Mitochondrial Neurogastrointestinal Encephalomyopathy
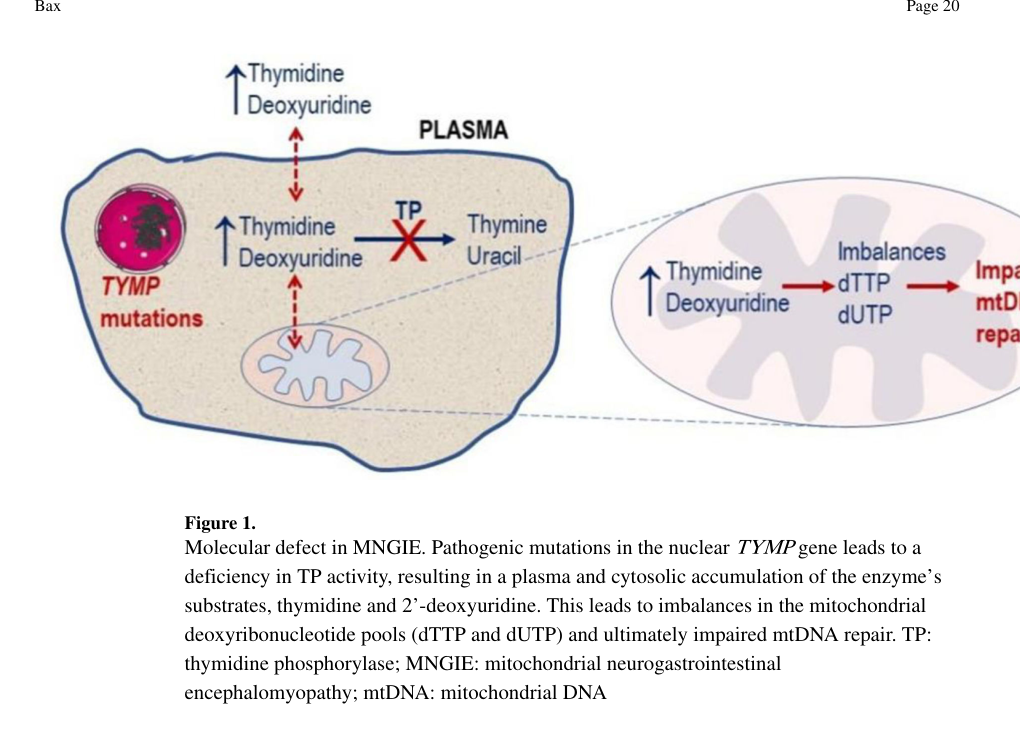
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE; MONDO:0017575) is an autosomal recessive nuclear-gene disorder of nucleoside metabolism. Classic MNGIE is caused by biallelic loss-of-function variants in TYMP, encoding thymidine phosphorylase (TP). Loss of TP activity produces systemic accumulation of the deoxyribonucleosides thymidine and 2'-deoxyuridine in plasma and tissues, which imbalances the mitochondrial deoxyribonucleotide (dNTP) pools and secondarily destabilizes the mitochondrial genome, causing mtDNA depletion, multiple deletions, and site-specific point mutations and ultimately respiratory-chain dysfunction in smooth muscle, peripheral nerve, and brain. The clinical picture comprises progressive gastrointestinal dysmotility (early satiety, postprandial emesis, pseudo-obstruction, diarrhea) with cachexia, ptosis and progressive external ophthalmoplegia, demyelinating sensorimotor peripheral neuropathy, and a largely asymptomatic diffuse leukoencephalopathy on brain MRI. Onset is usually between the first and fifth decades and the disorder is progressive and frequently fatal in early adulthood. Rare MNGIE-like phenotypes have been reported in association with POLG and RRM2B, which are also nuclear genes required for mtDNA maintenance.
Ask OpenScientist
Ask a research question about Mitochondrial Neurogastrointestinal Encephalomyopathy. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (1 reference)
Subtypes
3Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Pathophysiology
5Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Pathograph
Phenotypes
10Digestive 1
Show evidence (1 reference)
Eye 1
Show evidence (1 reference)
Musculoskeletal 1
Show evidence (1 reference)
Nervous System 2
Show evidence (1 reference)
Show evidence (1 reference)
Other 5
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
3Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Medical Actions
6Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Biochemical Markers
2Show evidence (1 reference)
Show evidence (1 reference)
Clinical Trials
1Show evidence (1 reference)
Source YAML
click to showname: Mitochondrial Neurogastrointestinal Encephalomyopathy
creation_date: "2026-06-05T12:00:00Z"
category: Mendelian
synonyms:
- MNGIE
- MNGIE disease
- mitochondrial neurogastrointestinal encephalopathy disease
- MNGIE-MTDPS1
- thymidine phosphorylase deficiency
- mitochondrial DNA depletion syndrome 1 (MNGIE type)
description: >-
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE; MONDO:0017575)
is an autosomal recessive nuclear-gene disorder of nucleoside metabolism.
Classic MNGIE is caused by biallelic loss-of-function variants in TYMP,
encoding thymidine phosphorylase (TP). Loss of TP activity produces systemic
accumulation of the deoxyribonucleosides thymidine and 2'-deoxyuridine in
plasma and tissues, which imbalances the mitochondrial deoxyribonucleotide
(dNTP) pools and secondarily destabilizes the mitochondrial genome, causing
mtDNA depletion, multiple deletions, and site-specific point mutations and
ultimately respiratory-chain dysfunction in smooth muscle, peripheral nerve,
and brain. The clinical picture comprises progressive gastrointestinal
dysmotility (early satiety, postprandial emesis, pseudo-obstruction, diarrhea)
with cachexia, ptosis and progressive external ophthalmoplegia, demyelinating
sensorimotor peripheral neuropathy, and a largely asymptomatic diffuse
leukoencephalopathy on brain MRI. Onset is usually between the first and fifth
decades and the disorder is progressive and frequently fatal in early
adulthood. Rare MNGIE-like phenotypes have been reported in association with
POLG and RRM2B, which are also nuclear genes required for mtDNA maintenance.
notes: >-
MONDO cross-check: MNGIE is anchored as MONDO:0017575 (Orphanet:298), defined
by the association of gastrointestinal dysmotility, peripheral neuropathy,
chronic progressive external ophthalmoplegia, and leukoencephalopathy.
The canonical molecular cause is TYMP (formerly ECGF1) thymidine phosphorylase
deficiency; OMIM phenotype 603041. POLG- and RRM2B-related MNGIE-like
phenotypes are modeled here as subtypes because the deep-research evidence and
the Filosto 2018 review explicitly describe them as distinct MNGIE-type
phenotypes, but classic TYMP-related MNGIE is the defining form.
disease_term:
preferred_term: Mitochondrial Neurogastrointestinal Encephalomyopathy
term:
id: MONDO:0017575
label: mitochondrial neurogastrointestinal encephalomyopathy
parents:
- Mitochondrial DNA Depletion Syndrome
- Mitochondrial Disease
references:
- reference: PMID:20301358
title: "Mitochondrial Neurogastrointestinal Encephalopathy Disease."
tags:
- GeneReviews
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
description: >-
MNGIE is inherited in an autosomal recessive manner; the parents of an
affected individual are obligate asymptomatic heterozygotes.
evidence:
- reference: PMID:20301358
reference_title: "Mitochondrial Neurogastrointestinal Encephalopathy Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
MNGIE disease is inherited in an autosomal recessive manner. The parents
of an affected individual are obligate heterozygotes and therefore carry
one mutated allele; heterozygotes are asymptomatic.
explanation: >-
GeneReviews establishes autosomal recessive inheritance with asymptomatic
carrier parents.
has_subtypes:
- name: Classic MNGIE
display_name: Classic TYMP-related MNGIE (MTDPS1)
description: >-
The defining form of MNGIE, caused by biallelic loss-of-function variants in
TYMP with thymidine phosphorylase deficiency and systemic accumulation of
thymidine and 2'-deoxyuridine. Presents with the classic constellation of
gastrointestinal dysmotility, cachexia, ptosis/ophthalmoplegia, peripheral
neuropathy, and leukoencephalopathy.
evidence:
- reference: PMID:9924029
reference_title: "Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Examination of 12 MNGIE probands revealed homozygous or
compound-heterozygous mutations in the gene specifying thymidine
phosphorylase (TP), located on chromosome 22q13.32-qter.
explanation: >-
The original gene-discovery paper established TYMP/TP loss-of-function as
the cause of classic MNGIE.
- name: MNGIE-like POLG
display_name: POLG-related MNGIE-like phenotype
description: >-
A rare MNGIE-type phenotype linked to mutations in POLG, the catalytic
subunit of mitochondrial DNA polymerase gamma. Distinct from classic
TYMP-related MNGIE; thymidine phosphorylase activity and plasma nucleosides
are not the defining biomarkers.
evidence:
- reference: PMID:30373120
reference_title: "Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Other two MNGIE-type phenotypes have been described so far, which are
linked to mutations in POLG and RRM2B genes.
explanation: >-
Filosto 2018 review documents POLG as one of the genes underlying an
MNGIE-type phenotype distinct from classic TYMP disease.
- name: MNGIE-like RRM2B
display_name: RRM2B-related MNGIE-like phenotype
description: >-
A rare MNGIE-type phenotype linked to mutations in RRM2B, the p53-inducible
small subunit of ribonucleotide reductase required for mitochondrial dNTP
supply. Distinct from classic TYMP-related MNGIE.
evidence:
- reference: PMID:30373120
reference_title: "Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Other two MNGIE-type phenotypes have been described so far, which are
linked to mutations in POLG and RRM2B genes.
explanation: >-
Filosto 2018 review documents RRM2B as one of the genes underlying an
MNGIE-type phenotype distinct from classic TYMP disease.
pathophysiology:
- name: Thymidine Phosphorylase Deficiency
description: >-
Biallelic loss-of-function variants in TYMP abolish thymidine phosphorylase
(TP) enzyme activity. TP normally catalyzes the reversible phosphorolysis of
the pyrimidine deoxyribonucleosides thymidine and 2'-deoxyuridine to their
bases (thymine and uracil) plus 2-deoxyribose-1-phosphate. Leukocyte TP
activity in patients is typically less than 5-18% of controls.
gene:
preferred_term: TYMP
term:
id: hgnc:3148
label: TYMP
molecular_functions:
- preferred_term: thymidine phosphorylase activity
modifier: DECREASED
term:
id: GO:0009032
label: thymidine phosphorylase activity
biological_processes:
- preferred_term: pyrimidine nucleobase metabolic process
term:
id: GO:0006206
label: pyrimidine nucleobase metabolic process
modifier: DECREASED
evidence:
- reference: PMID:9924029
reference_title: "Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
TP activity in leukocytes from MNGIE patients was less than 5 percent of
controls, indicating that loss-of-function mutations in TP cause the
disease.
explanation: >-
Demonstrates that biallelic TYMP mutations cause near-complete loss of TP
enzyme activity, the primary biochemical lesion in MNGIE.
downstream:
- target: Systemic Nucleoside Accumulation
description: >-
Loss of TP activity prevents catabolism of thymidine and 2'-deoxyuridine,
which accumulate systemically in plasma and tissues.
causal_link_type: DIRECT
evidence:
- reference: PMID:30373120
reference_title: "Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
mutations in TYMP, which cause a loss of function of thymidine
phosphorylase (TP), nucleoside accumulation in plasma and tissues, and
mitochondrial dysfunction.
explanation: >-
Directly links TP loss of function to systemic nucleoside accumulation.
- name: Systemic Nucleoside Accumulation
description: >-
In the absence of TP activity, the deoxyribonucleosides thymidine and
2'-deoxyuridine accumulate to high concentrations in plasma and tissues.
