RPE65-Related Retinopathy
RPE65-related retinopathy is an inherited retinal disease caused by pathogenic variants in RPE65, encoding the all-trans-retinyl ester isomerohydrolase essential for regenerating the photopigment chromophore 11-cis-retinal within the retinal pigment epithelium (RPE). Loss of this enzyme activity arrests the visual (retinoid) cycle, chronically depleting photoreceptors of the chromophore required for phototransduction, and drives progressive rod-dominant retinal degeneration. Biallelic loss-of-function variants cause a clinical spectrum ranging from Leber congenital amaurosis type 2 (LCA2), with onset in the first year of life and severe visual impairment, through early-onset severe retinal dystrophy (EOSRD) to juvenile retinitis pigmentosa. Rare heterozygous dominant-acting variants produce a typically milder, later-onset retinitis pigmentosa phenotype. RPE65-related recessive retinopathy is distinguished by being the first inherited disease to receive FDA-approved gene therapy: voretigene neparvovec (Luxturna), a subretinal AAV2 vector delivering functional RPE65 cDNA, approved in December 2017.
Ask OpenScientist
Ask a research question about RPE65-Related Retinopathy. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Classifications
Inheritance
2Show evidence (1 reference)
Show evidence (1 reference)
Subtypes
1Show evidence (1 reference)
Pathophysiology
3Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Pathograph
- Target 'Severely reduced visual function and legal blindness' (from 'Progressive Photoreceptor Degeneration') not found in named elements
Phenotypes
6Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (2 references)
Genetic Associations
1Show evidence (2 references)
Medical Actions
3Show evidence (6 references)
Show evidence (1 reference)
Show evidence (1 reference)
Clinical Trials
1Show evidence (1 reference)
Source YAML
click to showname: RPE65-Related Retinopathy
creation_date: "2026-06-18T00:00:00Z"
category: Mendelian
description: >-
RPE65-related retinopathy is an inherited retinal disease caused by pathogenic
variants in RPE65, encoding the all-trans-retinyl ester isomerohydrolase essential
for regenerating the photopigment chromophore 11-cis-retinal within the retinal
pigment epithelium (RPE). Loss of this enzyme activity arrests the visual (retinoid)
cycle, chronically depleting photoreceptors of the chromophore required for
phototransduction, and drives progressive rod-dominant retinal degeneration. Biallelic
loss-of-function variants cause a clinical spectrum ranging from Leber congenital
amaurosis type 2 (LCA2), with onset in the first year of life and severe visual
impairment, through early-onset severe retinal dystrophy (EOSRD) to juvenile
retinitis pigmentosa. Rare heterozygous dominant-acting variants produce a typically
milder, later-onset retinitis pigmentosa phenotype. RPE65-related recessive
retinopathy is distinguished by being the first inherited disease to receive FDA-approved
gene therapy: voretigene neparvovec (Luxturna), a subretinal AAV2 vector delivering
functional RPE65 cDNA, approved in December 2017.
disease_term:
preferred_term: RPE65-related recessive retinopathy
term:
id: MONDO:0100368
label: RPE65-related recessive retinopathy
synonyms:
- Leber congenital amaurosis type 2
- LCA2
- RPE65-related early-onset severe retinal dystrophy
- retinitis pigmentosa type 20
- RPE65-related LCA
parents:
- Ophthalmological Disease
- Retinal Dystrophy
- Inherited retinal dystrophy
notes: >-
The three clinical phenotypes of the autosomal recessive form — LCA2 (onset
infancy, most severe), EOSRD (onset early childhood), and juvenile RP (onset late
childhood/adolescence, mildest) — represent a single genetic entity under
MONDO:0100368 and are distinguished by allele severity and residual RPE65 activity
rather than by separate disease mechanisms. MONDO:0100452 (RPE65-related dominant
retinopathy) is captured as a subtype; its molecular mechanism (dominant-negative
or gain-of-function) is less well characterized than the recessive loss-of-function
form. The gene therapy approval (voretigene neparvovec) applies specifically to
biallelic RPE65 mutations and does not cover the dominant form. Epidemiologically,
biallelic RPE65 mutations account for approximately 8% of Leber congenital amaurosis
cases and 2% of retinitis pigmentosa cases. Clinical presentation is highly variable
and fundus abnormalities can be minimal in early childhood, making genetic testing
essential for diagnosis.
has_subtypes:
- name: RPE65-related dominant retinopathy
subtype_term:
preferred_term: RPE65-related dominant retinopathy
term:
id: MONDO:0100452
label: RPE65-related dominant retinopathy
description: >-
Rare heterozygous dominant-acting RPE65 variants produce a retinitis pigmentosa
phenotype that is generally milder and later in onset than the recessive LCA2/EOSRD
form. Night blindness and progressive peripheral visual field loss are the
predominant features; onset may extend into adulthood. Whether the dominant
mechanism reflects dominant-negative interference with RPE65 enzyme complex
assembly, gain-of-function toxicity, or haploinsufficiency is not fully resolved
for most reported families.
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "Biallelic mutations in this gene, which disrupt the visual cyde, can be described as Leber congenital amaurosis type 2, retinitis pigmentosa type 20, early-onset retinal dystrophy, and other clinical labels for severe rod-mediated inherited retinal dystrophies, which all eventually progress to complete blindness."
explanation: >-
The phase 3 trial paper contextualizes the RPE65 mutation phenotypic spectrum.
Retinitis pigmentosa type 20 (RP20) reflects the less severe, adult-onset
end of RPE65-related disease; the dominant retinopathy subtype (MONDO:0100452)
represents this rarer inheritance context alongside the biallelic recessive
forms.
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
description: >-
The vast majority of RPE65-related retinopathy is caused by biallelic
loss-of-function variants, producing a range of severity from LCA2 (most
severe) to juvenile RP (mildest) depending on residual RPE65 enzymatic activity.
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "GENETIC COUNSELING: RPE65-related retinal degeneration is inherited in an autosomal recessive manner. If both parents of a child diagnosed with autosomal recessive RPE65-related retinal degeneration are known to be heterozygous for an RPE65 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier."
explanation: >-
The GeneReview confirms autosomal recessive inheritance as the primary mode
for RPE65-related retinal degeneration and provides the expected offspring
risk ratios.
- name: Autosomal dominant
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
description: >-
Rare heterozygous dominant-acting RPE65 variants cause a less severe retinitis
pigmentosa phenotype (MONDO:0100452). The dominant form is substantially rarer
than the biallelic recessive form.
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "Biallelic mutations in this gene, which disrupt the visual cyde, can be described as Leber congenital amaurosis type 2, retinitis pigmentosa type 20, early-onset retinal dystrophy, and other clinical labels for severe rod-mediated inherited retinal dystrophies, which all eventually progress to complete blindness."
explanation: >-
The trial paper notes the spectrum of RPE65-associated presentations;
the dominant form (MONDO:0100452) is the inheritance context in which
heterozygous pathogenic RPE65 variants cause RP-like disease.
pathophysiology:
- name: RPE65 Isomerohydrolase Deficiency
description: >-
Pathogenic variants in RPE65 reduce or abolish the enzymatic activity of
all-trans-retinyl ester isomerohydrolase in RPE cells. RPE65 normally
catalyzes the conversion of all-trans-retinyl esters to 11-cis-retinol
in the retinoid (visual) cycle; loss
of this activity depletes the supply of 11-cis-retinal available for photopigment
regeneration in adjacent rod and cone photoreceptors. The RPE cell is the primary
site of defect; photoreceptors themselves do not express RPE65.
gene:
preferred_term: RPE65
modifier: DECREASED
term:
id: hgnc:10294
label: RPE65
cell_types:
- preferred_term: retinal pigment epithelial cell
term:
id: CL:0002586
label: retinal pigment epithelial cell
biological_processes:
- preferred_term: retinol metabolic process
modifier: DECREASED
term:
id: GO:0042572
label: retinol metabolic process
- preferred_term: retinoid metabolic process
modifier: DECREASED
term:
id: GO:0001523
label: retinoid metabolic process
downstream:
- target: Visual Cycle Arrest and 11-cis-Retinal Depletion
description: >-
Failure to regenerate 11-cis-retinol from all-trans-retinyl esters prevents
supply of the photopigment chromophore 11-cis-retinal to rod and cone
photoreceptors, arresting the visual cycle.
evidence:
- reference: PMID:24732759
reference_title: General pathophysiology in retinal degeneration.
supports: SUPPORT
evidence_source: OTHER
snippet: "Within the RPE cells, stored all-trans retinal is isomerized to 11-cis retinal by retinal pigment epithelial 65-kDa (RPE65) protein. 11-cis retinal then passes out of the RPE and into the OS of the photoreceptor cell to bind to opsin and make RHO."
explanation: >-
This review confirms that RPE65 is responsible for the key isomerization
step supplying 11-cis-retinal to photoreceptor outer segments; loss of
RPE65 activity blocks this chromophore supply.
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The RPE65 gene encodes all-trans retinyl ester isomerase, an enzyme crucial to the retinoid cycle."
explanation: >-
The phase 3 voretigene trial paper defines RPE65 as the retinoid cycle
isomerase, establishing enzyme deficiency as the proximal mechanistic lesion.
- reference: PMID:24732759
reference_title: General pathophysiology in retinal degeneration.
supports: SUPPORT
evidence_source: OTHER
snippet: "Within the RPE cells, stored all-trans retinal is isomerized to 11-cis retinal by retinal pigment epithelial 65-kDa (RPE65) protein."
explanation: >-
This review identifies RPE65 as the isomerization enzyme in RPE cells,
localizing the primary defect to the RPE rather than the photoreceptors
themselves.
- name: Visual Cycle Arrest and 11-cis-Retinal Depletion
description: >-
Absent or severely reduced RPE65 activity arrests the retinoid cycle at the
all-trans-retinyl ester stage, preventing regeneration of 11-cis-retinol and
ultimately 11-cis-retinal. Without this chromophore, opsin proteins in rod and
cone outer segments remain in an unliganded (apo-opsin) state. Apo-opsin cannot
form functional photopigment and may signal constitutively, generating abnormal
phototransduction cascade activity and metabolic stress in photoreceptor cells.
cell_types:
- preferred_term: retinal pigment epithelial cell
term:
id: CL:0002586
label: retinal pigment epithelial cell
- preferred_term: retinal rod cell
term:
id: CL:0000604
label: retinal rod cell
- preferred_term: retinal cone cell
term:
id: CL:0000573
label: retinal cone cell
biological_processes:
- preferred_term: retinoid metabolic process
modifier: DECREASED
term:
id: GO:0001523
label: retinoid metabolic process
- preferred_term: photoreceptor cell maintenance
modifier: DECREASED
term:
id: GO:0045494
label: photoreceptor cell maintenance
downstream:
- target: Progressive Photoreceptor Degeneration
description: >-
Chronic 11-cis-retinal depletion produces ongoing rod and cone metabolic
stress and drives progressive photoreceptor loss, starting with rod-dominant
degeneration that eventually extends to cone loss and outer retinal atrophy.
