Krabbe Disease Due To Saposin A Deficiency
Krabbe disease due to saposin A deficiency is a rare atypical Krabbe disease (globoid cell leukodystrophy) caused by biallelic PSAP variants that abolish saposin A, the lysosomal sphingolipid activator protein required by galactosylceramidase (GALC) to degrade galactosylceramide. Although the GALC gene is normal, the missing cofactor leads to galactosylceramide accumulation and progressive central and peripheral demyelination, producing a clinical picture almost identical to classic Krabbe disease.
Ask OpenScientist
Ask a research question about Krabbe Disease Due To Saposin A Deficiency. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Mappings
Definitions
1Show evidence (1 reference)
Inheritance
1Pathophysiology
3Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Pathograph
Phenotypes
5Musculoskeletal 1
Nervous System 3
Show evidence (1 reference)
Other 1
Show evidence (1 reference)
Genetic Associations
1Show evidence (1 reference)
Medical Actions
1Differential Diagnoses
1Conditions with similar clinical presentations that must be differentiated from Krabbe Disease Due To Saposin A Deficiency:
- Caused by GALC enzyme deficiency (deficient in both leukocytes and fibroblasts), whereas saposin A deficiency spares fibroblast enzyme activity.
Show evidence (1 reference)
Source YAML
click to showname: Krabbe Disease Due To Saposin A Deficiency
creation_date: "2026-06-13T00:00:00Z"
description: >-
Krabbe disease due to saposin A deficiency is a rare atypical Krabbe disease (globoid
cell leukodystrophy) caused by biallelic PSAP variants that abolish saposin A, the
lysosomal sphingolipid activator protein required by galactosylceramidase (GALC) to
degrade galactosylceramide. Although the GALC gene is normal, the missing cofactor leads
to galactosylceramide accumulation and progressive central and peripheral demyelination,
producing a clinical picture almost identical to classic Krabbe disease.
category: Mendelian
disease_term:
preferred_term: Krabbe disease due to saposin A deficiency
term:
id: MONDO:0012720
label: Krabbe disease due to saposin A deficiency
mappings:
mondo_mappings:
- term:
id: MONDO:0012720
label: Krabbe disease due to saposin A deficiency
mapping_predicate: skos:exactMatch
mapping_source: MONDO
mapping_justification: Primary MONDO disease identifier for this saposin A deficiency Krabbe disease entry.
synonyms:
- Saposin A deficiency
- Atypical Krabbe disease due to saposin A deficiency
- Globoid cell leukodystrophy due to saposin A deficiency
parents:
- Sphingolipidosis
- Lysosomal Storage Disorder
pathophysiology:
- name: Saposin A Deficiency from PSAP Variants
conforms_to: "lysosomal_substrate_accumulation#Lysosomal Hydrolase or Cofactor Deficiency"
description: >-
Saposin A arises from proteolytic cleavage of prosaposin (encoded by PSAP). Biallelic
PSAP variants affecting the saposin A domain abolish saposin A, the in vivo activator
that galactosylceramidase requires to degrade galactosylceramide. The GALC gene and
enzyme are intact; the defect is a missing activator cofactor.
gene:
preferred_term: PSAP
term:
id: hgnc:9498
label: PSAP
cell_types:
- preferred_term: oligodendrocyte
term:
id: CL:0000128
label: oligodendrocyte
evidence:
- reference: PMID:29995202
reference_title: "The Second Case of Saposin A Deficiency and Altered Autophagy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Both the enzyme galactosylceramidase and its in vivo activator molecule, saposin A, are essential during GalCer degradation."
explanation: "Galactosylceramidase requires the saposin A activator (from PSAP) to degrade galactosylceramide."
- reference: PMID:15773042
reference_title: "A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A three base pair deletion was found in the saposin A coding sequence of the prosaposin gene"
explanation: "A PSAP saposin A-domain deletion was the first reported human cause of saposin A deficiency."
downstream:
- target: Galactosylceramide Accumulation and Demyelination
description: Without the activator, galactosylceramide cannot be degraded and accumulates.