Plasma thymidine >3 micromol/L and 2'-deoxyuridine >5 micromol/L are
characteristic, whereas healthy individuals have undetectable or very low
levels. This systemic accumulation is the disease-defining biochemical
phenotype.
chemical_entities:
- preferred_term: thymidine
modifier: INCREASED
term:
id: CHEBI:17748
label: thymidine
- preferred_term: 2'-deoxyuridine
modifier: INCREASED
term:
id: CHEBI:16450
label: 2'-deoxyuridine
evidence:
- reference: PMID:30959750
reference_title: "Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in MNGIE."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
There is a systemic accumulation of thymidine and deoxyuridine in the
absence of thymidine phosphorylase activity, which then generates
imbalances within the mitochondrial deoxyribonucleotide pools
explanation: >-
Establishes systemic thymidine/deoxyuridine accumulation as the
consequence of TP deficiency and the trigger for dNTP-pool imbalance.
downstream:
- target: Mitochondrial dNTP Pool Imbalance
description: >-
Elevated thymidine and deoxyuridine distort the mitochondrial
deoxyribonucleotide pools, raising dTTP and depleting dCTP.
causal_link_type: DIRECT
evidence:
- reference: PMID:30959750
reference_title: "Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in MNGIE."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
which then generates imbalances within the mitochondrial
deoxyribonucleotide pools, causing mitochondrial DNA (mtDNA) point
mutations, depletion and deletion abnormalities, and ultimately
mitochondrial dysfunction
explanation: >-
Links nucleoside accumulation to dNTP-pool imbalance and downstream
mtDNA instability.
- name: Mitochondrial dNTP Pool Imbalance
description: >-
Excess thymidine and deoxyuridine are salvaged inside mitochondria, raising
dTTP and depleting dCTP and thereby unbalancing the mitochondrial
deoxyribonucleotide pools required for faithful mitochondrial DNA
replication.
biological_processes:
- preferred_term: pyrimidine nucleotide metabolic process
term:
id: GO:0006220
label: pyrimidine nucleotide metabolic process
modifier: ABNORMAL
cellular_components:
- preferred_term: mitochondrion
term:
id: GO:0005739
label: mitochondrion
evidence:
- reference: PMID:30959750
reference_title: "Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in MNGIE."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
imbalances within the mitochondrial deoxyribonucleotide pools, causing
mitochondrial DNA (mtDNA) point mutations, depletion and deletion
abnormalities, and ultimately mitochondrial dysfunction
explanation: >-
Establishes the dNTP-pool imbalance as the mechanistic bridge between
nucleoside accumulation and mtDNA instability.
downstream:
- target: mtDNA Instability
description: >-
Imbalanced dNTP pools impair mitochondrial DNA replication and
maintenance, producing mtDNA depletion, multiple deletions, and
site-specific point mutations.
causal_link_type: DIRECT
evidence:
- reference: PMID:9924029
reference_title: "Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The pathogenic mechanism may be related to aberrant thymidine
metabolism, leading to impaired replication or maintenance of mtDNA, or
both.
explanation: >-
Links aberrant thymidine metabolism to impaired mtDNA
replication/maintenance.
- name: mtDNA Instability
description: >-
Disturbed mitochondrial dNTP supply impairs mitochondrial DNA replication,
leading over time to accumulation of mtDNA point mutations, multiple
deletions, and eventual mtDNA depletion in affected tissues. These are
secondary (acquired) mtDNA defects rather than primary inherited mtDNA
mutations.
biological_processes:
- preferred_term: mitochondrial DNA replication
term:
id: GO:0006264
label: mitochondrial DNA replication
modifier: ABNORMAL
- preferred_term: mitochondrial DNA metabolic process
term:
id: GO:0032042
label: mitochondrial DNA metabolic process
modifier: ABNORMAL
evidence:
- reference: PMID:32173240
reference_title: "Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
This leads to an imbalance of the mitochondrial deoxyribonucleotide supply
and subsequently impairs mitochondrial DNA replication. Over time, there
is a build-up of mutations, deletions, and eventual depletion of mtDNA
resulting in impaired mitochondrial function
explanation: >-
Documents the progression from dNTP-pool imbalance to mtDNA mutations,
deletions, depletion and mitochondrial dysfunction.
downstream:
- target: Respiratory Chain Dysfunction
description: >-
mtDNA depletion and deletions reduce the cell's capacity to synthesize
functional respiratory-chain subunits, impairing oxidative
phosphorylation in smooth muscle, peripheral nerve, and brain.
causal_link_type: DIRECT
evidence:
- reference: PMID:32173240
reference_title: "Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
eventual depletion of mtDNA resulting in impaired mitochondrial
function
explanation: >-
Links mtDNA depletion to impaired mitochondrial (respiratory-chain)
function.
- name: Respiratory Chain Dysfunction
description: >-
Secondary mtDNA abnormalities cause respiratory-chain (oxidative
phosphorylation) dysfunction that preferentially injures high-energy-demand
tissues. In gastrointestinal smooth muscle and the enteric nervous system
this produces the hallmark dysmotility; in peripheral nerve it produces a
demyelinating sensorimotor neuropathy; and in the central nervous system it
produces a diffuse leukoencephalopathy.
cell_types:
- preferred_term: gastrointestinal smooth muscle cell
term:
id: CL:0000192
label: smooth muscle cell
- preferred_term: peripheral neuron
term:
id: CL:0000540
label: neuron
cellular_components:
- preferred_term: mitochondrion
term:
id: GO:0005739
label: mitochondrion
evidence:
- reference: PMID:30373120
reference_title: "Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The clinical picture includes progressive gastrointestinal dysmotility,
cachexia, ptosis and ophthalmoparesis, peripheral neuropathy, and diffuse
leukoencephalopathy, which usually lead to death in early adulthood.
explanation: >-
Links the mitochondrial dysfunction to the multisystem clinical
manifestations of MNGIE.
phenotypes:
- name: Gastrointestinal dysmotility
description: >-
Progressive gastrointestinal dysmotility is the hallmark feature, manifesting
as early satiety, nausea, dysphagia, gastroesophageal reflux, postprandial
emesis, episodic abdominal pain and distention, and diarrhea, often
progressing to chronic intestinal pseudo-obstruction.
phenotype_term:
preferred_term: Gastrointestinal dysmotility
term:
id: HP:0002579
label: Gastrointestinal dysmotility
clinical_course: PROGRESSIVE
frequency: VERY_FREQUENT
evidence:
- reference: PMID:20301358
reference_title: "Mitochondrial Neurogastrointestinal Encephalopathy Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Mitochondrial neurogastrointestinal encephalopathy (MNGIE) disease is
characterized by progressive gastrointestinal dysmotility (manifesting as
early satiety, nausea, dysphagia, gastroesophageal reflux, postprandial
emesis, episodic abdominal pain and/or distention, and diarrhea)
explanation: >-
GeneReviews lists progressive gastrointestinal dysmotility as a defining
feature of MNGIE.
- name: Intestinal pseudo-obstruction
description: >-
Severe enteric dysmotility frequently produces episodes of chronic
intestinal pseudo-obstruction (CIPO)-like disease with abdominal distention
and sub-occlusive crises.
phenotype_term:
preferred_term: Intestinal pseudo-obstruction
term:
id: HP:0004389
label: Intestinal pseudo-obstruction
evidence:
- reference: PMID:26264513
reference_title: "Allogeneic haematopoietic stem cell transplantation for MNGIE."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
disease characteristics (liver disease, history of gastrointestinal
pseudo-obstruction or both)
explanation: >-
Gastrointestinal pseudo-obstruction is documented as a disease
characteristic in the international MNGIE transplant cohort.
- name: Cachexia
description: >-
Severe weight loss and cachexia result from chronic gastrointestinal
dysfunction and malnutrition and are major contributors to morbidity and
mortality.
phenotype_term:
preferred_term: Cachexia
term:
id: HP:0004326
label: Cachexia
clinical_course: PROGRESSIVE
frequency: VERY_FREQUENT
evidence:
- reference: PMID:20301358
reference_title: "Mitochondrial Neurogastrointestinal Encephalopathy Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
cachexia; ptosis/ophthalmoplegia or ophthalmoparesis; leukoencephalopathy;
and demyelinating peripheral neuropathy
explanation: >-
GeneReviews lists cachexia among the cardinal features of MNGIE.
- name: Ptosis
description: >-
Drooping of the upper eyelid is a common, often early, ocular sign,
frequently accompanying ophthalmoplegia.
phenotype_term:
preferred_term: Ptosis
term:
id: HP:0000508
label: Ptosis
frequency: VERY_FREQUENT
evidence:
- reference: PMID:20301358
reference_title: "Mitochondrial Neurogastrointestinal Encephalopathy Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
cachexia; ptosis/ophthalmoplegia or ophthalmoparesis; leukoencephalopathy;
and demyelinating peripheral neuropathy
explanation: >-
GeneReviews lists ptosis among the cardinal features of MNGIE.
- name: Progressive external ophthalmoplegia
description: >-
Chronic progressive external ophthalmoplegia / ophthalmoparesis, with
impaired ocular motility, is part of the classic MNGIE phenotype.
phenotype_term:
preferred_term: Progressive external ophthalmoplegia
term:
id: HP:0000590
label: Progressive external ophthalmoplegia
clinical_course: PROGRESSIVE
frequency: VERY_FREQUENT
evidence:
- reference: PMID:32173240
reference_title: "Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Characteristic clinical findings of patients with MNGIE include
gastrointestinal dysmotility leading to malnutrition and cachexia,
peripheral neuropathy, skeletal myopathy, progressive external
ophthalmoparesis (PEO), and ptosis
explanation: >-
Documents progressive external ophthalmoparesis as a characteristic
finding in MNGIE.
- name: Demyelinating peripheral neuropathy
description: >-
A demyelinating sensorimotor peripheral neuropathy manifests as paresthesias
(tingling, numbness, pain) and symmetric distal weakness more prominently
affecting the lower extremities.
phenotype_term:
preferred_term: Demyelinating peripheral neuropathy
term:
id: HP:0007108
label: Demyelinating peripheral neuropathy
clinical_course: PROGRESSIVE
frequency: VERY_FREQUENT
evidence:
- reference: PMID:20301358
reference_title: "Mitochondrial Neurogastrointestinal Encephalopathy Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
demyelinating peripheral neuropathy (manifesting as paresthesias
(tingling, numbness, and pain) and symmetric and distal weakness more
prominently affecting the lower extremities)
explanation: >-
GeneReviews characterizes the neuropathy in MNGIE as demyelinating and
sensorimotor.
- name: Leukoencephalopathy
description: >-
Diffuse leukoencephalopathy is observed on brain MRI as widespread
T2-hyperintense / T1-hypointense white-matter abnormality, characteristically
extensive yet often clinically asymptomatic with relative cognitive sparing.
phenotype_term:
preferred_term: Leukoencephalopathy
term:
id: HP:0002352
label: Leukoencephalopathy
frequency: VERY_FREQUENT
evidence:
- reference: PMID:20301358
reference_title: "Mitochondrial Neurogastrointestinal Encephalopathy Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
asymptomatic leukoencephalopathy as observed on brain MRI
explanation: >-
GeneReviews documents asymptomatic leukoencephalopathy on brain MRI as a
diagnostic feature.
- name: Multiple mitochondrial DNA deletions
description: >-
Skeletal muscle and other tissues show secondary mtDNA abnormalities,
including multiple deletions, point mutations, and depletion, reflecting the
downstream consequence of dNTP-pool imbalance.
phenotype_term:
preferred_term: Multiple mitochondrial DNA deletions
term:
id: HP:0003689
label: Multiple mitochondrial DNA deletions
evidence:
- reference: PMID:9924029
reference_title: "Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
an autosomal recessive human disease associated with multiple deletions of
skeletal muscle mitochondrial DNA (mtDNA)
explanation: >-
Documents multiple mtDNA deletions in skeletal muscle as a defining
molecular feature of MNGIE.
- name: Skeletal myopathy
description: >-
A skeletal myopathy with muscle weakness and exercise intolerance reflects
secondary mitochondrial dysfunction; muscle biopsy may show ragged-red and
cytochrome c oxidase-deficient fibers.
phenotype_term:
preferred_term: Skeletal myopathy
term:
id: HP:0003198
label: Myopathy
evidence:
- reference: PMID:32173240
reference_title: "Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Characteristic clinical findings of patients with MNGIE include
gastrointestinal dysmotility leading to malnutrition and cachexia,
peripheral neuropathy, skeletal myopathy, progressive external
ophthalmoparesis (PEO), and ptosis
explanation: >-
Documents skeletal myopathy as a characteristic finding in MNGIE.