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "despite the absence of functional RPE65 isomerohydrolase and the subsequent inability of the retinal pigment epithelium cells to provide sufficient 11-cis retinal to the photoreceptors, the photoreceptors degenerate slowly, so that phenotypic recovery is possible through restoration of the missing enzyme to the retinal pigment epithelium cells."
explanation: >-
This clinical study establishes that photoreceptors degenerate slowly in
the absence of RPE65 activity; the slow degeneration rate explains why
gene therapy can rescue function even in older patients with residual
viable retina.
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "The three phenotypes of autosomal recessive RPE65-related retinal degeneration, from most severe to mildest, are Leber congenital amaurosis (LCA), early-onset severe retinal dystrophy (EOSRD), and juvenile retinitis pigmentosa (RP)."
explanation: >-
The clinical spectrum from LCA to juvenile RP reflects graded severity of
visual cycle arrest — more complete RPE65 loss produces earlier, more severe
disease while residual activity delays and moderates photoreceptor stress.
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Biallelic mutations in this gene, which disrupt the visual cyde, can be described as Leber congenital amaurosis type 2, retinitis pigmentosa type 20, early-onset retinal dystrophy, and other clinical labels for severe rod-mediated inherited retinal dystrophies, which all eventually progress to complete blindness."
explanation: >-
The trial paper confirms that all clinical labels for RPE65 biallelic disease
share a single visual cycle disruption mechanism and converge on progressive
blindness, supporting the unified visual cycle arrest model.
- name: Progressive Photoreceptor Degeneration
conforms_to: "photoreceptor_degeneration#Rod Photoreceptor Apoptosis"
description: >-
Chronic visual cycle arrest drives progressive rod photoreceptor apoptosis
followed by secondary cone degeneration and outer retinal atrophy. The
slow rate of degeneration reflects the ability of photoreceptors to survive
for years or decades without adequate 11-cis-retinal, but the trajectory
is inexorably toward complete blindness without intervention. Gene therapy
can interrupt this cascade by restoring enzymatic activity in residual viable RPE.
cell_types:
- preferred_term: retinal rod cell
term:
id: CL:0000604
label: retinal rod cell
- preferred_term: retinal cone cell
term:
id: CL:0000573
label: retinal cone cell
- preferred_term: retinal pigment epithelial cell
term:
id: CL:0002586
label: retinal pigment epithelial cell
biological_processes:
- preferred_term: photoreceptor cell maintenance
modifier: DECREASED
term:
id: GO:0045494
label: photoreceptor cell maintenance
- preferred_term: neuron apoptotic process
modifier: INCREASED
term:
id: GO:0051402
label: neuron apoptotic process
downstream:
- target: Severely reduced visual function and legal blindness
description: >-
Progressive rod then cone loss produces night blindness, peripheral field
constriction, and ultimately loss of central vision; most LCA2 individuals
are legally blind by the fourth decade.
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "all affected individuals are legally blind by the fourth decade and many have complete loss of vision (i.e., no light perception)."
explanation: >-
The GeneReview confirms that the natural history of RPE65-related LCA
inevitably progresses to legal blindness and profound visual loss by
adulthood.
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "despite the absence of functional RPE65 isomerohydrolase and the subsequent inability of the retinal pigment epithelium cells to provide sufficient 11-cis retinal to the photoreceptors, the photoreceptors degenerate slowly, so that phenotypic recovery is possible through restoration of the missing enzyme to the retinal pigment epithelium cells."
explanation: >-
This establishes that progressive photoreceptor loss in RPE65-related disease
is slow enough that residual viable retina persists for years to decades,
opening a therapeutic window for gene augmentation.
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "while vision is relatively stable in the first decade of life, it begins to decline again starting in the teens, with 50% of individuals being legally blind (visual acuity of 20/200 and/or visual fields extending <20 degrees from fixation) by age 20 years."
explanation: >-
The GeneReview natural-history data show the progressive character of
photoreceptor loss in the LCA2 form, with accelerating decline from the
teen years onward.
phenotypes:
- category: Ophthalmological
name: Nyctalopia
frequency: VERY_FREQUENT
description: >-
Night blindness is commonly the first symptom, often apparent in infancy or
early childhood in the LCA2/EOSRD forms, reflecting the rod-dominant initial
functional deficit from 11-cis-retinal depletion.
phenotype_term:
preferred_term: Night blindness
term:
id: HP:0000662
label: Nyctalopia
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "Individuals typically experience progressive decline in central vision, and many meet the criteria for legal blindness by the fourth decade; some progress to profound visual loss (including no light perception) by mid-adulthood. In RPE65-related juvenile RP, onset of visual manifestations typically occurs in late childhood or adolescence. Individuals often present with nyctalopia, progressive peripheral visual field loss, and relatively preserved central visual acuity in earlier stages."
explanation: >-
The GeneReview identifies nyctalopia as a consistent presenting feature
across the RPE65 recessive spectrum, most prominent in the juvenile RP
end of the spectrum.
- reference: DOI:10.3341/kjo.2023.0008
reference_title: "Voretigene Neparvovec for the Treatment of RPE65-associated Retinal Dystrophy: Consensus and Recommendations from the Korea RPE65-IRD Consensus Paper Committee"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "early-onset severe nyctalopia, nystagmus, low vision, and progressive visual field constriction, with retinitis pigmentosa by other genetic mutations, appropriate genetic testing is essential to make a correct diagnosis"
explanation: >-
This Korean consensus review confirms early-onset severe nyctalopia as a
characteristic shared feature of RPE65-associated retinal dystrophy, alongside
nystagmus and progressive visual field constriction.
- category: Ophthalmological
name: Severely reduced visual acuity
frequency: VERY_FREQUENT
description: >-
Visual acuity is typically severely impaired from early life in LCA2/EOSRD
and progressively declines to legal blindness in the second to fourth decade.
phenotype_term:
preferred_term: Reduced visual acuity
term:
id: HP:0007663
label: Reduced visual acuity
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "children ages four to ten years in the largest case series had severe visual impairment (mean visual acuity: 20/126)."
explanation: >-
The GeneReview reports severely reduced mean visual acuity in LCA2 children,
confirming early and marked visual impairment as a hallmark phenotype.
- category: Ophthalmological
name: Nystagmus
frequency: FREQUENT
description: >-
Nystagmus is common in LCA2/EOSRD, reflecting the early and severe impairment
of visual function from infancy. It may be the first sign prompting ophthalmological
evaluation in affected neonates.
phenotype_term:
preferred_term: Nystagmus
term:
id: HP:0000639
label: Nystagmus
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A less common but more severe inherited retinal dystrophy, Leber congenital amaurosis, a retinitis pigmentosa subtype, is further characterised by earlier onset, more rapid progression, and nystagmus."
explanation: >-
The phase 3 voretigene trial introduction identifies nystagmus as a defining
clinical feature of LCA, directly supporting its characteristic presence in
RPE65-LCA2.
- category: Ophthalmological
name: Abnormal electroretinogram
frequency: VERY_FREQUENT
description: >-
Electroretinography typically shows markedly reduced or absent rod responses
on dark-adapted testing and impaired cone responses on light-adapted testing,
consistent with the panretinal visual cycle defect affecting both rod and
cone photoreceptors.
phenotype_term:
preferred_term: Abnormal electroretinogram
term:
id: HP:0000512
label: Abnormal electroretinogram
evidence:
- reference: PMID:24732759
reference_title: General pathophysiology in retinal degeneration.
supports: SUPPORT
evidence_source: OTHER
snippet: "Electroretinogram examination shows an attenuated visual response in the cone cells and a complete loss of visual response under scotopic, dark-adapted conditions"
explanation: >-
This pathophysiology review describes the hallmark ERG findings in retinal
degeneration (including RPE65-related disease): absent dark-adapted rod
responses and attenuated light-adapted cone responses, reflecting the
panretinal visual cycle defect.
- category: Ophthalmological
name: Progressive peripheral visual field loss
frequency: VERY_FREQUENT
description: >-
Constriction of the peripheral visual field accompanies rod degeneration and
is progressive; severe constriction is present by early adulthood in most
LCA2/EOSRD patients.
phenotype_term:
preferred_term: Peripheral visual field constriction
term:
id: HP:0001133
label: Constriction of peripheral visual field
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Mean sum total degrees of Goldmann visual field (III4e) nearly doubled in the intervention group and decreased in the control group"
explanation: >-
The phase 3 trial measured Goldmann visual field as an outcome; the control
group's decreasing visual field confirms that peripheral visual field loss
is a natural history feature of RPE65-related retinopathy.
- category: Ophthalmological
name: Photophobia
description: >-
Light sensitivity/photoaversion is a variable feature of the retinal dystrophy
spectrum that includes RPE65-associated disease, most often reported in the
mid-stage of rod-cone degeneration. It is a less consistent feature than
nyctalopia and visual field constriction in RPE65-related retinopathy, and its
reported frequency varies across cohorts.
phenotype_term:
preferred_term: Photophobia
term:
id: HP:0000613
label: Photophobia
evidence:
- reference: PMID:24732759
reference_title: General pathophysiology in retinal degeneration.
supports: SUPPORT
evidence_source: OTHER
snippet: "patients may become photophobic, especially in response to diffuse light, such as occurs with cloudy weather"
explanation: >-
This retinal degeneration pathophysiology review documents that patients with
rod-cone dystrophy become photophobic in the mid-stage of disease, supporting
photophobia as a variable feature of the degeneration spectrum that includes
RPE65-associated retinopathy.
- reference: PMID:35129589
reference_title: "RPE65-Associated Retinopathies in the Italian Population: A Longitudinal Natural History Study."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
explanation: >-
Testa et al. 2022 is a multicenter Italian natural history study of 43 patients
with RPE65-associated inherited retinal dystrophy. The abstract (the extent of
the cached record) does not itemize photophobia; any symptom-level frequency
would derive from the inaccessible full-text tables, so this item only partially
substantiates photophobia as a documented feature of an RPE65-specific cohort.
genetic:
- name: RPE65 pathogenic variants
gene_term:
preferred_term: RPE65
term:
id: hgnc:10294
label: RPE65
association: Causative
features: >-
Biallelic (compound heterozygous or homozygous) loss-of-function variants cause
the recessive LCA2/EOSRD/juvenile RP spectrum. Missense variants that reduce but
do not abolish RPE65 enzymatic activity tend to produce milder EOSRD or juvenile
RP, while severe truncating variants in both alleles produce the LCA2 phenotype.
