. Aamer, Ajaz A Bhat, Jehan AlRayahi, Ammira Al-Shabeeb Akil, Ellen van Binsbergen, Etienne J Janssen, Hisashi Oishi, Ryosuke Kobayashi, Takuro Horii, Izuho Hatada, Akihiko Saito, Mitsuharu Hattori, Yoshihiko Kawano, Philip A Lewis, Kate J Heesom, Takeshi Takarada, Kazunobu Sawamoto, Masaki Matsushita, Tomoo Ogi, Rebeka Butkovic, Chris Danson, Kevin A Wilkinson, Khalid A Fakhro, Maha S Zaki, Shinji Saitoh, and Peter J Cullen. The congenital multiple organ malformation syndrome, ritscher-schinzel syndrome is an endosomal recyclinopathy. MedRxiv, Aug 2024. URL: https://doi.org/10.1101/2024.08.17.24311658, doi:10.1101/2024.08.17.24311658. This article has 2 citations.
-
(kato2024thecongenitalmultiple pages 6-9): Kohji Kato, Yosuke Nishio, Kirsty J McMillan, Aljazi Al-Maraghi, Hester Y Kroes, Mohamed S Abdel-Hamid, Emma Jones, Shrestha Shaw, Aya Yoshida, Shiomi Otsuji, Yuka Murofushi, Waleed H. O. Aamer, Ajaz A Bhat, Jehan AlRayahi, Ammira Al-Shabeeb Akil, Ellen van Binsbergen, Etienne J Janssen, Hisashi Oishi, Ryosuke Kobayashi, Takuro Horii, Izuho Hatada, Akihiko Saito, Mitsuharu Hattori, Yoshihiko Kawano, Philip A Lewis, Kate J Heesom, Takeshi Takarada, Kazunobu Sawamoto, Masaki Matsushita, Tomoo Ogi, Rebeka Butkovic, Chris Danson, Kevin A Wilkinson, Khalid A Fakhro, Maha S Zaki, Shinji Saitoh, and Peter J Cullen. The congenital multiple organ malformation syndrome, ritscher-schinzel syndrome is an endosomal recyclinopathy. MedRxiv, Aug 2024. URL: https://doi.org/10.1101/2024.08.17.24311658, doi:10.1101/2024.08.17.24311658. This article has 2 citations.
-
(kato2024thecongenitalmultiple pages 14-16): Kohji Kato, Yosuke Nishio, Kirsty J McMillan, Aljazi Al-Maraghi, Hester Y Kroes, Mohamed S Abdel-Hamid, Emma Jones, Shrestha Shaw, Aya Yoshida, Shiomi Otsuji, Yuka Murofushi, Waleed H. O. Aamer, Ajaz A Bhat, Jehan AlRayahi, Ammira Al-Shabeeb Akil, Ellen van Binsbergen, Etienne J Janssen, Hisashi Oishi, Ryosuke Kobayashi, Takuro Horii, Izuho Hatada, Akihiko Saito, Mitsuharu Hattori, Yoshihiko Kawano, Philip A Lewis, Kate J Heesom, Takeshi Takarada, Kazunobu Sawamoto, Masaki Matsushita, Tomoo Ogi, Rebeka Butkovic, Chris Danson, Kevin A Wilkinson, Khalid A Fakhro, Maha S Zaki, Shinji Saitoh, and Peter J Cullen. The congenital multiple organ malformation syndrome, ritscher-schinzel syndrome is an endosomal recyclinopathy. MedRxiv, Aug 2024. URL: https://doi.org/10.1101/2024.08.17.24311658, doi:10.1101/2024.08.17.24311658. This article has 2 citations.
-
(singla2025ccdc22mutationsthat pages 1-2): Amika Singla, Carolyn Rogers, Mary-Joe Touma, Yassin El-Najjar, Alison Colley, Daniel J. Boesch, Daniel D. Billadeau, Jozef Gecz, Baoyu Chen, and Ezra Burstein. Ccdc22 mutations that impair commd binding cause attenuated 3c/ritscher-schinzel syndrome. BMC Medical Genomics, May 2025. URL: https://doi.org/10.1186/s12920-025-02168-7, doi:10.1186/s12920-025-02168-7. This article has 1 citations and is from a peer-reviewed journal.
-
(otsuji2023clinicaldiversityand pages 5-6): Shiomi Otsuji, Yosuke Nishio, Maki Tsujita, Marlene Rio, Céline Huber, Carlos Antón-Plágaro, Seiji Mizuno, Yoshihiko Kawano, Satoko Miyatake, Marleen Simon, Ellen van Binsbergen, Richard H van Jaarsveld, Naomichi Matsumoto, Valerie Cormier-Daire, Peter J.Cullen, Shinji Saitoh, and Kohji Kato. Clinical diversity and molecular mechanism of vps35l-associated ritscher-schinzel syndrome. Journal of Medical Genetics, 60:359-367, Sep 2023. URL: https://doi.org/10.1136/jmg-2022-108602, doi:10.1136/jmg-2022-108602. This article has 29 citations and is from a domain leading peer-reviewed journal.
-
(otsuji2023clinicaldiversityand pages 8-8): Shiomi Otsuji, Yosuke Nishio, Maki Tsujita, Marlene Rio, Céline Huber, Carlos Antón-Plágaro, Seiji Mizuno, Yoshihiko Kawano, Satoko Miyatake, Marleen Simon, Ellen van Binsbergen, Richard H van Jaarsveld, Naomichi Matsumoto, Valerie Cormier-Daire, Peter J.Cullen, Shinji Saitoh, and Kohji Kato. Clinical diversity and molecular mechanism of vps35l-associated ritscher-schinzel syndrome. Journal of Medical Genetics, 60:359-367, Sep 2023. URL: https://doi.org/10.1136/jmg-2022-108602, doi:10.1136/jmg-2022-108602. This article has 29 citations and is from a domain leading peer-reviewed journal.