- name: Increased CSF protein concentration
category: Laboratory
description: >-
Cerebrospinal fluid protein is often elevated, a supportive though
nonspecific finding in the diagnostic workup.
phenotype_term:
preferred_term: Increased CSF protein concentration
term:
id: HP:0002922
label: Increased CSF protein concentration
evidence:
- reference: PMID:30373120
reference_title: "Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1)."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
it is essential that clinicians be warned about the clinical features and
diagnostic procedures useful to suspect diagnosis of MNGIE-MTDPS1
explanation: >-
The Filosto review covers the diagnostic features of MNGIE; elevated CSF
protein is a recognized supportive finding. Evidence marked PARTIAL because
the abstract does not quantify the CSF abnormality directly.
biochemical:
- name: Elevated plasma thymidine
presence: INCREASED
notes: >-
The disease-defining biochemical abnormality is systemic accumulation of
thymidine, with a characteristic plasma threshold greater than 3 micromol/L;
healthy individuals have very low or undetectable levels.
biomarker_term:
preferred_term: thymidine
term:
id: CHEBI:17748
label: thymidine
evidence:
- reference: PMID:30959750
reference_title: "Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in MNGIE."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
plasma thymidine and deoxyuridine concentrations greater than 3 µmol/L and
5 µmol/L, respectively
explanation: >-
Establishes the diagnostic plasma thymidine threshold used to confirm
MNGIE.
- name: Elevated plasma 2'-deoxyuridine
presence: INCREASED
notes: >-
Plasma 2'-deoxyuridine is systemically elevated, with a characteristic
threshold greater than 5 micromol/L; together with elevated thymidine it
forms the disease-defining biochemical signature.
biomarker_term:
preferred_term: 2'-deoxyuridine
term:
id: CHEBI:16450
label: 2'-deoxyuridine
evidence:
- reference: PMID:30959750
reference_title: "Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in MNGIE."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
plasma thymidine and deoxyuridine concentrations greater than 3 µmol/L and
5 µmol/L, respectively
explanation: >-
Establishes the diagnostic plasma 2'-deoxyuridine threshold used to confirm
MNGIE.
genetic:
- name: TYMP pathogenic variants
gene_term:
preferred_term: TYMP
term:
id: hgnc:3148
label: TYMP
association: Causative
relationship_type: CAUSATIVE
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:20301358
reference_title: "Mitochondrial Neurogastrointestinal Encephalopathy Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
MNGIE disease is inherited in an autosomal recessive manner. The parents
of an affected individual are obligate heterozygotes and therefore carry
one mutated allele; heterozygotes are asymptomatic.
explanation: >-
GeneReviews establishes autosomal recessive inheritance for TYMP-related
MNGIE.
features: >-
Biallelic (homozygous or compound-heterozygous) loss-of-function variants in
TYMP (formerly ECGF1), encoding thymidine phosphorylase, cause classic
MNGIE. The gene is located on chromosome 22q13.32-qter.
evidence:
- reference: PMID:9924029
reference_title: "Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Examination of 12 MNGIE probands revealed homozygous or
compound-heterozygous mutations in the gene specifying thymidine
phosphorylase (TP), located on chromosome 22q13.32-qter.
explanation: >-
Establishes biallelic TYMP variants as the cause of classic MNGIE.
- name: POLG variants (MNGIE-like)
gene_term:
preferred_term: POLG
term:
id: hgnc:9179
label: POLG
association: Causative
relationship_type: CAUSATIVE
subtype: MNGIE-like POLG
features: >-
Mutations in POLG, the catalytic subunit of mitochondrial DNA polymerase
gamma, underlie a rare MNGIE-type phenotype distinct from classic
TYMP-related disease.
evidence:
- reference: PMID:30373120
reference_title: "Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Other two MNGIE-type phenotypes have been described so far, which are
linked to mutations in POLG and RRM2B genes.
explanation: >-
Documents POLG as a cause of an MNGIE-type phenotype.
- name: RRM2B variants (MNGIE-like)
gene_term:
preferred_term: RRM2B
term:
id: hgnc:17296
label: RRM2B
association: Causative
relationship_type: CAUSATIVE
subtype: MNGIE-like RRM2B
features: >-
Mutations in RRM2B, the p53-inducible small subunit of ribonucleotide
reductase required for mitochondrial dNTP supply, underlie a rare MNGIE-type
phenotype distinct from classic TYMP-related disease.
evidence:
- reference: PMID:30373120
reference_title: "Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Other two MNGIE-type phenotypes have been described so far, which are
linked to mutations in POLG and RRM2B genes.
explanation: >-
Documents RRM2B as a cause of an MNGIE-type phenotype.
treatments:
- name: Allogeneic hematopoietic stem cell transplantation
description: >-
Allogeneic HSCT replaces deficient thymidine phosphorylase via
donor-derived hematopoietic cells, restoring TP enzyme activity and lowering
circulating thymidine and deoxyuridine. In an international retrospective
cohort, TP activity rose from undetectable to normal in all survivors and
long-term engrafted patients showed clinical improvement, but
transplant-related mortality is high, so it is reserved for selected
patients with an optimal donor and is most beneficial before advanced
irreversible damage.
therapeutic_modality: CELL_THERAPY
treatment_term:
preferred_term: allogeneic hematopoietic stem cell transplantation
term:
id: MAXO:0001479
label: allogeneic hematopoietic stem cell transplantation
evidence:
- reference: PMID:26264513
reference_title: "Allogeneic haematopoietic stem cell transplantation for MNGIE."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Allogeneic haematopoietic stem cell transplantation can restore thymidine
phosphorylase enzyme function in patients with mitochondrial
neurogastrointestinal encephalomyopathy and improve clinical
manifestations of mitochondrial neurogastrointestinal encephalomyopathy in
the long term.
explanation: >-
Demonstrates that allogeneic HSCT restores TP enzyme function and improves
clinical manifestations long term.
- reference: PMID:26264513
reference_title: "Allogeneic haematopoietic stem cell transplantation for MNGIE."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
Overall, 9 of 24 patients (37.5%) were alive at last follow-up with a
median follow-up of these surviving patients of 1430 days.
explanation: >-
Documents the substantial transplant-related mortality that limits HSCT to
carefully selected patients.
- name: Liver transplantation
description: >-
Because the liver is a major systemic source of thymidine phosphorylase,
orthotopic liver transplantation supplies TP and lowers toxic plasma
nucleosides. In a series of four additional patients, plasma thymidine nearly
normalized in all and symptoms stabilized, with a more favorable safety
profile than HSCT, especially when liver disease is already present.
therapeutic_modality: SURGERY
treatment_term:
preferred_term: liver transplantation
term:
id: MAXO:0001175
label: liver transplantation
evidence:
- reference: PMID:32173240
reference_title: "Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Following LT, thymidine levels nearly normalized in all four patients and
remained low for the duration of follow-up. Disease symptoms stabilized in
all patients, with some manifesting improvements, including intestinal
function. No patient died, and LT appeared to have a more favorable safety
profile than HSCT
explanation: >-
Demonstrates near-normalization of thymidine and symptom stabilization
after liver transplantation with a favorable safety profile.
- name: Erythrocyte-encapsulated thymidine phosphorylase (EE-TP)
description: >-
EE-TP is an experimental enzyme-replacement therapy in which the patient's
autologous erythrocytes are loaded ex vivo with E. coli thymidine
phosphorylase; circulating loaded red cells metabolize thymidine and
deoxyuridine taken up via the equilibrative nucleoside transporter ENT1. In a
three-patient compassionate-use study, EE-TP was well tolerated and reduced
plasma thymidine and deoxyuridine in all patients, with clinical improvement
in two. There are no approved therapies for MNGIE; a planned phase 2 trial
(NCT03866954) was withdrawn before enrollment.
therapeutic_modality: PROTEIN_REPLACEMENT
treatment_term:
preferred_term: enzyme replacement or supplementation therapy
term:
id: MAXO:0000933
label: enzyme replacement or supplementation therapy
evidence:
- reference: PMID:30959750
reference_title: "Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in MNGIE."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
EE-TP was well tolerated and reductions in the disease-associated plasma
metabolites, thymidine, and deoxyuridine were observed in all three
patients. Clinical improvements, including weight gain and improved disease
scores, were observed in two patients
explanation: >-
Demonstrates biochemical and clinical efficacy of EE-TP enzyme-replacement
therapy in MNGIE.
- name: Peritoneal dialysis / hemodialysis
description: >-
Dialysis physically removes circulating thymidine and deoxyuridine to
transiently reduce the nucleoside burden, but the metabolites rapidly
reaccumulate, so dialysis is at best a temporizing supportive measure
without durable neurologic benefit.
therapeutic_modality: OTHER
treatment_term:
preferred_term: peritoneal dialysis
term:
id: MAXO:0000603
label: peritoneal dialysis
evidence:
- reference: PMID:30959750
reference_title: "Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in MNGIE."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
a number of experimental therapeutic approaches have been investigated,
which either directly remove the deoxyribonucleosides (haemodialysis and
peritoneal dialysis) or introduce the deficient thymidine phosphorylase
explanation: >-
Documents dialysis as an experimental approach to directly remove the toxic
deoxyribonucleosides; benefit is transient.
- name: Nutritional support (parenteral / enteral nutrition)
description: >-
Supportive management of cachexia and gastrointestinal failure includes
gastrostomy and parenteral/enteral feeding for nutritional support, together
with antibiotics for intestinal bacterial overgrowth and neuropathic-pain
agents.
therapeutic_modality: OTHER
treatment_term:
preferred_term: total parenteral nutrition intake
term:
id: MAXO:0000114
label: total parenteral nutrition intake
evidence:
- reference: PMID:20301358
reference_title: "Mitochondrial Neurogastrointestinal Encephalopathy Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
gastrostomy, and parenteral feeding for nutritional support; antibiotics
for intestinal bacterial overgrowth
explanation: >-
GeneReviews recommends gastrostomy and parenteral feeding for nutritional
support in MNGIE.
- name: Gene therapy (investigational)
description: >-
Hematopoietic stem cell gene therapy and AAV/liver-directed gene-therapy
approaches aim to restore endogenous TYMP/TP activity and normalize
nucleoside levels without allogeneic-donor risk. These remain preclinical /
investigational, with efficacy and safety demonstrated in Tymp/Upp1
double-knockout mouse models but no human efficacy data yet.
therapeutic_modality: GENE_THERAPY
treatment_term:
preferred_term: gene therapy
term:
id: MAXO:0001001
label: gene therapy
evidence:
- reference: PMID:30373120
reference_title: "Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1)."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
newer, promising therapies are expected in the near future
explanation: >-
The Filosto review notes emerging therapies, including gene-therapy
approaches, are anticipated; these remain investigational. Marked PARTIAL
because the abstract does not report human gene-therapy outcomes.
clinical_trials:
- name: NCT03866954
phase: PHASE_II
status: WITHDRAWN
description: >-
Open-label phase 2 trial of erythrocyte-encapsulated thymidine phosphorylase
(EE-TP) in MNGIE, sponsored by St George's, University of London. Withdrawn
before enrollment due to a change of circumstances with the commercial
partner.
target_phenotypes:
- preferred_term: Gastrointestinal dysmotility
term:
id: HP:0002579
label: Gastrointestinal dysmotility
evidence:
- reference: clinicaltrials:NCT03866954
reference_title: "The Safety, Tolerability, Pharmacodynamics, and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase (EE-TP) in Patients With MNGIE"
supports: SUPPORT
snippet: >-
The purpose of this study is to determine the safety, tolerability, action
and effectiveness of repeated doses of Erythrocyte Encapsulated Thymidine
Phosphorylase (EE-TP) for the treatment of patients with Mitochondrial
Neurogastrointestinal Encephalomyopathy (MNGIE).
explanation: >-
Registered phase 2 trial of EE-TP enzyme-replacement therapy for MNGIE.
prevalence:
- population: General population
notes: >-
MNGIE is an ultra-rare disorder. Affected individuals have a markedly reduced
life expectancy, with premature death reported at an average age of about
37.6 years.
evidence:
- reference: PMID:30959750
reference_title: "Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in MNGIE."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
It causes relentless and progressive morbidity, followed by premature
death at an average age of 37.6 years
explanation: >-
Documents the ultra-rare, progressive, and frequently fatal natural
history of MNGIE.
datasets: []
References & Deep Research
References
1Deep Research
11. Disease Information
1.1 Concise overview (definition)
MNGIE is an ultra‑rare, progressive autosomal recessive metabolic mitochondrial disorder most commonly caused by biallelic TYMP variants leading to thymidine phosphorylase (TP) deficiency, systemic accumulation of thymidine (dThd) and 2′‑deoxyuridine (dUrd), and downstream secondary mitochondrial DNA (mtDNA) instability (multiple deletions, depletion, point mutations) with multisystem gastrointestinal and neurologic disease. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3, bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
A frequently cited disease description emphasizes the canonical clinical constellation: gastrointestinal dysmotility and cachexia together with peripheral neuropathy, ophthalmoplegia/ptosis, and leukoencephalopathy. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
1.2 Key identifiers and controlled vocabulary
- MONDO: MONDO:0017575 (OpenTargets Search: Mitochondrial neurogastrointestinal encephalomyopathy)
- MeSH / ICD-10 / ICD-11 / Orphanet / OMIM: Not retrievable in this tool run (gap).