Rare heterozygous dominant-acting variants cause the milder RP-like dominant form
(MONDO:0100452).
inheritance:
- name: Autosomal recessive
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "individuals aged 3 years or older with, in each eye, best corrected visual acuity of 20/60 or worse, or visual field less than 20 degrees in any meridian, or both, with confirmed genetic diagnosis of biallelic RPE65 mutations"
explanation: >-
The pivotal trial required biallelic RPE65 mutations as an eligibility
criterion, directly confirming that the approved gene therapy indication
targets biallelic (autosomal recessive) RPE65-mediated disease.
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The RPE65 gene encodes all-trans retinyl ester isomerase, an enzyme crucial to the retinoid cycle."
explanation: >-
This paper directly identifies RPE65 as the causative gene encoding a
critical retinoid cycle enzyme, confirming the gene-disease relationship.
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "The diagnosis of autosomal recessive RPE65-related retinal degeneration is established in a proband with suggestive findings and biallelic pathogenic variants in RPE65 identified by molecular genetic testing."
explanation: >-
The GeneReview confirms RPE65 as the causative gene for the autosomal
recessive form and establishes molecular genetic testing as the diagnostic
standard.
treatments:
- name: Voretigene Neparvovec Gene Therapy
therapeutic_modality: GENE_THERAPY
description: >-
Subretinal AAV2-mediated gene supplementation therapy delivering a codon-optimized
human RPE65 cDNA under control of a hybrid promoter, administered bilaterally by
subretinal injection. Indicated for biallelic RPE65 mutation-associated retinal
dystrophy in patients aged 12 months or older with sufficient viable retina. FDA
approved December 2017 (Luxturna, Spark Therapeutics) — the first FDA-approved
gene therapy for an inherited disease. Perioperative oral prednisone is used to
attenuate immune responses.
treatment_term:
preferred_term: gene therapy
term:
id: MAXO:0001001
label: gene therapy
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Voretigene neparvovec gene replacement improved functional vision in RPE65-mediated inherited retinal dystrophy previously medically untreatable."
explanation: >-
The phase 3 randomised controlled trial demonstrated that voretigene
neparvovec significantly improved functional vision (multi-luminance
mobility testing) and full-field light sensitivity in patients with
RPE65-mediated retinal dystrophy, forming the basis of FDA approval.
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "13 (65%) of 20 intervention participants, but no control participants, passed MLMT at the lowest luminance level tested (1 lux), demonstrating maximum possible improvement"
explanation: >-
The primary efficacy endpoint showed that 65% of voretigene-treated participants
achieved maximum possible improvement on the multi-luminance mobility test
(passing at 1 lux), while no control participants did, demonstrating the
clinical magnitude of gene therapy benefit.
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "Subretinal gene supplementation therapy for individuals between ages 12 months and 65 years with retinal degeneration known to be associated with biallelic RPE65 pathogenic variants."
explanation: >-
The GeneReview confirms the approved gene therapy indication and age range,
consistent with the subretinal delivery approach established in the
phase 3 trial.
- reference: DOI:10.1159/000526317
reference_title: "Gene Therapy for Inherited Retinal Disease: Long-Term Durability of Effect"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "sustained results for up to 7.5 years for the full-field light sensitivity threshold test and 5 years for the multi-luminance mobility test in the Phase I and Phase III trials, respectively"
explanation: >-
This systematic review of voretigene neparvovec long-term outcomes documents
sustained functional improvements for up to 7.5 years (FST) and 5 years
(MLMT) across Phase I and Phase III clinical trials, confirming durable efficacy.
- reference: DOI:10.3390/biom14010122
reference_title: "Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Thirty-five patients (34%) experienced ocular treatment-emergent adverse events (TEAEs), most frequently related to chorioretinal atrophy"
explanation: >-
The prospective PERCEIVE real-world registry (n=103 patients) documents the
real-world safety profile of voretigene neparvovec; chorioretinal atrophy is
the most frequent ocular adverse event, occurring in 12.6% of patients, which
is important for post-treatment monitoring.
- reference: DOI:10.1038/s41433-024-03065-6
reference_title: "Voretigene neparvovec for inherited retinal dystrophy due to RPE65 mutations: a scoping review of eligibility and treatment challenges from clinical trials to real practice"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "paediatric patients retain more viable cells, present a less severe disease stage and show the highest potential for improvements, making them the most suitable candidates for treatment"
explanation: >-
This scoping review of real-world eligibility for voretigene neparvovec
identifies pediatric patients as optimal candidates due to greater photoreceptor
viability, supporting early evaluation and intervention in children with
confirmed biallelic RPE65 mutations.
- name: Supportive care and low vision rehabilitation
therapeutic_modality: OTHER
description: >-
Low vision aids, orientation and mobility training, and educational supports for
the visually impaired are core components of management for patients not eligible
for or not yet treated with gene therapy. Dietary interventions including
docosahexaenoic acid and lutein supplements have been considered supportively, and
children should be monitored with routine ophthalmological follow-up.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "Healthy balanced diet to reach the minimum reference daily intake (RDI) for nutrients. Docosahexaenoic acid / eicosapentaenoic acid supplements up to 500 mg/day as well as lutein supplements up to 10 mg/day can be considered if dietary intake is not sufficient."
explanation: >-
The GeneReview describes supportive management including dietary supplements
as standard of care alongside or in lieu of gene therapy, alongside early
referral to low vision services.
- name: Genetic counseling
therapeutic_modality: OTHER
description: >-
Genetic counseling is indicated for affected individuals and their families to
clarify the inheritance pattern (autosomal recessive), reproductive risk (25%
recurrence for parents of an affected child), and to identify at-risk siblings
who may benefit from early evaluation and timely gene therapy consideration.
treatment_term:
preferred_term: genetic counseling
term:
id: MAXO:0000079
label: genetic counseling
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "Evaluation of relatives at risk: It is appropriate to clarify the genetic status of apparently unaffected older and younger at-risk sibs in order to identify those who have biallelic RPE65 pathogenic variants and may benefit from early evaluation, counseling, and timely consideration of subretinal gene supplementation therapy."
explanation: >-
The GeneReview explicitly recommends genetic status clarification in
at-risk siblings to enable early evaluation and gene therapy consideration,
establishing genetic counseling as a core management component.
classifications:
harrisons_chapter:
- classification_value: GENETICS_ENVIRONMENT_DISEASE
- classification_value: NEUROLOGIC
diagnosis:
- name: Molecular genetic testing
diagnosis_term:
preferred_term: diagnostic procedure
term:
id: MAXO:0000003
label: diagnostic procedure
description: >-
Molecular confirmation of biallelic pathogenic RPE65 variants is required to
establish the diagnosis, guide prognosis, and determine eligibility for voretigene
neparvovec gene therapy (which requires confirmed biallelic RPE65 mutations).
Next-generation sequencing panels covering inherited retinal disease genes or
whole exome/genome sequencing are the primary diagnostic tools.
results: >-
Identification of biallelic pathogenic RPE65 variants confirms the diagnosis
and qualifies the patient for gene therapy evaluation; detection of a single
heterozygous variant in a clinically affected patient should prompt consideration
of a dominant form (MONDO:0100452) or a missed second allele.
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "The diagnosis of autosomal recessive RPE65-related retinal degeneration is established in a proband with suggestive findings and biallelic pathogenic variants in RPE65 identified by molecular genetic testing."
explanation: >-
The GeneReview specifies that molecular genetic testing confirming biallelic
RPE65 variants is the standard for establishing the diagnosis.
- name: Functional visual assessment and retinal imaging
diagnosis_term:
preferred_term: diagnostic procedure
term:
id: MAXO:0000003
label: diagnostic procedure
description: >-
Full-field light sensitivity threshold (FST) testing, Goldmann visual field
perimetry, electroretinography (ERG), and optical coherence tomography (OCT)
together characterize the degree of photoreceptor dysfunction and remaining viable
retinal tissue, both for diagnosis and for assessing gene therapy eligibility
(minimum retinal thickness on OCT is required).
results: >-
Dark-adapted ERG shows severely reduced or absent rod responses; light-adapted
ERG is also impaired. FST demonstrates markedly elevated light sensitivity
thresholds. OCT shows outer retinal layer preservation in gene-therapy-eligible
patients. Improvement in FST after voretigene neparvovec is an established
biomarker of restored RPE65 enzymatic activity.
evidence:
- reference: PMID:28712537
reference_title: "Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "in the intervention group showed a rapid, greater than 2 log units improvement by day 30 in light sensitivity that remained stable over 1 year"
explanation: >-
The phase 3 trial establishes that FST improvement of more than 2 log units
after gene therapy directly reflects restoration of RPE65 enzymatic activity
in the RPE, validating FST as a key functional biomarker for this disease.
epidemiology:
- name: Prevalence among inherited retinal dystrophy subtypes
description: >-
Biallelic RPE65 mutations account for approximately 8% of Leber congenital
amaurosis cases and 2% of retinitis pigmentosa cases, making it one of the
more common single-gene causes within these rare inherited retinal dystrophy
categories. Absolute prevalence is rare; the gene therapy approval has made
genetic diagnosis of greater clinical relevance.
evidence:
- reference: DOI:10.1038/s41433-024-03065-6
reference_title: "Voretigene neparvovec for inherited retinal dystrophy due to RPE65 mutations: a scoping review of eligibility and treatment challenges from clinical trials to real practice"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Biallelic mutations in the RPE65 gene affect nearly 8% of Leber Congenital Amaurosis and 2% of Retinitis Pigmentosa cases."
explanation: >-
This scoping review of voretigene neparvovec eligibility provides the
prevalence figures for RPE65 mutations among the LCA and RP subtypes,
establishing the relative frequency of this treatable genetic cause.
progression:
- phase: Early-onset severe visual impairment with relative stability in first decade
age_range: Infancy to age 10
notes: >-
In the LCA2 form, visual manifestations arise in infancy or the first year.
Children aged 4–10 have severe mean visual acuity (20/126). Vision is
relatively stable through the first decade but already severely impaired.