-
(kato2024thecongenitalmultiple pages 4-6): Kohji Kato, Yosuke Nishio, Kirsty J McMillan, Aljazi Al-Maraghi, Hester Y Kroes, Mohamed S Abdel-Hamid, Emma Jones, Shrestha Shaw, Aya Yoshida, Shiomi Otsuji, Yuka Murofushi, Waleed H. O. Aamer, Ajaz A Bhat, Jehan AlRayahi, Ammira Al-Shabeeb Akil, Ellen van Binsbergen, Etienne J Janssen, Hisashi Oishi, Ryosuke Kobayashi, Takuro Horii, Izuho Hatada, Akihiko Saito, Mitsuharu Hattori, Yoshihiko Kawano, Philip A Lewis, Kate J Heesom, Takeshi Takarada, Kazunobu Sawamoto, Masaki Matsushita, Tomoo Ogi, Rebeka Butkovic, Chris Danson, Kevin A Wilkinson, Khalid A Fakhro, Maha S Zaki, Shinji Saitoh, and Peter J Cullen. The congenital multiple organ malformation syndrome, ritscher-schinzel syndrome is an endosomal recyclinopathy. MedRxiv, Aug 2024. URL: https://doi.org/10.1101/2024.08.17.24311658, doi:10.1101/2024.08.17.24311658. This article has 2 citations.
-
(singla2025ccdc22mutationsthat pages 4-5): Amika Singla, Carolyn Rogers, Mary-Joe Touma, Yassin El-Najjar, Alison Colley, Daniel J. Boesch, Daniel D. Billadeau, Jozef Gecz, Baoyu Chen, and Ezra Burstein. Ccdc22 mutations that impair commd binding cause attenuated 3c/ritscher-schinzel syndrome. BMC Medical Genomics, May 2025. URL: https://doi.org/10.1186/s12920-025-02168-7, doi:10.1186/s12920-025-02168-7. This article has 1 citations and is from a peer-reviewed journal.
-
(healy2023structureofthe media 9aee47de): Michael D. Healy, Kerrie E. McNally, Rebeka Butkovič, Molly Chilton, Kohji Kato, Joanna Sacharz, Calum McConville, Edmund R.R. Moody, Shrestha Shaw, Vicente J. Planelles-Herrero, Sathish K.N. Yadav, Jennifer Ross, Ufuk Borucu, Catherine S. Palmer, Kai-En Chen, Tristan I. Croll, Ryan J. Hall, Nikeisha J. Caruana, Rajesh Ghai, Thi H.D. Nguyen, Kate J. Heesom, Shinji Saitoh, Imre Berger, Christiane Schaffitzel, Tom A. Williams, David A. Stroud, Emmanuel Derivery, Brett M. Collins, and Peter J. Cullen. Structure of the endosomal commander complex linked to ritscher-schinzel syndrome. Cell, 186:2219-2237.e29, May 2023. URL: https://doi.org/10.1016/j.cell.2023.04.003, doi:10.1016/j.cell.2023.04.003. This article has 88 citations and is from a highest quality peer-reviewed journal.
-
(healy2023structureofthe media 5bc4c3b6): Michael D. Healy, Kerrie E. McNally, Rebeka Butkovič, Molly Chilton, Kohji Kato, Joanna Sacharz, Calum McConville, Edmund R.R. Moody, Shrestha Shaw, Vicente J. Planelles-Herrero, Sathish K.N. Yadav, Jennifer Ross, Ufuk Borucu, Catherine S. Palmer, Kai-En Chen, Tristan I. Croll, Ryan J. Hall, Nikeisha J. Caruana, Rajesh Ghai, Thi H.D. Nguyen, Kate J. Heesom, Shinji Saitoh, Imre Berger, Christiane Schaffitzel, Tom A. Williams, David A. Stroud, Emmanuel Derivery, Brett M. Collins, and Peter J. Cullen. Structure of the endosomal commander complex linked to ritscher-schinzel syndrome. Cell, 186:2219-2237.e29, May 2023. URL: https://doi.org/10.1016/j.cell.2023.04.003, doi:10.1016/j.cell.2023.04.003. This article has 88 citations and is from a highest quality peer-reviewed journal.
-
(healy2023structureofthe media 274803a4): Michael D. Healy, Kerrie E. McNally, Rebeka Butkovič, Molly Chilton, Kohji Kato, Joanna Sacharz, Calum McConville, Edmund R.R. Moody, Shrestha Shaw, Vicente J. Planelles-Herrero, Sathish K.N. Yadav, Jennifer Ross, Ufuk Borucu, Catherine S. Palmer, Kai-En Chen, Tristan I. Croll, Ryan J. Hall, Nikeisha J. Caruana, Rajesh Ghai, Thi H.D. Nguyen, Kate J. Heesom, Shinji Saitoh, Imre Berger, Christiane Schaffitzel, Tom A. Williams, David A. Stroud, Emmanuel Derivery, Brett M. Collins, and Peter J. Cullen. Structure of the endosomal commander complex linked to ritscher-schinzel syndrome. Cell, 186:2219-2237.e29, May 2023. URL: https://doi.org/10.1016/j.cell.2023.04.003, doi:10.1016/j.cell.2023.04.003. This article has 88 citations and is from a highest quality peer-reviewed journal.