1.3 Synonyms and alternative names
Commonly used alternative names include: - “MNGIE” - “mitochondrial neurogastrointestinal encephalopathy” (abbreviated identically as MNGIE in some sources) - “MNGIE‑MTDPS1” (MNGIE as a mitochondrial DNA depletion syndrome subtype terminology used in some clinical genetics literature) (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, OpenTargets Search: Mitochondrial neurogastrointestinal encephalomyopathy)
1.4 Structured identifiers/nomenclature summary
| Disease name | Synonyms / alternative names | MONDO ID | Associated gene(s) | Inheritance | Key defining biomarker thresholds | Notes |
|---|---|---|---|---|---|---|
| Mitochondrial neurogastrointestinal encephalomyopathy | MNGIE; mitochondrial neurogastrointestinal encephalopathy; MNGIE-MTDPS1 | MONDO:0017575 | TYMP (primary causal gene); MNGIE-like phenotypes also reported with POLG, RRM2B, LIG3 | Autosomal recessive | Plasma thymidine >3 µmol/L; plasma 2'-deoxyuridine >5 µmol/L | Canonical MNGIE is the TYMP-related form with thymidine phosphorylase deficiency and systemic thymidine/deoxyuridine accumulation; healthy individuals typically have plasma thymidine and deoxyuridine <0.05 µmol/L (OpenTargets Search: Mitochondrial neurogastrointestinal encephalomyopathy, bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3, bax2020mitochondrialneurogastrointestinalencephalomyopathy media 7e94c311) |
| Mitochondrial neurogastrointestinal encephalomyopathy (TYMP-associated form) | TYMP-related MNGIE; thymidine phosphorylase deficiency MNGIE | MONDO:0017575 | TYMP | Autosomal recessive | Same defining plasma thresholds: thymidine >3 µmol/L, deoxyuridine >5 µmol/L | TYMP encodes thymidine phosphorylase; disease-defining biochemistry reflects loss of TP activity and nucleoside accumulation (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3) |
| MNGIE-like phenotype | MNGIE-like disease; mitochondrial DNA depletion syndrome, MNGIE type | MONDO:0030696* | POLG, RRM2B, LIG3 reported in evidence context | Usually autosomal recessive in reported examples | No single TYMP-style biomarker threshold established in the provided evidence context | Distinct from classic TYMP-associated MNGIE; included because the evidence context explicitly notes genetically heterogeneous MNGIE-like phenotypes (OpenTargets Search: Mitochondrial neurogastrointestinal encephalomyopathy, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3) |
Table: This table summarizes the core disease naming conventions, MONDO mapping, causal genes, inheritance, and hallmark plasma biomarker thresholds for classic TYMP-associated MNGIE and related MNGIE-like entities.
2. Etiology
2.1 Disease causal factors (genetic and mechanistic)
Primary cause (classic MNGIE): biallelic loss‑of‑function variants in TYMP (thymidine phosphorylase), causing TP deficiency and systemic accumulation of dThd and dUrd. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3)
MNGIE-like phenotypes: Similar neurogastrointestinal/encephalopathic presentations can also be caused by defects in other mtDNA maintenance genes (e.g., POLG, RRM2B), and OpenTargets also links LIG3 to an “MNGIE type” mtDNA depletion syndrome concept (MONDO:0030696). (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, OpenTargets Search: Mitochondrial neurogastrointestinal encephalomyopathy)
2.2 Risk factors
- Genetic: autosomal recessive inheritance implies risk is concentrated in individuals with biallelic pathogenic TYMP variants; heterozygous carriers have partial TP activity (reported ~35% in reviews) without detectable plasma deoxyribonucleoside accumulation. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
- Environmental/lifestyle: No specific environmental risk factors or gene–environment interactions were identified in the retrieved evidence.
2.3 Protective factors
No validated protective genetic or environmental factors were identified in the retrieved evidence.
3. Phenotypes
3.1 Core phenotype spectrum (with HPO suggestions)
The canonical phenotype includes: - Gastrointestinal dysmotility that can resemble chronic intestinal pseudo‑obstruction, and contributes to severe nutritional compromise. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3) - Cachexia/weight loss and progressive denutrition. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3) - Peripheral neuropathy (often sensorimotor). (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6) - Ophthalmoplegia/ophthalmoparesis and ptosis. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3) - Diffuse leukoencephalopathy on brain MRI, often extensive while cognition may be relatively preserved. (halter2015allogeneichaematopoieticstem pages 2-3, bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6) - Hearing loss is also reported in some syntheses (frequency reported as 61% in one source in this evidence set). (yadak2017lentiviralhematopoieticstem pages 21-25)
A structured phenotype-to-HPO scaffold is provided here: | Clinical phenotype | Phenotype type | Suggested HPO term(s) | Typical onset/course | Brief notes | Supporting evidence snippet | |---|---|---|---|---|---| | Gastrointestinal dysmotility / chronic intestinal pseudo-obstruction (CIPO)-like disease | Symptom/sign | HP:0002579 Intestinal pseudo-obstruction; HP:0002014 Diarrhea; HP:0002017 Nausea; HP:0002015 Dysphagia; HP:0002027 Abdominal pain; HP:0002242 Vomiting | Usually begins in childhood, adolescence, or early adulthood; progressive and often severe | Hallmark feature of MNGIE; includes dysmotility, recurrent pseudo-obstruction, abdominal pain, vomiting, diarrhea, dysphagia, and severe nutritional compromise | “progressive gastrointestinal dysmotility”; “CIPO-like”; “gastrointestinal and ocular involvement are usually initial features” | | Cachexia / severe weight loss | Symptom/sign | HP:0004326 Cachexia; HP:0001824 Weight loss; HP:0001510 Growth delay | Progressive over years; often worsens with GI disease | Major contributor to morbidity and mortality; may require enteral or parenteral nutritional support | “cachexia”; “severe denutrition”; “4 kg weight gain” after treatment indicates baseline wasting | | Peripheral neuropathy | Clinical sign / electrophysiologic abnormality | HP:0009830 Peripheral neuropathy; HP:0003401 Sensory neuropathy; HP:0003323 Peripheral axonal neuropathy; HP:0001270 Motor delay/weakness-related terms as applicable | Typically develops by teens to early adulthood; progressive | Often sensorimotor, causing paresthesias, weakness, gait impairment; supported by NCS/EMG | “peripheral neuropathy”; “stocking-glove paresthesia and weakness”; “electrophysiologically confirmed peripheral neuropathy” | | External ophthalmoplegia / ophthalmoparesis | Clinical sign | HP:0000602 Ophthalmoplegia; HP:0000601 Impaired ocular motility | Common early neurologic feature; slowly progressive | Part of the classic phenotype; may accompany ptosis and chronic progressive external ophthalmoplegia-like presentation | “ophthalmoplegia”; “external ophthalmoparesis”; “CPEO” | | Ptosis | Clinical sign | HP:0000508 Ptosis | Often early; progressive | Frequently co-occurs with ophthalmoplegia and may be a presenting clue | “ptosis” | | Diffuse leukoencephalopathy on brain MRI | Imaging abnormality | HP:0002352 Leukoencephalopathy; HP:0002500 Abnormal cerebral white matter morphology | Often present by time of diagnosis; usually progressive radiologically but may be clinically silent | Characteristic MRI pattern with white-matter T2/FLAIR hyperintensities; cognition often relatively spared | “diffuse leukoencephalopathy”; “MRI shows leukoencephalopathy while cognition is generally spared” | | Hearing loss / sensorineural deafness | Symptom/sign | HP:0000407 Sensorineural hearing impairment; HP:0000365 Hearing impairment | Variable; can appear early or later in disease course | Not universal but repeatedly reported; may broaden suspicion beyond classic tetrad | “hearing loss”; “deafness”; “reported in 61% of patients” | | Skeletal myopathy / exercise intolerance / weakness | Symptom/sign / pathology-supported feature | HP:0003323 Myopathy; HP:0001324 Muscle weakness; HP:0003546 Exercise intolerance | Progressive, often alongside neuropathy | Reflects secondary mitochondrial dysfunction; may show ragged-red or COX-deficient fibers on muscle biopsy | “skeletal myopathy”; “improved strength and mobility”; “ragged red fibers” | | Elevated CSF protein | Laboratory abnormality | HP:0012116 Abnormal cerebrospinal fluid protein level; HP:0002922 Increased CSF protein | Can be detected during workup; not necessarily symptomatic | Helpful supportive biomarker in differential diagnosis | “CSF protein is often elevated (typically 60–100 mg/dL)” | | Lactic acidosis / elevated lactate | Laboratory abnormality | HP:0003128 Lactic acidosis; HP:0011013 Abnormality of metabolic homeostasis | Variable; may fluctuate with disease burden | Supports mitochondrial dysfunction but is not specific to MNGIE | “lactic acidosis”; “elevated lactate” | | Cognitive preservation despite MRI abnormalities | Clinical characteristic | HP:0000729 Autistic behavior not applicable; no precise HPO for preserved cognition; consider phenotype note only | Often persistent across course | Important distinguishing feature: marked white-matter disease may occur with limited overt encephalopathy | “cognition is generally spared” | | Mitochondrial muscle pathology | Pathology finding | HP:0003200 Rigid spine not applicable; use pathology note with HP:0008322 Abnormal muscle biopsy | Appears in established disease | Muscle biopsy may show ragged-red fibers, COX-deficient fibers, and mtDNA deletions/depletion | “ragged red fibers, cytochrome c oxidase-deficient fibers”; “mtDNA deletions/depletions” | | Gastrointestinal bacterial overgrowth / nutritional failure complications | Complication | HP:0011968 Malnutrition; HP:0004395 Small intestinal bacterial overgrowth | Usually later-stage or secondary to severe dysmotility | Contributes to poor quality of life and treatment complexity | “small intestinal bacterial overgrowth”; need for “TPN” / nutritional support |
Table: This table summarizes the main clinical and laboratory phenotypes reported for MNGIE, with suggested HPO mappings and concise notes on onset and progression. It is useful as a phenotype-to-ontology scaffold for disease knowledge base curation.
3.2 Temporal phenotype characteristics (onset, severity, progression)
- Typical onset: often in the second or third decade, although “first signs often appear in childhood/adolescence.” (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3)
- Course: relentlessly progressive over 2–4 decades, with severe GI complications and cachexia prominent as causes of morbidity/mortality. (halter2015allogeneichaematopoieticstem pages 2-3)
3.3 Quality of life impact
The disease’s GI dysmotility, cachexia, neuropathy, and progressive multisystem disability commonly necessitate intensive supportive care (e.g., tube feeding or parenteral nutrition) and leads to substantial functional impairment; clinical trial endpoints explicitly include BMI, quality of life, and function (EQ‑5D, PROMIS GI) reflecting the major QoL burden. (NCT03866954 chunk 2, NCT06784453 chunk 2)
4. Genetic / Molecular Information
4.1 Causal genes and molecular defect
- Causal gene (classic MNGIE): TYMP (encodes thymidine phosphorylase, TP). (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3)
- Mechanistic biochemical defect: TP deficiency leads to systemic accumulation of thymidine and 2′‑deoxyuridine, which perturbs mitochondrial dNTP pools and drives mtDNA replication/repair defects with mtDNA depletion and multiple deletions. (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3)
A review summarizing TP’s role notes that TP “normally catabolizes dThd and dUrd” and that TP deficiency causes “dNTP pool imbalance with mtDNA depletion, multiple deletions, and point mutations.” (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3)
4.2 Pathogenic variants and variant spectrum
- A diagnostic review notes that HGMD listed 97 reported TYMP mutations (at the time of that review). (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
- A 2024 case series reported a novel TYMP variant c.877T>C (p.Cys293Arg) and additionally estimated “550 number of cases of MNGIE … published … from 1983–2023,” providing a literature-derived case-count statistic (not prevalence). (yadak2017lentiviralhematopoieticstem pages 25-27)
4.3 Modifier genes / epigenetics / chromosomal abnormalities
No robust modifier-gene, epigenetic, or chromosomal abnormality evidence specific to MNGIE was identified in the retrieved sources.