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "In RPE65-related LCA, onset of visual manifestations frequently occurs in infancy during the first year of life. Central vision can be variable, but children ages four to ten years in the largest case series had severe visual impairment (mean visual acuity: 20/126). While vision is relatively stable in the first decade of life, it begins to decline again starting in the teens"
explanation: >-
The GeneReview documents the natural history of RPE65-LCA: early-onset
severe impairment with relative plateau in the first decade before
the accelerating teenage decline.
- phase: Accelerating decline leading to legal blindness in second and third decade
age_range: Age 10–40
notes: >-
Vision loss accelerates from the teenage years onward. By age 20, 50% of
LCA2 individuals meet legal blindness criteria. After age 20, progressive
loss leads to complete blindness by the fourth decade in virtually all cases.
Gene therapy can interrupt this trajectory when viable retina remains.
evidence:
- reference: PMID:31725251
reference_title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
supports: SUPPORT
evidence_source: OTHER
snippet: "with 50% of individuals being legally blind (visual acuity of 20/200 and/or visual fields extending <20 degrees from fixation) by age 20 years. After age 20 years, vision loss is more rapidly progressive: all affected individuals are legally blind by the fourth decade and many have complete loss of vision (i.e., no light perception)."
explanation: >-
The GeneReview natural history data establish the biphasic decline pattern
with the critical therapeutic window — the decades before complete photoreceptor
loss — in which gene therapy can restore function.
clinical_trials:
- name: NCT00999609
phase: PHASE_III
status: COMPLETED
description: >-
Pivotal randomised, controlled, open-label Phase 3 trial of voretigene
neparvovec (AAV2-hRPE65v2) in participants aged 3 years or older with biallelic
RPE65 mutations and sufficient viable retina. Primary endpoint: 1-year change
in bilateral multi-luminance mobility test (MLMT) performance. Results
published as PMID:28712537 (Russell et al., Lancet 2017); the trial was the
basis for FDA approval of voretigene neparvovec (Luxturna) in December 2017.
target_phenotypes:
- preferred_term: Reduced visual acuity
term:
id: HP:0007663
label: Reduced visual acuity
- preferred_term: Constriction of peripheral visual field
term:
id: HP:0001133
label: Constriction of peripheral visual field
evidence:
- reference: clinicaltrials:NCT00999609
reference_title: "A Safety and Efficacy Study in Subjects With Leber Congenital Amaurosis (LCA) Using Adeno-Associated Viral Vector to Deliver the Gene for Human RPE65 to the Retinal Pigment Epithelium (RPE) [AAV2-hRPE65v2-301]"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The study is a Phase 3, open-label, randomized controlled trial of gene therapy intervention by subretinal administration of AAV2-hRPE65v2 (voretigene neparvovec-rzyl). At least twenty-four subjects, three years of age or older, will be recruited."
explanation: >-
ClinicalTrials.gov record for the pivotal Phase 3 voretigene neparvovec
trial that established efficacy and safety supporting FDA approval for
biallelic RPE65-mediated retinal dystrophy.
references:
- reference: PMID:31725251
title: "Autosomal Recessive RPE65-Related Retinal Degeneration."
tags:
- GeneReviews
References & Deep Research
References
1Deep Research
1Executive Summary
RPE65-related retinopathy is an inherited retinal degeneration caused predominantly by biallelic pathogenic variants in RPE65, which encodes the retinoid isomerohydrolase required for 11-cis-retinoid regeneration in the visual cycle in retinal pigment epithelium (RPE). Biochemically, loss of RPE65 reduces 11-cis-retinoids and leads to retinyl-ester accumulation, resulting in rod dysfunction (early nyctalopia) and progressive photoreceptor degeneration with severe early-onset visual impairment. Clinically it spans Leber congenital amaurosis (LCA), early-onset severe retinal dystrophy (EOSRD), and early/severe retinitis pigmentosa (RP) phenotypes. The main real-world therapy is voretigene neparvovec (Luxturna®), a subretinal AAV gene-augmentation treatment, which improves light sensitivity and functional vision in selected patients with viable retinal cells, with durability measured to 5 years (Phase III MLMT) and 7.5 years (Phase I FST) in follow-up publications and reviews. (stepanova2023amoleculargenetic pages 1-2, han2023voretigeneneparvovecfor pages 1-2, leroy2023genetherapyfor pages 1-2)
1. Disease Information
1.1 Definition and overview
RPE65-associated retinal dystrophy/retinopathy refers to retinal degenerations caused by RPE65 mutations and presenting clinically as LCA, EOSRD, and early/severe RP. (han2023voretigeneneparvovecfor pages 1-2)
Direct abstract quote (2023 consensus): “Mutations in the RPE65 gene… share common clinical characteristics, such as early-onset severe nyctalopia, nystagmus, low vision, and progressive visual field constriction…” (han2023voretigeneneparvovecfor pages 1-2)
1.2 Key identifiers (available from retrieved evidence)
- MONDO (OpenTargets):
- RPE65-related recessive retinopathy: MONDO:0100368 (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65)
- RPE65-related dominant retinopathy: MONDO:0100452 (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65)
- Leber congenital amaurosis: MONDO:0018998 (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65)
- OMIM (explicitly cited in retrieved text):
- LCA (OMIM 204100) (stepanova2023amoleculargenetic pages 1-2)
- Early-onset RP20 (OMIM 613794) (stepanova2023amoleculargenetic pages 1-2)
Not retrieved in the available excerpts: Orphanet IDs, ICD-10/ICD-11 codes, and MeSH identifiers specific to “RPE65-related retinopathy” (trial excerpts did include MeSH terms for LCA and RP but without stable identifiers in the extracted snippet). (NCT00999609 chunk 2)
1.3 Common synonyms / alternative names (as used in the literature)
- “RPE65-associated retinal dystrophy” (han2023voretigeneneparvovecfor pages 1-2)
- “RPE65-mediated inherited retinal dystrophy” (testa2024voretigeneneparvovecfor pages 1-2, fischer2024realworldsafetyand pages 1-2)
- “RPE65-associated retinopathy/retinopathies” (stepanova2023amoleculargenetic pages 1-2, testa2022rpe65associatedretinopathiesin pages 1-2)
- “LCA2” / “LCA type 2” (RPE65-related LCA subtype) (chiu2021anupdateon pages 5-6)
1.4 Evidence sources: aggregated vs individual
Most information in this report is derived from aggregated disease-level sources: consensus statements and scoping reviews (2023–2024), registry-based post-authorization studies (2024), and multicenter natural history cohorts (2022), rather than EHR-only single-patient sources. (han2023voretigeneneparvovecfor pages 1-2, testa2024voretigeneneparvovecfor pages 1-2, fischer2024realworldsafetyand pages 1-2, testa2022rpe65associatedretinopathiesin pages 1-2)
| Concept | Identifier system | Identifier | Evidence/notes | Source (with year and URL) |
|---|---|---|---|---|
| RPE65-related recessive retinopathy | MONDO | MONDO:0100368 | OpenTargets lists disease-target association for RPE65; useful umbrella disease mapping term for biallelic RPE65 disease (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65) | OpenTargets, accessed in this session: https://platform.opentargets.org |
| RPE65-related dominant retinopathy | MONDO | MONDO:0100452 | OpenTargets also lists a distinct dominant entity; relevant for differential classification because most therapeutic literature here concerns recessive/biallelic disease (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65) | OpenTargets, accessed in this session: https://platform.opentargets.org |
| Leber congenital amaurosis | MONDO | MONDO:0018998 | OpenTargets lists LCA as associated with RPE65; many RPE65 cases present clinically as LCA/LCA2 (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65, chiu2021anupdateon pages 5-6) | OpenTargets; Chiu et al. 2021, https://doi.org/10.3390/ijms22094534 |
| Leber congenital amaurosis | OMIM | OMIM:204100 | Russian cohort review explicitly states that biallelic RPE65 variants cause LCA (OMIM 204100) (stepanova2023amoleculargenetic pages 1-2) | Stepanova et al. 2023, https://doi.org/10.3390/genes14112056 |
| Severe early-onset retinitis pigmentosa / RP20 | OMIM | OMIM:613794 | Same review explicitly maps severe early-onset RP due to RPE65 to RP20 (OMIM 613794) (stepanova2023amoleculargenetic pages 1-2) | Stepanova et al. 2023, https://doi.org/10.3390/genes14112056 |
| LCA type 2 / LCA2 | Disease subtype term | Not explicitly identified in evidence by OMIM/MONDO | Review states “LCA type 2 (LCA2) is caused by the mutation in the RPE65 gene on chromosome 1p31” (chiu2021anupdateon pages 5-6) | Chiu et al. 2021, https://doi.org/10.3390/ijms22094534 |
| RPE65-associated retinal dystrophy | Synonym / disease label | — | Used in Korean consensus; encompasses LCA, EOSRD, and early/severe RP phenotypes due to RPE65 mutations (han2023voretigeneneparvovecfor pages 1-2) | Han et al. 2023, https://doi.org/10.3341/kjo.2023.0008 |
| RPE65-mediated inherited retinal dystrophy | Synonym / disease label | — | Used in treatment and registry literature, especially for voretigene neparvovec eligibility and outcomes (testa2024voretigeneneparvovecfor pages 1-2, fischer2024realworldsafetyand pages 1-2) | Testa et al. 2024, https://doi.org/10.1038/s41433-024-03065-6; Fischer et al. 2024, https://doi.org/10.3390/biom14010122 |
| RPE65-associated retinopathy / retinopathies | Synonym / disease label | — | Used in natural history and molecular epidemiology studies for biallelic RPE65 disease spectrum (stepanova2023amoleculargenetic pages 1-2, testa2022rpe65associatedretinopathiesin pages 1-2) | Stepanova et al. 2023, https://doi.org/10.3390/genes14112056; Testa et al. 2022, https://doi.org/10.1167/iovs.63.2.13 |
| Early-onset severe retinal dystrophy | Phenotypic classification term | EOSRD | Frequently used clinical classification overlapping with LCA in RPE65 disease (han2023voretigeneneparvovecfor pages 1-2, testa2022rpe65associatedretinopathiesin pages 1-2) | Han et al. 2023, https://doi.org/10.3341/kjo.2023.0008; Testa et al. 2022, https://doi.org/10.1167/iovs.63.2.13 |
| Safety and Efficacy Study in Subjects With Leber Congenital Amaurosis | ClinicalTrials.gov | NCT00999609 | Pivotal phase 3 voretigene neparvovec study; trial excerpt specifies molecular confirmation of RPE65 mutations and viable retinal cells (NCT00999609 chunk 2) | ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT00999609 |
| Safety Study in Subjects With Leber Congenital Amaurosis | ClinicalTrials.gov | NCT00516477 | Phase 1 RPE65 gene therapy study referenced in trial search results (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65) | ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT00516477 |
| Phase 1 Follow-on Study of AAV2-hRPE65v2 Vector in Subjects With LCA2 | ClinicalTrials.gov | NCT01208389 | Follow-on bilateral/second-eye study after initial phase 1 treatment (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65) | ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT01208389 |
| Long-term Follow-up Study in Subjects Who Received Voretigene Neparvovec-rzyl | ClinicalTrials.gov | NCT03602820 | Long-term observational follow-up after VN treatment (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65) | ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT03602820 |
| Patient Registry Study for Patients Treated With Voretigene Neparvovec in US | ClinicalTrials.gov | NCT03597399 | US registry-based observational study of treated patients (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65) | ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT03597399 |
| Study of Efficacy and Safety of Voretigene Neparvovec in Japanese Patients With Biallelic RPE65 Mutation-associated Retinal Dystrophy | ClinicalTrials.gov | NCT04516369 | Japanese phase 3 study of VN in genetically confirmed biallelic RPE65 disease (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65) | ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT04516369 |
Table: This table compiles the main disease names and formal identifiers explicitly present in the retrieved evidence for RPE65-related retinopathy. It is useful for harmonizing terminology across natural history studies, treatment trials, and ontology-based knowledge bases.