5. Environmental Information
No disease-specific infectious, toxin, or lifestyle etiologies were identified in the retrieved evidence; MNGIE is primarily a genetic metabolic disease driven by TYMP deficiency. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3)
6. Mechanism / Pathophysiology
6.1 Canonical causal chain (upstream → downstream)
- Upstream trigger: biallelic TYMP variants → TP deficiency. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3)
- Metabolic consequence: systemic accumulation of dThd/dUrd (circulating and tissue). (halter2015allogeneichaematopoieticstem pages 2-3, bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
- Mitochondrial genome consequence: imbalance of intramitochondrial dNTP pools → mtDNA replication defects with site‑specific point mutations, multiple deletions, and depletion. (halter2015allogeneichaematopoieticstem pages 2-3)
- Cellular/tissue consequence: progressive OXPHOS dysfunction and multisystem manifestations (GI dysmotility, neuropathy, ophthalmoplegia, leukoencephalopathy, cachexia). (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3)
A visual overview of the core biochemical defect and nucleoside accumulation is captured in Figure 1 of Bax 2020 (cropped). (bax2020mitochondrialneurogastrointestinalencephalomyopathy media 7e94c311)
Suggested GO biological process terms (examples): mitochondrial DNA replication; mitochondrial DNA metabolic process; pyrimidine nucleoside metabolic process; nucleoside catabolic process.
Suggested GO cellular component term: mitochondrion.
6.2 Recent developments (2024): lysosomal dysfunction as an additional mechanistic layer
A 2024 mechanistic study expanded the pathophysiology beyond mtDNA instability, proposing that TP deficiency also induces lysosomal dysfunction and organelle cross‑talk disruption: - Patient muscle showed reduced LAMP1 and increased mitochondrial content. - Patient fibroblasts showed decreased LAMP2, lowered lysosomal acidity, reduced lysosomal enzyme activity, and impaired protein degradation. - TYMP knockout or TP inhibition reproduced similar lysosomal defects. - Lysosome immunoprecipitation (Lyso‑IP) indicated accumulation of nucleosides within lysosomes and changes in lysosomal proteome (e.g., decreased V‑ATPase components), suggesting impaired acidification and nucleoside overload as a driver of downstream mitochondrial homeostasis disruption. (du2024lysosomaldysfunctionand pages 1-3, du2024lysosomaldysfunctionand pages 4-6)
This constitutes a notable 2024 advance because it frames MNGIE as involving widespread organelle disruption (lysosome–mitochondria axis) rather than being solely explained by mtDNA replication stress. (du2024lysosomaldysfunctionand pages 1-3)
Suggested GO terms (examples): lysosome organization; lysosomal acidification; autophagy; mitophagy.
Suggested CL cell types (examples): enteric neuron; Schwann cell; skeletal muscle fiber cell; fibroblast.
7. Anatomical Structures Affected
7.1 Organ and system level
- Digestive system: small intestine and broader GI tract dysmotility is central (pseudo-obstruction–like) and drives malnutrition/cachexia. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3)
- Peripheral nervous system: peripheral neuropathy. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
- Central nervous system: diffuse cerebral white matter involvement (leukoencephalopathy) on MRI, often with relative cognitive sparing. (halter2015allogeneichaematopoieticstem pages 2-3)
- Eye/extraocular muscles: ptosis and ophthalmoparesis. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3)
7.2 Tissue/cell level and pathology
- Skeletal muscle biopsy may show ragged-red fibers and cytochrome c oxidase‑deficient fibers, with mtDNA deletions/depletion as downstream consequences. (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, du2024lysosomaldysfunctionand pages 4-6)
- The 2024 mechanistic study implicates lysosomal defects in patient fibroblasts and muscle, suggesting broader cellular compartment involvement. (du2024lysosomaldysfunctionand pages 1-3)
Suggested UBERON terms (examples): small intestine; peripheral nerve; brain white matter; skeletal muscle.
Suggested GO cellular component terms: lysosome; mitochondrion.
8. Temporal Development
8.1 Onset and progression
- Diagnosis is typically made in the 2nd–3rd decade, but signs may begin earlier (childhood/adolescence). (halter2015allogeneichaematopoieticstem pages 2-3)
- Disease progresses over decades and is “relentlessly progressive,” with death commonly due to cachexia and infections. (halter2015allogeneichaematopoieticstem pages 2-3)
8.2 Disease course patterns
The disease generally follows a progressive rather than relapsing course; key complications (GI failure, infections) emerge with advancing malnutrition and disability. (halter2015allogeneichaematopoieticstem pages 2-3)
9. Inheritance and Population
9.1 Inheritance
Classic MNGIE is autosomal recessive due to biallelic TYMP variants. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3)
9.2 Epidemiology
Robust prevalence/incidence estimates were not found in the retrieved sources. However, a 2024 report performed a literature search and estimated ~550 published cases from 1983–2023, which is a useful statistic for rarity characterization but is not a population prevalence estimate. (yadak2017lentiviralhematopoieticstem pages 25-27)
9.3 Penetrance/expressivity
- Expressivity appears variable; biochemical severity correlates with TP activity in some sources (e.g., <10% typical MNGIE; 10–20% milder/late onset), though exceptions exist. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9)
10. Diagnostics
10.1 Biochemical and enzymatic diagnostics (key statistics)
A key diagnostic review provides quantitative thresholds: - Healthy plasma thymidine and deoxyuridine are typically <0.05 µmol/L; MNGIE patients often have thymidine >3 µmol/L and 2′‑deoxyuridine >5 µmol/L. (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6) - Leukocyte TP activity is markedly reduced in patients (0–46 nmol thymidine formed/hour/mg protein) compared with controls (253–1000), supporting enzyme deficiency as an orthogonal diagnostic confirmation. (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
10.2 Imaging and electrophysiology
- Brain MRI typically shows diffuse leukoencephalopathy (T2 hyperintensity, T1 hypointensity). (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
- EMG/NCS supports peripheral neuropathy as part of the diagnostic workup. (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
10.3 Consolidated diagnostics table
| Diagnostic modality | What to test / finding | Specimen / source | Typical MNGIE result / threshold | Comparator / notes | Suggested ontology terms | Citation |
|---|---|---|---|---|---|---|
| Genetic testing | TYMP sequencing (biallelic pathogenic variants) | Genomic DNA from blood or other clinical specimen | Diagnostic benchmark is identification of homozygous or compound heterozygous TYMP variants | Primary causal gene for classic MNGIE; MNGIE-like phenotypes also reported with POLG and RRM2B | Gene: TYMP; MONDO: MONDO_0017575 | (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, OpenTargets Search: Mitochondrial neurogastrointestinal encephalomyopathy) |
| Plasma nucleosides | Thymidine (dThd) and deoxyuridine (dUrd) quantification | Plasma | dThd >3 µmol/L and dUrd >5 µmol/L are characteristic; in some series patients have ~10–20 µM plasma nucleosides | Healthy individuals: both typically <0.05 µmol/L | CHEBI: thymidine, deoxyuridine | (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, bax2020mitochondrialneurogastrointestinalencephalomyopathy media 7e94c311) |
| Urine nucleosides | Thymidine and deoxyuridine quantification | Urine / 24 h urine | Elevated urinary dThd/dUrd; reported diagnostic clues include urine dThd >3 µmol/L and dUrd >5 µmol/L | Can be followed longitudinally for treatment response | CHEBI: thymidine, deoxyuridine | (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9, yadak2017lentiviralhematopoieticstem pages 19-21) |
| Enzyme assay | Thymidine phosphorylase (TP) activity | Buffy coat leukocytes / leukocytes | Markedly reduced: reported 0–46 nmol thymidine formed/h/mg protein or <8–10% of controls | Controls reported 253–1000 nmol/h/mg; one source cites control mean ~634 nmol thymine formed/h/mg protein; heterozygotes may retain ~35% activity | GO: thymidine phosphorylase activity; Protein: TP | (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9, yadak2017lentiviralhematopoieticstem pages 19-21, yadak2017mitochondrialneurogastrointestinalencephalomyopathy pages 2-3) |
| Neuroimaging | Brain MRI | Brain white matter | Diffuse leukoencephalopathy; typically T1 hypointense and T2 hyperintense white-matter abnormalities | Often extensive and may be clinically asymptomatic | UBERON: brain white matter; SNOMED/term suggestion: leukoencephalopathy | (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, halter2015allogeneichaematopoieticstem pages 2-3, yadak2017lentiviralhematopoieticstem pages 21-25) |
| Electrodiagnostics | EMG / nerve conduction studies (NCS) | Peripheral nerves / muscle | Supports peripheral neuropathy; electrophysiologically confirmed neuropathy is a common diagnostic feature | Used as part of workup with clinical suspicion | UBERON: peripheral nerve, skeletal muscle | (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, NCT01694953 chunk 1) |
| Muscle biopsy / histopathology | Ragged-red fibers, COX-deficient fibers, mtDNA abnormalities | Skeletal muscle biopsy | Mitochondrial pathology may show ragged-red fibers, cytochrome c oxidase-deficient fibers, and mtDNA deletions/depletion | Supportive rather than strictly required if biochemical/genetic diagnosis is established | UBERON: skeletal muscle; GO: mitochondrion; GO: mitochondrial DNA metabolic process | (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, yadak2017lentiviralhematopoieticstem pages 21-25, du2024lysosomaldysfunctionand pages 4-6) |
| mtDNA analysis in muscle | mtDNA deletions / depletion testing | Skeletal muscle DNA | Secondary mitochondrial genome defects including multiple deletions, point mutations, and mtDNA depletion | Reflect downstream consequence of nucleoside imbalance rather than primary inherited mtDNA mutation | GO: mitochondrial DNA replication; GO: mitochondrion | (halter2015allogeneichaematopoieticstem pages 2-3, yadak2017lentiviralhematopoieticstem pages 25-27, du2024lysosomaldysfunctionand pages 4-6) |
| CSF analysis | Cerebrospinal fluid protein | CSF | Often elevated; typically 60–100 mg/dL | Supportive but nonspecific | UBERON: cerebrospinal fluid | (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9, yadak2017lentiviralhematopoieticstem pages 21-25) |
| Analytical platform notes | Nucleoside measurement methodology | Plasma / urine | Reported methods include HPLC-UV and LC-MS/MS/UPLC approaches for dThd/dUrd | Useful for reproducible biomarker monitoring in diagnosis and follow-up | (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, levene2020safetyandefficacy pages 9-14) |
Table: This table summarizes the principal diagnostic modalities for MNGIE, including genetic confirmation, hallmark nucleoside biomarkers, enzyme deficiency testing, imaging, electrophysiology, biopsy findings, and supportive CSF abnormalities. It is useful as a concise disease-knowledge-base artifact linking each test to specimen type, expected results, and ontology suggestions.