2. Etiology
2.1 Disease causal factors
Primary cause: germline RPE65 variants, typically biallelic (autosomal recessive) causing RPE65-associated retinopathies, including LCA and early-onset RP. (stepanova2023amoleculargenetic pages 1-2, testa2024voretigeneneparvovecfor pages 1-2)
2.2 Risk factors
- Genetic risk factors (causal variants): biallelic pathogenic/likely pathogenic variants in RPE65. (stepanova2023amoleculargenetic pages 1-2)
- Genotype–phenotype trend (severity timing): individuals with two missense alleles tend to present later (≥1 year) than those with one/two truncating variants (<1 year). (han2023voretigeneneparvovecfor pages 2-4)
Environmental risk factors: no specific environmental toxins/lifestyle factors were identified as causal in the retrieved evidence; disease is primarily monogenic.
2.3 Protective factors
No definitive genetic or environmental “protective factors” for preventing disease onset were identified in the retrieved clinical evidence. (Limit: not exhaustively searched beyond retrieved sources.)
2.4 Gene–environment interactions
No gene–environment interaction evidence specific to RPE65 retinopathy was retrieved.
3. Phenotypes
RPE65-related retinopathy typically presents with night blindness, nystagmus, severe early visual impairment, and progressive visual field constriction; ERG is often markedly reduced/absent, and fundus findings may be minimal early but evolve to retinal degeneration. (han2023voretigeneneparvovecfor pages 1-2, testa2024voretigeneneparvovecfor pages 1-2, kumaran2017lebercongenitalamaurosisearlyonset pages 1-2)
| Phenotype (plain language) | Phenotype type | Typical onset | Progression | Frequency/notes with quantitative values when available | Suggested HPO term(s) |
|---|---|---|---|---|---|
| Severe visual impairment / low visual acuity | Symptom/sign | Birth, infancy, or early childhood; mean self-reported symptom onset 2.2 ± 2.1 years in one natural-history cohort | Usually progressive, though acuity decline may be slow early; median age to low vision 33.8 years and blindness 41.4 years by BCVA in Italian cohort (testa2022rpe65associatedretinopathiesin pages 3-4, testa2022rpe65associatedretinopathiesin pages 1-2, testa2022rpe65associatedretinopathiesin pages 4-6) | Reported in 32/43 (74.4%) in Italian cohort; phase/phenotype labels include LCA and EOSRD; BCVA often severely reduced, but some residual vision may persist into adulthood (testa2022rpe65associatedretinopathiesin pages 3-4, testa2022rpe65associatedretinopathiesin pages 1-2, kumaran2017lebercongenitalamaurosisearlyonset pages 1-2) | HP:0000505 Visual impairment; HP:0000518 Cataract not primary; HP:0000572 Reduced visual acuity |
| Night blindness / severe nyctalopia | Symptom | Early childhood to infancy; often among earliest symptoms | Progressive, reflecting early rod dysfunction | Reported in 28/43 (65.1%) in Italian cohort; described as a characteristic early feature across RPE65 disease and often severe (testa2022rpe65associatedretinopathiesin pages 3-4, han2023voretigeneneparvovecfor pages 1-2, testa2024voretigeneneparvovecfor pages 1-2) | HP:0000662 Nyctalopia |
| Nystagmus / roving eye movements | Sign | Congenital or infancy | Often persistent; may accompany severe early vision loss | Reported in 24/43 (55.8%) in Italian cohort; classic early LCA/EOSRD sign noted in foundational reviews (testa2022rpe65associatedretinopathiesin pages 3-4, kumaran2017lebercongenitalamaurosisearlyonset pages 1-2, testa2022rpe65associatedretinopathiesin pages 1-2) | HP:0000639 Nystagmus |
| Constricted peripheral visual fields / visual field loss | Symptom/test | Childhood to adolescence, sometimes recognized later than nyctalopia | Progressive constriction | Reported in 18/43 (41.9%) in Italian cohort; Korean/Testa reviews describe progressive visual field constriction as a core feature; pivotal VN studies also used residual field as part of viability/eligibility assessment (testa2022rpe65associatedretinopathiesin pages 3-4, han2023voretigeneneparvovecfor pages 1-2, testa2024voretigeneneparvovecfor pages 1-2, NCT00999609 chunk 2) | HP:0001133 Constricted visual field |
| Poor pupillary light responses / abnormal pupils | Sign | Infancy | Usually persistent | Classic LCA/EOSRD feature in broader review literature; often accompanies severe congenital/early visual dysfunction (kumaran2017lebercongenitalamaurosisearlyonset pages 1-2, testa2022rpe65associatedretinopathiesin pages 1-2) | HP:0000613 Photophobia overlaps; HP:0007690 Abnormal pupillary light reflex |
| Photophobia / photoaversion | Symptom | Childhood | Variable; may persist | Reported in 20/43 (46.5%) in Italian cohort; also included among variable LCA manifestations in review literature (testa2022rpe65associatedretinopathiesin pages 3-4, chiu2021anupdateon pages 5-6) | HP:0000613 Photophobia |
| Markedly reduced or absent ERG | Test abnormality | Detectable at diagnostic testing in infancy/childhood | Typically severe and persistent; reflects generalized rod-cone dysfunction | ERG undetectable in 26/34 (76.5%) in Italian cohort; Kumaran review describes ERG as typically undetectable or severely abnormal in LCA/EOSRD; Testa 2024 notes reduced/non-detectable ERG as typical in RPE65 disease (testa2022rpe65associatedretinopathiesin pages 1-2, testa2022rpe65associatedretinopathiesin pages 4-6, kumaran2017lebercongenitalamaurosisearlyonset pages 1-2, testa2024voretigeneneparvovecfor pages 1-2) | HP:0030533 Abnormal electroretinogram; HP:0000550 Reduced retinal function |
| Minimal or normal early fundus, later retinal degeneration | Sign/imaging | Early childhood may have minimal abnormalities; later childhood/adulthood show degeneration | Progressive | Early fundus may be normal/minimally abnormal; later findings can include vessel attenuation, disc pallor, peripheral pigmentary change, salt-and-pepper change, or RP-like fundus (han2023voretigeneneparvovecfor pages 1-2, kumaran2017lebercongenitalamaurosisearlyonset pages 1-2, testa2022rpe65associatedretinopathiesin pages 7-8, testa2022rpe65associatedretinopathiesin pages 1-2) | HP:0000520 Prolonged dark adaptation not fundus; HP:0001103 Abnormality of the retina; HP:0000548 Retinal degeneration |
| Reduced/absent fundus autofluorescence | Imaging finding | Childhood to adulthood when imaged | Usually reflects progressive retinal/RPE dysfunction | Testa 2024 review describes markedly reduced/absent FAF as typical; useful in structural assessment and treatment selection (testa2024voretigeneneparvovecfor pages 1-2) | HP:0030610 Abnormal fundus autofluorescence |
| Retinal thinning / reduced central foveal thickness | Imaging finding | Usually documented from childhood onward | Progressive overall; cross-sectional decline with age | Central foveal thickness declined at about −0.6%/year cross-sectionally in Italian natural history study; ONL thinning common (~79% of eyes in excerpted analysis) (testa2022rpe65associatedretinopathiesin pages 1-2, testa2022rpe65associatedretinopathiesin pages 4-6) | HP:0030829 Retinal thinning; HP:0000546 Retinal atrophy |
| Epiretinal membrane | Imaging/sign | Later childhood to adulthood | Variable | Seen in 5/31 (16.1%) on OCT in Italian cohort; secondary rather than defining phenotype (testa2022rpe65associatedretinopathiesin pages 1-2) | HP:0011505 Epiretinal membrane |
| Oculodigital sign / eye-poking behavior | Behavioral sign | Infancy/early childhood | Can persist | Classic LCA/EOSRD feature emphasized in foundational review, though not quantified in RPE65-specific natural-history excerpt (kumaran2017lebercongenitalamaurosisearlyonset pages 1-2) | HP:0000657 Oculodigital sign |
Table: This table summarizes the main clinical phenotypes reported for RPE65-related retinopathy, including onset, progression, and quantitative natural-history details where available. It is useful for structuring phenotype annotations and mapping them to HPO terms.