11. Outcome / Prognosis
11.1 Survival statistics
- Mean/median life expectancy is frequently cited around ~37 years (“mean age of death ~37 years” in a review; “median life expectancy … 37 years” in an HSCT outcomes paper). (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3)
11.2 Complications and morbidity
- GI complications, severe malnutrition/cachexia, and infections are key contributors to mortality. (halter2015allogeneichaematopoieticstem pages 2-3)
11.3 Prognostic factors (evidence-limited)
Multiple sources emphasize that earlier diagnosis and intervention before irreversible GI/neuromuscular damage improves the plausibility of benefit from disease-modifying approaches. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9)
12. Treatment
12.1 Current applications and real-world implementations
The therapeutic landscape includes:
12.1.1 Liver transplantation (LT)
A 2020 case series supports LT as a disease-modifying strategy based on the liver being a major source of TP: - Pediatric case: pre‑LT plasma thymidine 25.61 µmol/L and deoxyuridine 25.9 µmol/L, with post‑LT levels “dropped to near normal levels,” alongside marked clinical improvement (diet tolerance, no evidence of GI dysmotility on follow‑up, stable MRI changes). (kripps2020successfullivertransplantation pages 2-3) - Across patients: thymidine reductions reported as >20×, 11×, 8×, and 24×, and post‑LT nucleosides could normalize to <0.7 µmol/L within 1–3 days and remain near‑normal in follow‑up intervals (months). (kripps2020successfullivertransplantation pages 4-5, kripps2020successfullivertransplantation pages 5-6) - Complications included infections (e.g., CMV viremia, pneumonia), thrombosis, metabolic issues (hypertriglyceridemia, diabetes), and immunosuppression-related events. (kripps2020successfullivertransplantation pages 4-5)
12.1.2 Allogeneic hematopoietic stem cell transplantation (HSCT)
An international HSCT series (Brain, 2015) positions HSCT as an attempt to provide TP through donor leukocytes/platelets: - 26 patients started conditioning; 24 evaluable transplants in the series excerpt. (halter2015allogeneichaematopoieticstem pages 2-3) - Reported survival in a retrospective series summarized elsewhere in the evidence set: 9/24 (37.5%) alive at last follow‑up (median follow‑up 1430 days), and 7/24 (29%) survived >2 years with reported clinical improvements, but transplant-related mortality is high and clinical benefit is uncertain in advanced disease. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9, halter2015allogeneichaematopoieticstem pages 2-3)
12.1.3 Erythrocyte-encapsulated thymidine phosphorylase (EE‑TP)
A compassionate-use/open-label evaluation in three adults used ex vivo loading of recombinant TP into autologous erythrocytes, with quantitative dosing and biomarker response: - Dose levels: 4, 9, 18, 29, 47, 108 U/kg (typically every 4 weeks; later some dosing every 2 weeks). (levene2020safetyandefficacy pages 5-9, levene2020safetyandefficacy pages 14-18) - Biomarker improvements: example Patient 1 plasma thymidine from 10 µmol/L to 2–6 µmol/L and deoxyuridine 20 → 3–13 µmol/L; urinary nucleosides fell substantially (e.g., thymidine 73 → 0–41 µmol/24h; deoxyuridine 118 → 0–49 µmol/24h). (levene2020safetyandefficacy pages 14-18) - Clinical improvements in some participants included weight gain and functional improvements (e.g., MRC motor score 56 → 74 by 23 months in one patient). (levene2020safetyandefficacy pages 18-25) - Safety: infusion reactions occurred in 2/3; overall described as tolerable with manageable AEs, but the approach requires repeated infusions and manufacturing logistics. (levene2020safetyandefficacy pages 18-25, levene2020safetyandefficacy pages 5-9)
12.2 Temporizing metabolic removal / bridging therapies
- Hemodialysis / CAPD: transiently reduce nucleosides but metabolites rapidly reaccumulate and durable neurologic benefit is limited. (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9)
- Platelet infusions: transient TP restoration with short-lived biomarker reduction. (halter2015allogeneichaematopoieticstem pages 2-3)
12.3 Experimental/advanced therapeutics
- Gene therapy (preclinical): reviews describe AAV-based and hematopoietic stem cell gene therapy approaches tested in mouse models (Tymp−/−Upp1−/−), but no human efficacy data were present in the retrieved context. (yadak2017mitochondrialneurogastrointestinalencephalomyopathy pages 2-3, yadak2017lentiviralhematopoieticstem pages 19-21)
12.4 Consolidated treatment table
| Modality | Mechanism / rationale | Key evidence / outcomes with quantitative data | Risks / limitations | Real-world status | Suggested MAXO term(s) |
|---|---|---|---|---|---|
| Allogeneic hematopoietic stem cell transplantation (HSCT) | Replaces deficient thymidine phosphorylase (TP) via donor-derived hematopoietic cells/platelets/leukocytes, aiming to clear circulating thymidine and deoxyuridine and restore nucleoside homeostasis (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) | International retrospective series: 26 patients started conditioning; 24 evaluable transplants; 9/24 (37.5%) alive at last follow-up (median follow-up 1430 days), and 7/24 (29%) survived >2 years with reported clinical improvements; biochemical correction can be achieved, but short-term intestinal pathology may not improve (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) | High transplant-related mortality; one mismatch in unrelated donor noted to increase mortality risk by ~9%; benefit appears greatest before advanced irreversible GI damage; intestinal neuropathology/Cajal cell loss may persist (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) | Clinical practice in selected centers for carefully chosen patients | |
| Liver transplantation (orthotopic LT) | Liver is a major systemic source of TP; graft supplies TP and lowers toxic plasma nucleosides, potentially with lower procedure-related mortality than HSCT (kripps2020successfullivertransplantation pages 2-3, kripps2020successfullivertransplantation pages 1-2) | Case series of 4 additional patients: plasma thymidine nearly normalized in all and remained low on follow-up; example pediatric case had pre-LT thymidine 25.61 umol/L and deoxyuridine 25.9 umol/L, dropping to near-normal after LT; another case had TP 6 nmol/mg/h pre-LT with thymidine 6.63 umol/L and deoxyuridine 11.17 umol/L, falling post-LT to 1.01 and 0.59 umol/L; reported thymidine reductions >20x, 11x, 8x, and 24x across four patients; symptom stabilization in all, with some improvements in intestinal function, mobility, cardiac function, and weight gain; no patient died in this series (kripps2020successfullivertransplantation pages 2-3, kripps2020successfullivertransplantation pages 4-5, kripps2020successfullivertransplantation pages 5-6, kripps2020successfullivertransplantation pages 1-2, kripps2020successfullivertransplantation pages 3-4) | Surgical and immunosuppression risks; complications reported included CMV viremia, pneumonia, bacteremia, thrombosis, rejection/immunosuppression-related diarrhea, hypertension, diabetes, hypertriglyceridemia, GI bleeding; some neurologic deficits and GI dysmotility may stabilize rather than reverse (kripps2020successfullivertransplantation pages 4-5, kripps2020successfullivertransplantation pages 5-6) | Clinical practice in limited expert centers; increasingly considered for suitable patients | |
| Erythrocyte-encapsulated thymidine phosphorylase (EE-TP) | Autologous erythrocytes are loaded ex vivo with TP; circulating loaded RBCs metabolize thymidine/deoxyuridine taken up via nucleoside transporters, extending enzyme exposure while reducing free-enzyme immunogenicity (levene2020safetyandefficacy pages 1-5, levene2020safetyandefficacy pages 9-14) | Open-label study in 3 adults: dose levels 4, 9, 18, 29, 47, and 108 U/kg every 4 weeks; Patient 1 received 31 cycles over 28 months, Patient 2 received 79 cycles over 76 months, Patient 3 escalated to 108 U/kg every 4 weeks; doses >=4 U/kg reduced urinary nucleosides in all patients; higher doses produced better plasma reductions, often below diagnostic thresholds within cycle; Patient 1 plasma thymidine fell from 10 to 2-6 umol/L and deoxyuridine from 20 to 3-13 umol/L; urinary thymidine/deoxyuridine fell from 73/118 umol/24h to 0-41/0-49 umol/24h; Patient 3 had plasma thymidine/deoxyuridine fall from 12/19 umol/L to <4/2 umol/L by ~60 days; clinical gains included weight gain, improved SF-36, improved gait/balance/fine finger function, and MRC score increase from 56 to 74 in one patient; 51Cr-labelled cells showed mean RBC life 108 days and half-life 32 days (levene2020safetyandefficacy pages 27-31, levene2020safetyandefficacy pages 14-18, levene2020safetyandefficacy pages 18-25, levene2020safetyandefficacy pages 5-9, levene2020safetyandefficacy pages 9-14) | Repeated lifelong infusions likely required; infusion reactions occurred in 2/3 patients; one patient developed progressive disease despite treatment and later died after cessation; biomarker rebound may occur; manufacturing/logistics are complex; phase 2 trial was withdrawn for commercial reasons before enrollment (levene2020safetyandefficacy pages 14-18, levene2020safetyandefficacy pages 18-25, NCT03866954 chunk 1) | Investigational / compassionate-use; no approved therapy identified in evidence | |
| Hemodialysis / continuous ambulatory peritoneal dialysis (CAPD) | Physical removal of circulating toxic nucleosides from blood/peritoneal compartment to transiently reduce thymidine/deoxyuridine burden (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) | Hemodialysis reported to transiently lower serum/urine nucleosides but without CSF or neurological benefit after 1 year; CAPD protocol reported for 22 months with 1000 mL exchanges, giving transient clinical benefit with relapse by ~15 months; reviews conclude nucleosides rapidly reaccumulate after dialysis (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) | Transient effect only; poor durability because metabolites reaccumulate quickly; limited impact on neurologic disease or advanced tissue pathology (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) | Supportive / temporizing measure, not definitive disease-modifying therapy | |
| Platelet infusions | Platelets are TP-rich and can transiently restore circulating TP activity, lowering nucleosides short-term (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) | Reported to produce transient TP restoration and short-lived reductions in nucleosides; considered proof-of-principle that enzyme replacement from blood components can work, but not durable enough as long-term standalone treatment (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) | Very short-lived benefit; repeated transfusions would be needed; not a durable correction strategy (halter2015allogeneichaematopoieticstem pages 2-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) | Experimental / occasional bridge strategy rather than standard long-term care | |
| Gene therapy (preclinical: hematopoietic stem cell gene therapy / AAV approaches) | Restores TYMP/TP activity using genetically modified cells or viral vectors; intended to provide sustained endogenous TP and normalize nucleoside levels without allogeneic donor risks (yadak2017lentiviralhematopoieticstem pages 19-21, yadak2017lentiviralhematopoieticstem pages 25-27) | Preclinical studies in Tymp-/- Upp1-/- mouse models showed efficacy/safety for hematopoietic stem cell gene therapy, and reviews describe AAV-based and lentiviral approaches as promising emerging strategies; no human efficacy trial results in provided evidence; 2024-2025 literature also notes ongoing interest in liver-directed/AAV concepts (yadak2017lentiviralhematopoieticstem pages 19-21, yadak2017lentiviralhematopoieticstem pages 25-27, yadak2017mitochondrialneurogastrointestinalencephalomyopathy pages 2-3) | Preclinical only in provided evidence; translational hurdles include vector delivery, conditioning requirements, durability, and safety; no clinical outcome data yet in provided context (yadak2017lentiviralhematopoieticstem pages 19-21, yadak2017lentiviralhematopoieticstem pages 25-27, yadak2017mitochondrialneurogastrointestinalencephalomyopathy pages 2-3) | Preclinical / investigational |
Table: This table summarizes disease-modifying and investigational treatment strategies for MNGIE, including mechanisms, quantitative outcomes, limitations, and current clinical status. It is useful for comparing which approaches have real-world clinical use versus those that remain experimental.
12.5 MAXO (Medical Action Ontology) mapping
MAXO terms were not available in the retrieved evidence context; therefore, MAXO mappings are left blank in the treatment artifact and should be added during ontology-curation using a MAXO reference.
13. Prevention
No primary prevention (e.g., vaccine) exists because MNGIE is genetic. Prevention focuses on: - Secondary prevention: early recognition and prompt genetic/biochemical diagnosis to enable early disease-modifying intervention consideration (LT/HSCT/EE‑TP) before irreversible damage. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) - Genetic counseling/cascade screening: implied by autosomal recessive inheritance and carrier state; not explicitly detailed in the retrieved evidence.
14. Other Species / Natural Disease
No naturally occurring non-human disease analogs were identified in the retrieved sources.