Quality-of-life impact (inferred from functional endpoints): Functional vision deficits are severe enough that pivotal and real-world studies use mobility and light-sensitivity endpoints (e.g., MLMT and FST) to quantify daily function changes after treatment. (leroy2023genetherapyfor pages 8-9, fischer2024realworldsafetyand pages 1-2)
4. Genetic / Molecular Information
4.1 Causal gene
- Gene: RPE65 (retinoid isomerohydrolase RPE65), chromosome region 1p31 (reported as 1p31.3 in consensus). (han2023voretigeneneparvovecfor pages 1-2, stepanova2023amoleculargenetic pages 1-2)
- Encodes a 533-aa (~65 kDa) RPE-specific protein. (han2023voretigeneneparvovecfor pages 1-2, stepanova2023amoleculargenetic pages 1-2)
4.2 Pathogenic variant landscape (recent database snapshots)
As of March 7, 2023, one consensus report summarizes: - ClinVar: 776 RPE65 variants (162 pathogenic, 65 likely pathogenic, 231 VUS); most are SNVs (n=671). (han2023voretigeneneparvovecfor pages 2-4) - LOVD: 364 variations (280 pathogenic/likely pathogenic, 60 VUS). (han2023voretigeneneparvovecfor pages 2-4) - HGMD: 292 disease-causing entries. (han2023voretigeneneparvovecfor pages 2-4)
4.3 Population-specific variant spectra (recent cohorts)
- Russian IRD cohort (2023): among 1053 unrelated IRD patients, 25/474 molecularly diagnosed IRD cases (5.3%) had RPE65-associated retinopathy; 26 variants detected, 9 novel; three common alleles (c.304G>T p.Glu102, c.370C>T p.Arg124, c.272G>A p.Arg91Gln) accounted for 41.8% of affected chromosomes. (stepanova2023amoleculargenetic pages 1-2)
- Danish LCA cohort: RPE65 was the most frequently mutated LCA gene (16%). Literature aggregation highlighted recurrent variants p.(R91W), p.(Y368H), and c.11+5G>A as major contributors; an estimate of RPE65 carrier frequency 1/158 was reported. (astuti2016comprehensivegenotypingreveals pages 6-7, astuti2016comprehensivegenotypingreveals pages 8-9)
4.4 Modifier genes / epigenetics / chromosomal abnormalities
No specific modifier genes, epigenetic mechanisms, or large chromosomal abnormalities were identified in the retrieved excerpts.
5. Environmental Information
No specific non-genetic environmental contributors were identified in the retrieved sources; the disorder is primarily monogenic.
6. Mechanism / Pathophysiology
6.1 Causal chain (molecular defect → clinical manifestations)
- Normal visual cycle: after photon absorption, 11-cis-retinal is converted to all-trans-retinal; all-trans-retinol is esterified in RPE; RPE65 converts all-trans-retinyl esters → 11-cis-retinol, later oxidized to 11-cis-retinal to regenerate photopigment. (stepanova2023amoleculargenetic pages 1-2)
- RPE65 loss of function: pathogenic variants reduce/abolish isomerohydrolase activity; biochemical consequences include accumulation of all-trans-retinyl esters and decrease/absence of visual pigment. (stepanova2023amoleculargenetic pages 1-2)
- Physiologic consequence: impaired photopigment regeneration produces rod-mediated night blindness and broader retinal dysfunction; over time, the consensus describes this disruption as leading to “progressive loss of photoreceptors.” (han2023voretigeneneparvovecfor pages 1-2)
Direct text quote (mechanism; 2023 cohort paper): “RPE65… plays a vital role in the regeneration of 11-cis-retinol in the visual cycle… [and] converts all-trans-retinyl esters into 11-cis-retinol…” (stepanova2023amoleculargenetic pages 1-2)
6.2 Cell types and anatomical substrates
- Primary cell type: retinal pigment epithelial cell (RPE) (RPE65 expression “exclusively in RPE”). (han2023voretigeneneparvovecfor pages 2-4)
- Downstream affected cells: rod and cone photoreceptors (functional loss and progressive degeneration are described; ERG often absent and nyctalopia prominent). (han2023voretigeneneparvovecfor pages 1-2, testa2024voretigeneneparvovecfor pages 1-2)
6.3 Ontology term suggestions
- GO (Biological Process) suggestions: visual perception; visual cycle; retinoid metabolic process; phototransduction-related processes (supported by RPE65’s role in 11-cis-retinoid regeneration). (stepanova2023amoleculargenetic pages 1-2, han2023voretigeneneparvovecfor pages 2-4)
- CL (Cell Ontology) suggestions: retinal pigment epithelial cell; rod photoreceptor cell; cone photoreceptor cell. (han2023voretigeneneparvovecfor pages 2-4, testa2024voretigeneneparvovecfor pages 1-2)
- UBERON suggestions: retina; retinal pigment epithelium; photoreceptor layer. (han2023voretigeneneparvovecfor pages 2-4, testa2024voretigeneneparvovecfor pages 1-2)
7. Anatomical Structures Affected
- Primary organ/system: eye/visual system; retina. (testa2024voretigeneneparvovecfor pages 1-2)
- Primary tissues: retina and retinal pigment epithelium. (han2023voretigeneneparvovecfor pages 2-4)
- Localization: typically bilateral retinal disease (implicitly in IRD cohorts and bilateral treatment paradigms). (kiraly2023outcomesandadverse pages 5-8, NCT00999609 chunk 2)
8. Temporal Development
8.1 Onset
- Frequently congenital/infantile (LCA) with severe visual loss from birth/early infancy and nystagmus. (kumaran2017lebercongenitalamaurosisearlyonset pages 1-2, testa2022rpe65associatedretinopathiesin pages 1-2)
- EOSRD onset overlaps but can present “between early childhood and age five,” often with milder residual function compared with classic LCA. (testa2022rpe65associatedretinopathiesin pages 1-2)
8.2 Progression
- Progressive constriction of visual fields and photoreceptor degeneration are core features. (han2023voretigeneneparvovecfor pages 1-2)
- Quantitative natural history (Italian cohort): median age to low vision 33.8 years and blindness 41.4 years (BCVA-based). (testa2022rpe65associatedretinopathiesin pages 1-2)
- Retinal structural decline: central foveal thickness decreased ~0.6% per year cross-sectionally with age. (testa2022rpe65associatedretinopathiesin pages 1-2)
9. Inheritance and Population
9.1 Inheritance
- Predominantly autosomal recessive (biallelic) in most clinical series and in Luxturna eligibility framing. (testa2024voretigeneneparvovecfor pages 1-2, stepanova2023amoleculargenetic pages 1-2)
- Rare dominant RPE65 retinopathy exists as a distinct MONDO entity. (OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65)
9.2 Epidemiology (recent quantitative statements)
- LCA prevalence reported as 1.20–2.37 per 100,000 in one consensus summary. (han2023voretigeneneparvovecfor pages 1-2)
- One scoping review states LCA prevalence ~1:300,000. (testa2024voretigeneneparvovecfor pages 1-2)
- Contribution of RPE65 to disease categories:
- “Nearly 8% of LCA and 2% of RP cases” in the 2024 scoping review. (testa2024voretigeneneparvovecfor pages 1-2)
- Estimated global prevalence among LCA ≈5–10%, versus <5% in RP, in 2023 consensus text. (han2023voretigeneneparvovecfor pages 1-2)
- Example regional frequency: in a Korean survey, biallelic RPE65 variants were found in 6/2,140 IRD patients (0.28%). (han2023voretigeneneparvovecfor pages 1-2)
9.3 Population genetics / founder effects
- Denmark: RPE65 was 16% of LCA in one national cohort; major recurrent variants p.(R91W), p.(Y368H), and c.11+5G>A were highlighted in literature aggregation; estimated RPE65 carrier frequency 1/158. (astuti2016comprehensivegenotypingreveals pages 6-7, astuti2016comprehensivegenotypingreveals pages 8-9)
- Russia: common alleles c.304G>T, c.370C>T, c.272G>A comprised 41.8% of affected chromosomes. (stepanova2023amoleculargenetic pages 1-2)
10. Diagnostics
10.1 Clinical/functional testing used in practice and trials
Common modalities include: - Visual acuity (BCVA), visual fields (e.g., Goldmann), OCT, ERG, fundus autofluorescence, and psychophysical tests such as full-field stimulus threshold (FST). (testa2022rpe65associatedretinopathiesin pages 1-2, testa2024voretigeneneparvovecfor pages 1-2, fischer2024realworldsafetyand pages 1-2)
10.2 Genetic testing
- Diagnostic emphasis: phenotypic overlap with other IRDs makes molecular diagnosis essential; “appropriate genetic testing is essential to make a correct diagnosis.” (han2023voretigeneneparvovecfor pages 1-2)
- In one consensus summary, genetic testing can identify underlying causes in “up to 76% of IRD cases.” (han2023voretigeneneparvovecfor pages 2-4)
- Example testing approach used in a 2023 national cohort: targeted massive-parallel sequencing (211-gene panel), confirmatory Sanger sequencing for biallelic status, and MLPA for exon-level copy number. (stepanova2023amoleculargenetic pages 1-2)
10.3 Eligibility/viability criteria for gene therapy (real-world implementations)
Trial inclusion criteria and real-world decisions use evidence of viable retinal cells, including OCT/ophthalmoscopy; one trial excerpt explicitly references thresholds such as >100 µm retinal thickness at the posterior pole or alternative criteria. (NCT00999609 chunk 2)