15. Model Organisms
Preclinical model systems include TYMP-deficient murine models (e.g., Tymp−/−Upp1−/−) used for gene therapy exploration, as noted in reviews; detailed phenotyping and model limitations were not extracted in the current evidence set. (yadak2017mitochondrialneurogastrointestinalencephalomyopathy pages 2-3, yadak2017lentiviralhematopoieticstem pages 19-21)
Recent developments and current research infrastructure (2020–2026)
Mechanistic advances (prioritized 2024)
- 2024 mechanistic primary research implicates lysosomal dysfunction and intralysosomal nucleoside overload as part of the disease process, supporting a broader organelle disruption framework (lysosome–mitochondria axis). (du2024lysosomaldysfunctionand pages 1-3)
Active/recent trials and real-world studies (ClinicalTrials.gov)
A structured trial summary is provided here: | NCT ID | Title | Study type | Status | Sponsor | Start date | Planned enrollment | Key endpoints | Notes | |---|---|---|---|---|---|---:|---|---| | NCT03866954 | Trial of Erythrocyte Encapsulated Thymidine Phosphorylase In Mitochondrial Neurogastrointestinal Encephalomyopathy | Interventional, Phase 2, open-label, multi-centre, sequential | Withdrawn | St George's, University of London; collaborator: Neovii Biotech | Estimated 2024-11 | 12 | Safety/tolerability; plasma and urine thymidine/deoxyuridine pharmacodynamics; efficacy measures including BMI; additional outcomes included anti-TP antibodies, TPN use, handgrip strength, RODS, 10 m walk, CGI-I, EQ-5D, PROMIS GI | EE-TP via autologous erythrocytes loaded with E. coli TP; dosing every 3 weeks until metabolic correction then every 2-4 weeks for planned 24 months plus 90-day follow-up; actual enrollment 0; withdrawn due to change of circumstances with commercial partner (NCT03866954 chunk 1, NCT03866954 chunk 2) | | NCT07627217 | MNGIE Natural History Study | Observational, retrospective natural history study/registry | Recruiting | University of Cambridge; collaborator: University of Bologna | 2026-03-17 | 50 | Primary: survival time at enrollment; additional data capture for genotype-phenotype subgroups, biomarkers/outcome measures, demographics, HPO-coded history, investigations, and prior treatments | International retrospective REDCap-based study; estimated primary/completion 2027-07-31; records prior EE-TP, HSCT, transplant, and dialysis exposures (NCT07627217 chunk 1, NCT07627217 chunk 2) | | NCT01694953 | The Natural History Study of Mitochondrial NeuroGastroIntestinal Encephalopathy (MNGIE) | Observational, multicenter, prospective cohort | Recruiting | Columbia University | 2013-02 | 20 | Primary: Timed Water Swallow; secondary: Degree of Neuropathy; other outcomes include MMSE, GI function, lean body mass, neuropsychological capability, quality of life, nutrition, motor function, biochemical parameters | Follow-up every 6 months for up to 5 years; includes patients age >=5 years with TYMP/TP defect and elevated plasma thymidine/deoxyuridine (NCT01694953 chunk 1) | | NCT04245917 | Natural History Study of MNGIE | Observational, prospective, multicenter, non-interventional | Suspended | Entrada Therapeutics, Inc. | 2020-08-06 | 60 | Primary: “MNGIE Clinical Course” over 5 years | Suspended for business reason; primary completion estimated 2025-02 and study completion estimated 2026-02 (NCT04245917 chunk 1) | | NCT06784453 | Evolution of Nutritional Status and Intestinal Function in Patients With MNGIE | Observational cohort | Completed | IRCCS Azienda Ospedaliero-Universitaria di Bologna | Not stated in extracted context | 8 | Primary nutritional-status measures: BMI, body composition, serum albumin; serial intestinal assessments including SIBO testing, fecal calprotectin, endoscopy, abdominal ultrasound, hepatic elastometry | Real-world natural history/supportive care study in genetically confirmed TYMP cases; records nutritional interventions and marrow/liver transplant exposures; follow-up every 4 months and at last follow-up/death (NCT06784453 chunk 2) |
Table: This table summarizes active or recent ClinicalTrials.gov studies relevant to MNGIE, including the withdrawn EE-TP interventional trial and major natural history cohorts. It is useful for identifying current research infrastructure, planned endpoints, and practical notes such as enrollment targets, follow-up duration, and reasons for suspension or withdrawal.
Key points: - A Phase 2 EE‑TP trial (NCT03866954) was withdrawn with 0 enrollment due to commercial circumstances, despite a detailed dosing plan and multi-domain outcomes (safety, PD nucleosides, BMI and function/QoL). (NCT03866954 chunk 1, NCT03866954 chunk 2) - Natural history efforts exist across academic (Columbia NCT01694953; Cambridge NCT07627217) and industry-sponsored (Entrada NCT04245917, suspended) programs, emphasizing biomarker/outcome measure discovery and trial readiness. (NCT01694953 chunk 1, NCT07627217 chunk 1, NCT04245917 chunk 1)
Expert opinions and analysis (authoritative synthesis)
Across authoritative reviews and clinical series, expert consensus themes include: 1. MNGIE is one of the few mitochondrial disorders where the molecular abnormality is metabolically accessible (circulating nucleosides), enabling monitoring and therapeutic manipulation. (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6) 2. Earlier intervention is critical, because irreversible GI/neuromuscular pathology may limit clinical recovery even when biochemical correction is achieved (e.g., HSCT biochemical correction with persistent intestinal pathology noted in related pathology work summarized in a review). (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9) 3. Transplant-based strategies (LT, HSCT) and enzyme replacement via EE‑TP represent leading disease-modifying approaches, but each has significant limitations (procedure risks and access for transplant; logistics/repeat dosing for EE‑TP). (kripps2020successfullivertransplantation pages 1-2, halter2015allogeneichaematopoieticstem pages 2-3, levene2020safetyandefficacy pages 18-25)
Key quantitative statistics (selected)
- Diagnostic plasma thresholds: thymidine >3 µmol/L, deoxyuridine >5 µmol/L (healthy typically <0.05 µmol/L). (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6, filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3)
- Leukocyte TP activity: patients 0–46 nmol/h/mg vs controls 253–1000 nmol/h/mg. (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
- Typical diagnosis window: 2nd–3rd decade, with childhood/adolescent onset of first signs. (halter2015allogeneichaematopoieticstem pages 2-3)
- Life expectancy: ~37 years (mean/median reported). (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3, halter2015allogeneichaematopoieticstem pages 2-3)
- HSCT outcomes (summarized): 9/24 (37.5%) alive at last follow-up; 7/24 (29%) alive >2 years with reported clinical improvements. (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9)
- LT biochemical response: post-LT thymidine and deoxyuridine can fall to near normal; example post-LT values include thymidine 1.01 µmol/L and deoxyuridine 0.59 µmol/L in one case; thymidine reductions up to >20×. (kripps2020successfullivertransplantation pages 4-5, kripps2020successfullivertransplantation pages 5-6)
- EE-TP dosing: 4–108 U/kg per 4-week cycles (with some later every-2-week dosing), producing measurable plasma/urine nucleoside reductions and functional improvements in some patients. (levene2020safetyandefficacy pages 5-9, levene2020safetyandefficacy pages 14-18)
- Literature case-count estimate: ~550 published cases (1983–2023). (yadak2017lentiviralhematopoieticstem pages 25-27)
Limitations and gaps
- OMIM/Orphanet/ICD and MeSH identifiers were not accessible with the current tool set.
- Robust prevalence/incidence estimates were not found in the retrieved evidence (case-count in literature is not a prevalence measure).
- MAXO term mapping and detailed LOINC/SNOMED mappings require dedicated ontology resources beyond the current evidence.
URLs and publication dates (selected high-value sources in this evidence set)
- Bax BE. Mitochondrial neurogastrointestinal encephalomyopathy: approaches to diagnosis and treatment. Mar 2020. https://doi.org/10.20517/jtgg.2020.08 (bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6)
- Filosto M, et al. MNGIE (MNGIE-MTDPS1). Oct 2018. https://doi.org/10.3390/jcm7110389 (filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3)
- Halter JP, et al. Allogeneic haematopoietic stem cell transplantation for MNGIE. Oct 2015. https://doi.org/10.1093/brain/awv226 (halter2015allogeneichaematopoieticstem pages 2-3)
- Kripps KA, et al. Successful liver transplantation in MNGIE. May 2020. https://doi.org/10.1016/j.ymgme.2020.03.001 (kripps2020successfullivertransplantation pages 2-3)
- Du J, et al. Lysosomal dysfunction and overload of nucleosides in thymidine phosphorylase deficiency of MNGIE. May 2024. https://doi.org/10.1186/s12967-024-05275-8 (du2024lysosomaldysfunctionand pages 1-3)
- ClinicalTrials.gov: NCT03866954 (EE‑TP Phase 2; withdrawn). Posted 2019; record version referenced 2024. (NCT03866954 chunk 1)
- ClinicalTrials.gov: NCT01694953 (Columbia natural history; started 2013-02; recruiting). (NCT01694953 chunk 1)
- ClinicalTrials.gov: NCT07627217 (Cambridge retrospective registry; start 2026-03-17; recruiting). (NCT07627217 chunk 1)
References
-
(OpenTargets Search: Mitochondrial neurogastrointestinal encephalomyopathy): Open Targets Query (Mitochondrial neurogastrointestinal encephalomyopathy, 12 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 1-3): Massimiliano Filosto, Stefano Cotti Piccinelli, Filomena Caria, Serena Gallo Cassarino, Enrico Baldelli, Anna Galvagni, Irene Volonghi, Mauro Scarpelli, and Alessandro Padovani. Mitochondrial neurogastrointestinal encephalomyopathy (mngie-mtdps1). Journal of Clinical Medicine, 7:389, Oct 2018. URL: https://doi.org/10.3390/jcm7110389, doi:10.3390/jcm7110389. This article has 66 citations.
-
(halter2015allogeneichaematopoieticstem pages 2-3): Joerg P. Halter, W. Michael, M. Schüpbach, Hanna Mandel, Carlo Casali, Kim Orchard, Matthew Collin, David Valcarcel, Attilio Rovelli, Massimiliano Filosto, Maria T. Dotti, Giuseppe Marotta, Guillem Pintos, Pere Barba, Anna Accarino, Christelle Ferra, Isabel Illa, Yves Beguin, Jaap A. Bakker, Jaap J. Boelens, Irenaeus F. M. de Coo, Keith Fay, Carolyn M. Sue, David Nachbaur, Heinz Zoller, Claudia Sobreira, Belinda Pinto Simoes, Simon R. Hammans, David Savage, Ramon Martí, Patrick F. Chinnery, Ronit Elhasid, Alois Gratwohl, and Michio Hirano. Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain : a journal of neurology, 138 Pt 10:2847-58, Oct 2015. URL: https://doi.org/10.1093/brain/awv226, doi:10.1093/brain/awv226. This article has 168 citations.
-
(bax2020mitochondrialneurogastrointestinalencephalomyopathy pages 4-6): Bridget E. Bax. Mitochondrial neurogastrointestinal encephalomyopathy: approaches to diagnosis and treatment. Journal of translational genetics and genomics, 4:1-16, Mar 2020. URL: https://doi.org/10.20517/jtgg.2020.08, doi:10.20517/jtgg.2020.08. This article has 39 citations.
-
(bax2020mitochondrialneurogastrointestinalencephalomyopathy media 7e94c311): Bridget E. Bax. Mitochondrial neurogastrointestinal encephalomyopathy: approaches to diagnosis and treatment. Journal of translational genetics and genomics, 4:1-16, Mar 2020. URL: https://doi.org/10.20517/jtgg.2020.08, doi:10.20517/jtgg.2020.08. This article has 39 citations.
-
(yadak2017lentiviralhematopoieticstem pages 21-25): Rana Yadak. Lentiviral hematopoietic stem cell gene therapy for mngie. Text, Jan 2017. URL: https://doi.org/10.13140/rg.2.2.11110.52807, doi:10.13140/rg.2.2.11110.52807. This article has 0 citations and is from a peer-reviewed journal.
-
(NCT03866954 chunk 2): Trial of Erythrocyte Encapsulated Thymidine Phosphorylase In Mitochondrial Neurogastrointestinal Encephalomyopathy. St George's, University of London. 2024. ClinicalTrials.gov Identifier: NCT03866954
-
(NCT06784453 chunk 2): Evolution of Nutritional Status and Intestinal Function in Patients With MNGIE. IRCCS Azienda Ospedaliero-Universitaria di Bologna. 2020. ClinicalTrials.gov Identifier: NCT06784453
-
(yadak2017lentiviralhematopoieticstem pages 25-27): Rana Yadak. Lentiviral hematopoietic stem cell gene therapy for mngie. Text, Jan 2017. URL: https://doi.org/10.13140/rg.2.2.11110.52807, doi:10.13140/rg.2.2.11110.52807. This article has 0 citations and is from a peer-reviewed journal.
-
(du2024lysosomaldysfunctionand pages 1-3): Jixiang Du, Fuchen Liu, Xihan Liu, Dandan Zhao, Dongdong Wang, Hongsheng Sun, Chuanzhu Yan, and Yuying Zhao. Lysosomal dysfunction and overload of nucleosides in thymidine phosphorylase deficiency of mngie. Journal of Translational Medicine, May 2024. URL: https://doi.org/10.1186/s12967-024-05275-8, doi:10.1186/s12967-024-05275-8. This article has 6 citations and is from a peer-reviewed journal.
-
(du2024lysosomaldysfunctionand pages 4-6): Jixiang Du, Fuchen Liu, Xihan Liu, Dandan Zhao, Dongdong Wang, Hongsheng Sun, Chuanzhu Yan, and Yuying Zhao. Lysosomal dysfunction and overload of nucleosides in thymidine phosphorylase deficiency of mngie. Journal of Translational Medicine, May 2024. URL: https://doi.org/10.1186/s12967-024-05275-8, doi:10.1186/s12967-024-05275-8. This article has 6 citations and is from a peer-reviewed journal.
-
(filosto2018mitochondrialneurogastrointestinalencephalomyopathy pages 7-9): Massimiliano Filosto, Stefano Cotti Piccinelli, Filomena Caria, Serena Gallo Cassarino, Enrico Baldelli, Anna Galvagni, Irene Volonghi, Mauro Scarpelli, and Alessandro Padovani. Mitochondrial neurogastrointestinal encephalomyopathy (mngie-mtdps1). Journal of Clinical Medicine, 7:389, Oct 2018. URL: https://doi.org/10.3390/jcm7110389, doi:10.3390/jcm7110389. This article has 66 citations.
-
(yadak2017lentiviralhematopoieticstem pages 19-21): Rana Yadak. Lentiviral hematopoietic stem cell gene therapy for mngie. Text, Jan 2017. URL: https://doi.org/10.13140/rg.2.2.11110.52807, doi:10.13140/rg.2.2.11110.52807. This article has 0 citations and is from a peer-reviewed journal.
-
(yadak2017mitochondrialneurogastrointestinalencephalomyopathy pages 2-3): Rana Yadak, Peter Sillevis Smitt, Marike W. van Gisbergen, Niek P. van Til, and Irenaeus F. M. de Coo. Mitochondrial neurogastrointestinal encephalomyopathy caused by thymidine phosphorylase enzyme deficiency: from pathogenesis to emerging therapeutic options. Frontiers in Cellular Neuroscience, Feb 2017. URL: https://doi.org/10.3389/fncel.2017.00031, doi:10.3389/fncel.2017.00031. This article has 65 citations.
-
(NCT01694953 chunk 1): The Natural History Study of Mitochondrial NeuroGastroIntestinal Encephalopathy (MNGIE). Columbia University. 2013. ClinicalTrials.gov Identifier: NCT01694953
-
(levene2020safetyandefficacy pages 9-14): Michelle Levene, Murray D Bain, Nicholas F Moran, Niran Nirmalananthan, Joanna Poulton, Mauro Scarpelli, Massimiliano Filosto, Hanna Mandel, Andrew D MacKinnon, Lynette Fairbanks, Dario Pacitti, and Bridget E Bax. Safety and efficacy of erythrocyte encapsulated thymidine phosphorylase in mitochondrial neurogastrointestinal encephalomyopathy. Prime Archives in Medicine, Jan 2020. URL: https://doi.org/10.37247/pamed.1.2020.2, doi:10.37247/pamed.1.2020.2. This article has 65 citations.
-
(kripps2020successfullivertransplantation pages 2-3): KimberlyA. Kripps, Warapan Nakayuenyongsuk, Brian J. Shayota, William Berquist, Natalia Gomez-Ospina, Carlos O. Esquivel, Waldo Concepcion, Jacinda B. Sampson, David J. Cristin, Whitney E. Jackson, Samuel Gilliland, Elizabeth A. Pomfret, Michael L. Kueht, Rowland W. Pettit, Youmna A. Sherif, Lisa T. Emrick, Sarah H. Elsea, Ryan Himes, Michio Hirano, Johan L.K. Van Hove, Fernando Scaglia, Gregory M. Enns, and Austin A. Larson. Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (mngie). May 2020. URL: https://doi.org/10.1016/j.ymgme.2020.03.001, doi:10.1016/j.ymgme.2020.03.001. This article has 43 citations and is from a peer-reviewed journal.
-
(kripps2020successfullivertransplantation pages 4-5): KimberlyA. Kripps, Warapan Nakayuenyongsuk, Brian J. Shayota, William Berquist, Natalia Gomez-Ospina, Carlos O. Esquivel, Waldo Concepcion, Jacinda B. Sampson, David J. Cristin, Whitney E. Jackson, Samuel Gilliland, Elizabeth A. Pomfret, Michael L. Kueht, Rowland W. Pettit, Youmna A. Sherif, Lisa T. Emrick, Sarah H. Elsea, Ryan Himes, Michio Hirano, Johan L.K. Van Hove, Fernando Scaglia, Gregory M. Enns, and Austin A. Larson. Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (mngie). May 2020. URL: https://doi.org/10.1016/j.ymgme.2020.03.001, doi:10.1016/j.ymgme.2020.03.001. This article has 43 citations and is from a peer-reviewed journal.
-
(kripps2020successfullivertransplantation pages 5-6): KimberlyA. Kripps, Warapan Nakayuenyongsuk, Brian J. Shayota, William Berquist, Natalia Gomez-Ospina, Carlos O. Esquivel, Waldo Concepcion, Jacinda B. Sampson, David J. Cristin, Whitney E. Jackson, Samuel Gilliland, Elizabeth A. Pomfret, Michael L. Kueht, Rowland W. Pettit, Youmna A. Sherif, Lisa T. Emrick, Sarah H. Elsea, Ryan Himes, Michio Hirano, Johan L.K. Van Hove, Fernando Scaglia, Gregory M. Enns, and Austin A. Larson. Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (mngie). May 2020. URL: https://doi.org/10.1016/j.ymgme.2020.03.001, doi:10.1016/j.ymgme.2020.03.001. This article has 43 citations and is from a peer-reviewed journal.
-
(levene2020safetyandefficacy pages 5-9): Michelle Levene, Murray D Bain, Nicholas F Moran, Niran Nirmalananthan, Joanna Poulton, Mauro Scarpelli, Massimiliano Filosto, Hanna Mandel, Andrew D MacKinnon, Lynette Fairbanks, Dario Pacitti, and Bridget E Bax. Safety and efficacy of erythrocyte encapsulated thymidine phosphorylase in mitochondrial neurogastrointestinal encephalomyopathy. Prime Archives in Medicine, Jan 2020. URL: https://doi.org/10.37247/pamed.1.2020.2, doi:10.37247/pamed.1.2020.2. This article has 65 citations.
-
(levene2020safetyandefficacy pages 14-18): Michelle Levene, Murray D Bain, Nicholas F Moran, Niran Nirmalananthan, Joanna Poulton, Mauro Scarpelli, Massimiliano Filosto, Hanna Mandel, Andrew D MacKinnon, Lynette Fairbanks, Dario Pacitti, and Bridget E Bax. Safety and efficacy of erythrocyte encapsulated thymidine phosphorylase in mitochondrial neurogastrointestinal encephalomyopathy. Prime Archives in Medicine, Jan 2020. URL: https://doi.org/10.37247/pamed.1.2020.2, doi:10.37247/pamed.1.2020.2. This article has 65 citations.
-
(levene2020safetyandefficacy pages 18-25): Michelle Levene, Murray D Bain, Nicholas F Moran, Niran Nirmalananthan, Joanna Poulton, Mauro Scarpelli, Massimiliano Filosto, Hanna Mandel, Andrew D MacKinnon, Lynette Fairbanks, Dario Pacitti, and Bridget E Bax. Safety and efficacy of erythrocyte encapsulated thymidine phosphorylase in mitochondrial neurogastrointestinal encephalomyopathy. Prime Archives in Medicine, Jan 2020. URL: https://doi.org/10.37247/pamed.1.2020.2, doi:10.37247/pamed.1.2020.2. This article has 65 citations.
-
(kripps2020successfullivertransplantation pages 1-2): KimberlyA. Kripps, Warapan Nakayuenyongsuk, Brian J. Shayota, William Berquist, Natalia Gomez-Ospina, Carlos O. Esquivel, Waldo Concepcion, Jacinda B. Sampson, David J. Cristin, Whitney E. Jackson, Samuel Gilliland, Elizabeth A. Pomfret, Michael L. Kueht, Rowland W. Pettit, Youmna A. Sherif, Lisa T. Emrick, Sarah H. Elsea, Ryan Himes, Michio Hirano, Johan L.K. Van Hove, Fernando Scaglia, Gregory M. Enns, and Austin A. Larson. Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (mngie). May 2020. URL: https://doi.org/10.1016/j.ymgme.2020.03.001, doi:10.1016/j.ymgme.2020.03.001. This article has 43 citations and is from a peer-reviewed journal.
-
(kripps2020successfullivertransplantation pages 3-4): KimberlyA. Kripps, Warapan Nakayuenyongsuk, Brian J. Shayota, William Berquist, Natalia Gomez-Ospina, Carlos O. Esquivel, Waldo Concepcion, Jacinda B. Sampson, David J. Cristin, Whitney E. Jackson, Samuel Gilliland, Elizabeth A. Pomfret, Michael L. Kueht, Rowland W. Pettit, Youmna A. Sherif, Lisa T. Emrick, Sarah H. Elsea, Ryan Himes, Michio Hirano, Johan L.K. Van Hove, Fernando Scaglia, Gregory M. Enns, and Austin A. Larson. Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (mngie). May 2020. URL: https://doi.org/10.1016/j.ymgme.2020.03.001, doi:10.1016/j.ymgme.2020.03.001. This article has 43 citations and is from a peer-reviewed journal.
-
(levene2020safetyandefficacy pages 1-5): Michelle Levene, Murray D Bain, Nicholas F Moran, Niran Nirmalananthan, Joanna Poulton, Mauro Scarpelli, Massimiliano Filosto, Hanna Mandel, Andrew D MacKinnon, Lynette Fairbanks, Dario Pacitti, and Bridget E Bax. Safety and efficacy of erythrocyte encapsulated thymidine phosphorylase in mitochondrial neurogastrointestinal encephalomyopathy. Prime Archives in Medicine, Jan 2020. URL: https://doi.org/10.37247/pamed.1.2020.2, doi:10.37247/pamed.1.2020.2. This article has 65 citations.
-
(levene2020safetyandefficacy pages 27-31): Michelle Levene, Murray D Bain, Nicholas F Moran, Niran Nirmalananthan, Joanna Poulton, Mauro Scarpelli, Massimiliano Filosto, Hanna Mandel, Andrew D MacKinnon, Lynette Fairbanks, Dario Pacitti, and Bridget E Bax. Safety and efficacy of erythrocyte encapsulated thymidine phosphorylase in mitochondrial neurogastrointestinal encephalomyopathy. Prime Archives in Medicine, Jan 2020. URL: https://doi.org/10.37247/pamed.1.2020.2, doi:10.37247/pamed.1.2020.2. This article has 65 citations.
-
(NCT03866954 chunk 1): Trial of Erythrocyte Encapsulated Thymidine Phosphorylase In Mitochondrial Neurogastrointestinal Encephalomyopathy. St George's, University of London. 2024. ClinicalTrials.gov Identifier: NCT03866954
-
(NCT07627217 chunk 1): Jelle van den Ameele. MNGIE Natural History Study. University of Cambridge. 2026. ClinicalTrials.gov Identifier: NCT07627217
-
(NCT07627217 chunk 2): Jelle van den Ameele. MNGIE Natural History Study. University of Cambridge. 2026. ClinicalTrials.gov Identifier: NCT07627217
-
(NCT04245917 chunk 1): Natural History Study of MNGIE. Entrada Therapeutics, Inc.. 2020. ClinicalTrials.gov Identifier: NCT04245917