11. Outcome / Prognosis
- Natural history suggests severe functional impairment early, with many patients meeting blindness criteria in adulthood; in the Italian cohort, 67.4% met blindness criteria at baseline. (testa2022rpe65associatedretinopathiesin pages 4-6)
- ERG is often nonrecordable: 76.5% (26/34) undetectable in one cohort. (testa2022rpe65associatedretinopathiesin pages 1-2)
- Genotype severity association: patients stratified by loss-of-function allele burden showed worse BCVA with more LoF alleles. (testa2022rpe65associatedretinopathiesin pages 1-2)
12. Treatment
12.1 Approved advanced therapeutic: voretigene neparvovec (Luxturna®)
Mechanism and delivery: AAV2-mediated delivery of human RPE65 cDNA by subretinal injection after vitrectomy, for patients with biallelic RPE65 mutations and sufficient viable retinal cells. (testa2024voretigeneneparvovecfor pages 1-2, fischer2024realworldsafetyand pages 1-2)
Recent real-world effectiveness (2024 PERCEIVE registry): - n=103 patients; mean age 19.5 years; mean follow-up 0.8 years (max 2.3). (fischer2024realworldsafetyand pages 1-2) - FST (white) mean change from baseline: −16.59 dB (month 1), −18.24 dB (month 6), −15.84 dB (year 1), −13.67 dB (year 2) in available eyes, indicating substantial light-sensitivity improvements through 2 years. (fischer2024realworldsafetyand pages 1-2) - Visual acuity change: “not clinically significant.” (fischer2024realworldsafetyand pages 1-2)
Safety (2024 PERCEIVE): - 34% experienced ocular TEAEs; most frequent was chorioretinal atrophy 12.6% (13/103). (fischer2024realworldsafetyand pages 1-2) - TEAEs of special interest in 17.5% (including procedure-related inflammation/infection). (fischer2024realworldsafetyand pages 1-2)
Single-center real-world safety signals (2023 Oxford cohort, 6 patients/12 eyes, mean follow-up 8.2 months): - Cataracts in 4 eyes, mild intraocular inflammation in 2 eyes, retinal atrophy in 10 eyes (some severe), and increased IOP in 6 eyes with glaucoma surgery in 4 eyes. (kiraly2023outcomesandadverse pages 1-2, kiraly2023outcomesandadverse pages 5-8)
12.2 Durability of effect (clinical trial follow-up)
A 2023 durability review reports sustained outcomes in human trials: - “sustained results for up to 7.5 years for the full-field light sensitivity threshold test and 5 years for the multi-luminance mobility test” in Phase I and Phase III trials. (leroy2023genetherapyfor pages 1-2) - Trial program summary: Phase I included 12 subjects with dose escalation and second-eye treatment; Phase III enrolled 31 randomized participants. (leroy2023genetherapyfor pages 7-8)
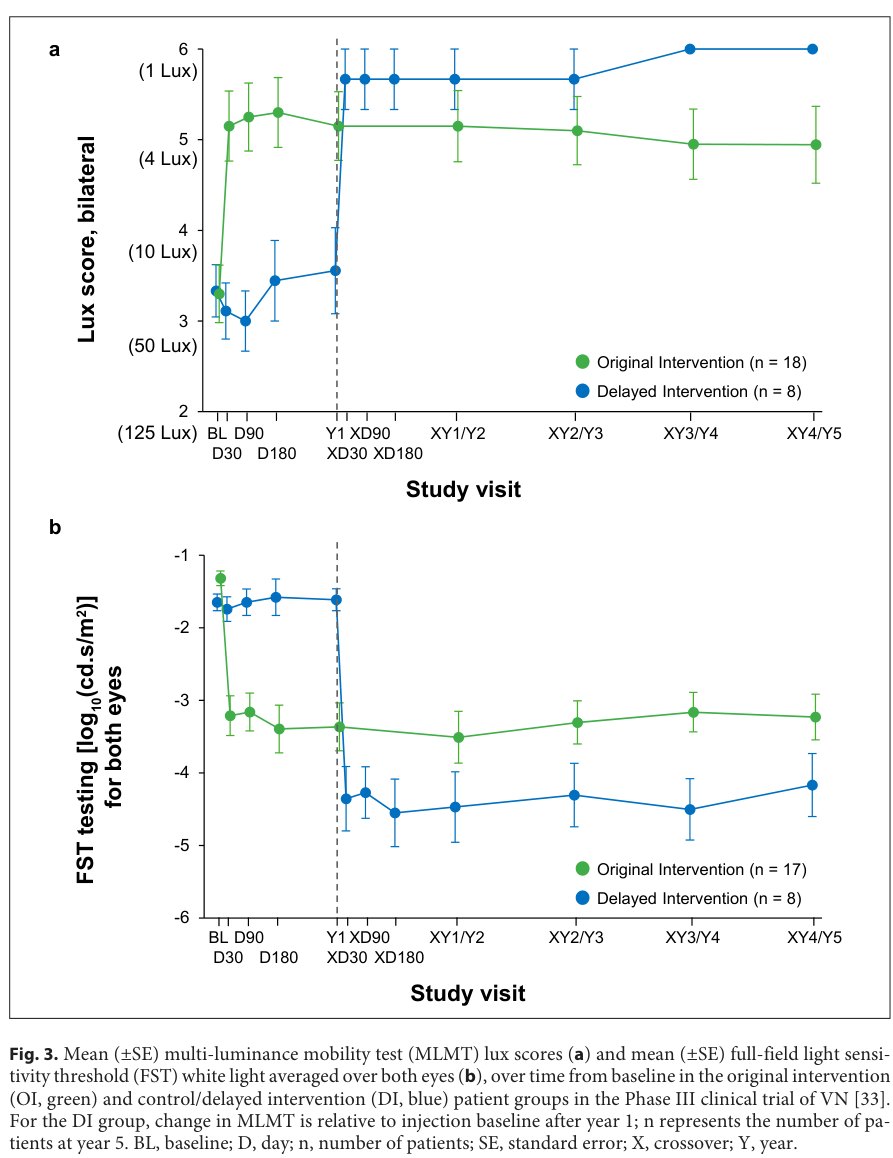
Figure evidence: Phase III durability of MLMT and FST trajectories through year 5 is shown in a reproduced figure. (leroy2023genetherapyfor media 51c99434)
12.3 Treatment strategy / patient selection (expert analysis)
A 2024 scoping review emphasizes that no single structural cutoff defines eligibility, but functional rescue is linked to photoreceptor preservation and that pediatric patients often have more viable cells and better potential for improvements. (testa2024voretigeneneparvovecfor pages 1-2)
12.4 Experimental / ongoing studies (examples)
ClinicalTrials.gov studies retrieved in this session include the pivotal trial and long-term follow-up/registry studies (e.g., NCT00999609, NCT03602820, NCT03597399, NCT04516369). (NCT00999609 chunk 2, OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65)
12.5 MAXO suggestions
- Gene replacement therapy / gene augmentation therapy (for AAV-mediated subretinal delivery of RPE65). (testa2024voretigeneneparvovecfor pages 1-2, fischer2024realworldsafetyand pages 1-2)
- Vitrectomy and subretinal injection as procedural components of delivery. (testa2024voretigeneneparvovecfor pages 1-2)
13. Prevention
No primary prevention (in the sense of preventing disease onset) is established for this monogenic condition in the retrieved evidence. Secondary/tertiary prevention centers on: - Genetic counseling and cascade testing in families (implied by autosomal recessive inheritance and diagnostic emphasis). (stepanova2023amoleculargenetic pages 1-2, han2023voretigeneneparvovecfor pages 1-2) - Early diagnosis and referral to assess eligibility for gene therapy while retinal cells remain viable. (testa2024voretigeneneparvovecfor pages 1-2, NCT00999609 chunk 2)
14. Other Species / Natural Disease
The retrieved evidence references long-duration treatment effects in canine disease models for RPE65 gene replacement (effects lasting nearly a decade), supporting comparative biology, but does not provide explicit taxonomy identifiers in the excerpt. (leroy2023genetherapyfor pages 1-2)
15. Model Organisms
The retrieved evidence indicates extensive animal-model work and reports long-term efficacy in canine models; a 2023 review included 71 animal-model gene-therapy publications and notes rAAV genome episomal persistence. (leroy2023genetherapyfor pages 1-2)
Recent Developments (2023–2024 highlights)
- Real-world registry scale-up: PERCEIVE provides the largest prospective post-authorization real-world dataset to date (103 patients) with quantified safety (chorioretinal atrophy 12.6%) and sustained FST improvements to 2 years. (fischer2024realworldsafetyand pages 1-2)
- Eligibility and implementation challenges: a 2024 scoping review synthesizes real-world variability and underscores that photoreceptor viability and pediatric timing are key determinants, with no universal structural cutoff for eligibility. (testa2024voretigeneneparvovecfor pages 1-2)
- Population-specific variant catalogs expanding: 2023 national cohort work adds novel variants and defines high-frequency alleles in Russia (three variants = 41.8% of affected chromosomes), supporting country-level screening strategies. (stepanova2023amoleculargenetic pages 1-2)
Limitations of this Report
- Some requested identifiers (Orphanet, ICD-10/ICD-11, MeSH) and several categories (environmental risk factors, epigenetics, modifier genes) were not explicitly present in the retrieved full-text excerpts, and are therefore not asserted here.
- This report emphasizes sources successfully retrieved in this tool session; it is not an exhaustive systematic review of all PubMed literature.
References
-
(stepanova2023amoleculargenetic pages 1-2): Anna Stepanova, Natalya Ogorodova, Vitaly Kadyshev, Olga Shchagina, Sergei Kutsev, and Aleksandr Polyakov. A molecular genetic analysis of rpe65-associated forms of inherited retinal degenerations in the russian federation. Genes, 14:2056, Nov 2023. URL: https://doi.org/10.3390/genes14112056, doi:10.3390/genes14112056. This article has 5 citations.
-
(han2023voretigeneneparvovecfor pages 1-2): Jinu Han, Kwangsic Joo, Ungsoo Samuel Kim, Se Joon Woo, Eun Kyoung Lee, Joo Yong Lee, Tae Kwann Park, Sang Jin Kim, and Suk Ho Byeon. Voretigene neparvovec for the treatment of rpe65-associated retinal dystrophy: consensus and recommendations from the korea rpe65-ird consensus paper committee. Korean Journal of Ophthalmology, 37:166-186, Apr 2023. URL: https://doi.org/10.3341/kjo.2023.0008, doi:10.3341/kjo.2023.0008. This article has 6 citations.
-
(leroy2023genetherapyfor pages 1-2): Bart P. Leroy, M. Dominik Fischer, John G. Flannery, Robert E. MacLaren, Deniz Dalkara, Hendrik P.N. Scholl, Daniel C. Chung, Claudio Spera, Daniel Viriato, and Judit Banhazi. Gene therapy for inherited retinal disease: long-term durability of effect. Ophthalmic Research, 66:179-196, Sep 2023. URL: https://doi.org/10.1159/000526317, doi:10.1159/000526317. This article has 81 citations and is from a peer-reviewed journal.
-
(OpenTargets Search: RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65): Open Targets Query (RPE65-related retinopathy,Leber congenital amaurosis,retinitis pigmentosa-RPE65, 3 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(NCT00999609 chunk 2): Safety and Efficacy Study in Subjects With Leber Congenital Amaurosis. Spark Therapeutics, Inc.. 2012. ClinicalTrials.gov Identifier: NCT00999609
-
(testa2024voretigeneneparvovecfor pages 1-2): Francesco Testa, Giacomo Bacci, Benedetto Falsini, Giancarlo Iarossi, Paolo Melillo, Dario Pasquale Mucciolo, Vittoria Murro, Anna Paola Salvetti, Andrea Sodi, Giovanni Staurenghi, and Francesca Simonelli. Voretigene neparvovec for inherited retinal dystrophy due to rpe65 mutations: a scoping review of eligibility and treatment challenges from clinical trials to real practice. Eye, 38:2504-2515, Apr 2024. URL: https://doi.org/10.1038/s41433-024-03065-6, doi:10.1038/s41433-024-03065-6. This article has 38 citations and is from a peer-reviewed journal.
-
(fischer2024realworldsafetyand pages 1-2): M. Dominik Fischer, Francesca Simonelli, Jayashree Sahni, Frank G. Holz, Rainer Maier, Christina Fasser, Andrea Suhner, Daniel P. Stiehl, Bee Chen, Isabelle Audo, and Bart P. Leroy. Real-world safety and effectiveness of voretigene neparvovec: results up to 2 years from the prospective, registry-based perceive study. Biomolecules, 14:122, Jan 2024. URL: https://doi.org/10.3390/biom14010122, doi:10.3390/biom14010122. This article has 64 citations.
-
(testa2022rpe65associatedretinopathiesin pages 1-2): Francesco Testa, Vittoria Murro, Sabrina Signorini, Leonardo Colombo, Giancarlo Iarossi, Francesco Parmeggiani, Benedetto Falsini, Anna Paola Salvetti, Raffaella Brunetti-Pierri, Giorgia Aprile, Chiara Bertone, Agnese Suppiej, Francesco Romano, Marianthi Karali, Simone Donati, Paolo Melillo, Andrea Sodi, Luciano Quaranta, Luca Rossetti, Luca Buzzonetti, Marzio Chizzolini, Stanislao Rizzo, Giovanni Staurenghi, Sandro Banfi, Claudio Azzolini, and Francesca Simonelli. rpe65-associated retinopathies in the italian population: a longitudinal natural history study. Investigative Opthalmology & Visual Science, 63:13, Feb 2022. URL: https://doi.org/10.1167/iovs.63.2.13, doi:10.1167/iovs.63.2.13. This article has 23 citations.
-
(chiu2021anupdateon pages 5-6): Wei Chiu, Ting-Yi Lin, Yun-Chia Chang, Henkie Isahwan-Ahmad Mulyadi Lai, Shen-Che Lin, Chun Ma, Aliaksandr A. Yarmishyn, Shiuan-Chen Lin, Kao-Jung Chang, Yu-Bai Chou, Chih-Chien Hsu, Tai-Chi Lin, Shih-Jen Chen, Yueh Chien, Yi-Ping Yang, and De-Kuang Hwang. An update on gene therapy for inherited retinal dystrophy: experience in leber congenital amaurosis clinical trials. International Journal of Molecular Sciences, 22:4534, Apr 2021. URL: https://doi.org/10.3390/ijms22094534, doi:10.3390/ijms22094534. This article has 128 citations.
-
(han2023voretigeneneparvovecfor pages 2-4): Jinu Han, Kwangsic Joo, Ungsoo Samuel Kim, Se Joon Woo, Eun Kyoung Lee, Joo Yong Lee, Tae Kwann Park, Sang Jin Kim, and Suk Ho Byeon. Voretigene neparvovec for the treatment of rpe65-associated retinal dystrophy: consensus and recommendations from the korea rpe65-ird consensus paper committee. Korean Journal of Ophthalmology, 37:166-186, Apr 2023. URL: https://doi.org/10.3341/kjo.2023.0008, doi:10.3341/kjo.2023.0008. This article has 6 citations.
-
(kumaran2017lebercongenitalamaurosisearlyonset pages 1-2): Neruban Kumaran, Anthony T Moore, Richard G Weleber, and Michel Michaelides. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. The British Journal of Ophthalmology, 101:1147-1154, Jul 2017. URL: https://doi.org/10.1136/bjophthalmol-2016-309975, doi:10.1136/bjophthalmol-2016-309975. This article has 412 citations.
-
(testa2022rpe65associatedretinopathiesin pages 3-4): Francesco Testa, Vittoria Murro, Sabrina Signorini, Leonardo Colombo, Giancarlo Iarossi, Francesco Parmeggiani, Benedetto Falsini, Anna Paola Salvetti, Raffaella Brunetti-Pierri, Giorgia Aprile, Chiara Bertone, Agnese Suppiej, Francesco Romano, Marianthi Karali, Simone Donati, Paolo Melillo, Andrea Sodi, Luciano Quaranta, Luca Rossetti, Luca Buzzonetti, Marzio Chizzolini, Stanislao Rizzo, Giovanni Staurenghi, Sandro Banfi, Claudio Azzolini, and Francesca Simonelli. rpe65-associated retinopathies in the italian population: a longitudinal natural history study. Investigative Opthalmology & Visual Science, 63:13, Feb 2022. URL: https://doi.org/10.1167/iovs.63.2.13, doi:10.1167/iovs.63.2.13. This article has 23 citations.
-
(testa2022rpe65associatedretinopathiesin pages 4-6): Francesco Testa, Vittoria Murro, Sabrina Signorini, Leonardo Colombo, Giancarlo Iarossi, Francesco Parmeggiani, Benedetto Falsini, Anna Paola Salvetti, Raffaella Brunetti-Pierri, Giorgia Aprile, Chiara Bertone, Agnese Suppiej, Francesco Romano, Marianthi Karali, Simone Donati, Paolo Melillo, Andrea Sodi, Luciano Quaranta, Luca Rossetti, Luca Buzzonetti, Marzio Chizzolini, Stanislao Rizzo, Giovanni Staurenghi, Sandro Banfi, Claudio Azzolini, and Francesca Simonelli. rpe65-associated retinopathies in the italian population: a longitudinal natural history study. Investigative Opthalmology & Visual Science, 63:13, Feb 2022. URL: https://doi.org/10.1167/iovs.63.2.13, doi:10.1167/iovs.63.2.13. This article has 23 citations.
-
(testa2022rpe65associatedretinopathiesin pages 7-8): Francesco Testa, Vittoria Murro, Sabrina Signorini, Leonardo Colombo, Giancarlo Iarossi, Francesco Parmeggiani, Benedetto Falsini, Anna Paola Salvetti, Raffaella Brunetti-Pierri, Giorgia Aprile, Chiara Bertone, Agnese Suppiej, Francesco Romano, Marianthi Karali, Simone Donati, Paolo Melillo, Andrea Sodi, Luciano Quaranta, Luca Rossetti, Luca Buzzonetti, Marzio Chizzolini, Stanislao Rizzo, Giovanni Staurenghi, Sandro Banfi, Claudio Azzolini, and Francesca Simonelli. rpe65-associated retinopathies in the italian population: a longitudinal natural history study. Investigative Opthalmology & Visual Science, 63:13, Feb 2022. URL: https://doi.org/10.1167/iovs.63.2.13, doi:10.1167/iovs.63.2.13. This article has 23 citations.
-
(leroy2023genetherapyfor pages 8-9): Bart P. Leroy, M. Dominik Fischer, John G. Flannery, Robert E. MacLaren, Deniz Dalkara, Hendrik P.N. Scholl, Daniel C. Chung, Claudio Spera, Daniel Viriato, and Judit Banhazi. Gene therapy for inherited retinal disease: long-term durability of effect. Ophthalmic Research, 66:179-196, Sep 2023. URL: https://doi.org/10.1159/000526317, doi:10.1159/000526317. This article has 81 citations and is from a peer-reviewed journal.
-
(astuti2016comprehensivegenotypingreveals pages 6-7): Galuh D N Astuti, Mette Bertelsen, Markus N Preising, Muhammad Ajmal, Birgit Lorenz, Sultana M H Faradz, Raheel Qamar, Rob W J Collin, Thomas Rosenberg, and Frans P M Cremers. Comprehensive genotyping reveals rpe65 as the most frequently mutated gene in leber congenital amaurosis in denmark. European Journal of Human Genetics, 24:1071-1079, Dec 2016. URL: https://doi.org/10.1038/ejhg.2015.241, doi:10.1038/ejhg.2015.241. This article has 98 citations and is from a domain leading peer-reviewed journal.
-
(astuti2016comprehensivegenotypingreveals pages 8-9): Galuh D N Astuti, Mette Bertelsen, Markus N Preising, Muhammad Ajmal, Birgit Lorenz, Sultana M H Faradz, Raheel Qamar, Rob W J Collin, Thomas Rosenberg, and Frans P M Cremers. Comprehensive genotyping reveals rpe65 as the most frequently mutated gene in leber congenital amaurosis in denmark. European Journal of Human Genetics, 24:1071-1079, Dec 2016. URL: https://doi.org/10.1038/ejhg.2015.241, doi:10.1038/ejhg.2015.241. This article has 98 citations and is from a domain leading peer-reviewed journal.
-
(kiraly2023outcomesandadverse pages 5-8): Peter Kiraly, Charles L. Cottriall, Laura J. Taylor, Jasleen K. Jolly, Jasmina Cehajic-Kapetanovic, Imran H. Yusuf, Cristina Martinez-Fernandez de la Camara, Morag Shanks, Susan M. Downes, Robert E. MacLaren, and M. Dominik Fischer. Outcomes and adverse effects of voretigene neparvovec treatment for biallelic rpe65-mediated inherited retinal dystrophies in a cohort of patients from a single center. Biomolecules, 13:1484, Oct 2023. URL: https://doi.org/10.3390/biom13101484, doi:10.3390/biom13101484. This article has 28 citations.
-
(kiraly2023outcomesandadverse pages 1-2): Peter Kiraly, Charles L. Cottriall, Laura J. Taylor, Jasleen K. Jolly, Jasmina Cehajic-Kapetanovic, Imran H. Yusuf, Cristina Martinez-Fernandez de la Camara, Morag Shanks, Susan M. Downes, Robert E. MacLaren, and M. Dominik Fischer. Outcomes and adverse effects of voretigene neparvovec treatment for biallelic rpe65-mediated inherited retinal dystrophies in a cohort of patients from a single center. Biomolecules, 13:1484, Oct 2023. URL: https://doi.org/10.3390/biom13101484, doi:10.3390/biom13101484. This article has 28 citations.
-
(leroy2023genetherapyfor pages 7-8): Bart P. Leroy, M. Dominik Fischer, John G. Flannery, Robert E. MacLaren, Deniz Dalkara, Hendrik P.N. Scholl, Daniel C. Chung, Claudio Spera, Daniel Viriato, and Judit Banhazi. Gene therapy for inherited retinal disease: long-term durability of effect. Ophthalmic Research, 66:179-196, Sep 2023. URL: https://doi.org/10.1159/000526317, doi:10.1159/000526317. This article has 81 citations and is from a peer-reviewed journal.
-
(leroy2023genetherapyfor media 51c99434): Bart P. Leroy, M. Dominik Fischer, John G. Flannery, Robert E. MacLaren, Deniz Dalkara, Hendrik P.N. Scholl, Daniel C. Chung, Claudio Spera, Daniel Viriato, and Judit Banhazi. Gene therapy for inherited retinal disease: long-term durability of effect. Ophthalmic Research, 66:179-196, Sep 2023. URL: https://doi.org/10.1159/000526317, doi:10.1159/000526317. This article has 81 citations and is from a peer-reviewed journal.