- name: Galactosylceramide Accumulation and Demyelination
conforms_to: "lysosomal_substrate_accumulation#Lysosomal Substrate Accumulation"
description: >-
Undegraded galactosylceramide accumulates and, with cytotoxic psychosine, drives
progressive central and peripheral demyelination (globoid cell leukodystrophy).
cell_types:
- preferred_term: oligodendrocyte
term:
id: CL:0000128
label: oligodendrocyte
cellular_components:
- preferred_term: lysosome
term:
id: GO:0005764
label: lysosome
biological_processes:
- preferred_term: galactosylceramide catabolic process
modifier: DECREASED
term:
id: GO:0006683
label: galactosylceramide catabolic process
evidence:
- reference: PMID:15773042
reference_title: "A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "sulphatide (stearoyl-1-14C) uptake indicated an abnormal storage of\ngalactosylceramide"
explanation: "Saposin A deficiency causes abnormal galactosylceramide storage."
downstream:
- target: Autophagic-Lysosomal Dysfunction
description: Substrate accumulation activates autophagy as a secondary cascade.
- name: Autophagic-Lysosomal Dysfunction
conforms_to: "lysosomal_substrate_accumulation#Autophagic-Lysosomal Dysfunction and Secondary Cascade"
description: >-
Galactosylceramide accumulation in patient fibroblasts is accompanied by activation of
autophagy, a secondary autophagic-lysosomal cascade documented for the first time in human
saposin A deficiency.
cell_types:
- preferred_term: fibroblast
term:
id: CL:0000057
label: fibroblast
biological_processes:
- preferred_term: autophagy
modifier: INCREASED
term:
id: GO:0006914
label: autophagy
evidence:
- reference: PMID:29995202
supports: SUPPORT
evidence_source: IN_VITRO
snippet: "Fibroblast studies showed GalCer accumulation and the activation of autophagy for the first time in a case of human saposin A deficiency"
explanation: "Autophagy activation is a documented secondary cascade in human saposin A deficiency."
phenotypes:
- name: Progressive encephalopathy
description: Progressive encephalopathy from leukodystrophy, presenting in infancy.
phenotype_term:
preferred_term: Progressive encephalopathy
term:
id: HP:0002448
label: Progressive encephalopathy
evidence:
- reference: PMID:15773042
reference_title: "A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A six-month-old infant girl presenting with progressive encephalopathy and"
explanation: "The first reported patient presented with progressive infantile encephalopathy."
- name: Leukodystrophy
description: Abnormal cerebral white matter myelination (globoid cell leukodystrophy).
phenotype_term:
preferred_term: Leukodystrophy
term:
id: HP:0002415
label: Leukodystrophy
evidence:
- reference: PMID:15773042
reference_title: "A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "abnormal myelination in the cerebral white matter"
explanation: "Abnormal cerebral white matter myelination (leukodystrophy) is the hallmark."
- name: Seizures
description: >-
Seizures are characteristic of the Krabbe-like infantile neurodegenerative course; a
specific quotable abstract snippet is not available for the two reported cases.
phenotype_term:
preferred_term: Seizure
term:
id: HP:0001250
label: Seizure
- name: Hypertonia
description: >-
Hypertonia/spasticity is typical of the Krabbe-like phenotype; a specific quotable
abstract snippet is not available for the two reported cases.
phenotype_term:
preferred_term: Hypertonia
term:
id: HP:0001276
label: Hypertonia
- name: Peripheral neuropathy
description: >-
Peripheral demyelinating neuropathy is expected from the globoid cell leukodystrophy
process; a specific quotable abstract snippet is not available for the two reported cases.
phenotype_term:
preferred_term: Peripheral neuropathy
term:
id: HP:0009830
label: Peripheral neuropathy
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
genetic:
- name: PSAP
association: Biallelic PSAP variants affecting the saposin A domain
relationship_type: CAUSATIVE
variant_origin: GERMLINE
gene_term:
preferred_term: PSAP
term:
id: hgnc:9498
label: PSAP
evidence:
- reference: PMID:15773042
reference_title: "A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A three base pair deletion was found in the saposin A coding sequence of the prosaposin gene"
explanation: "A PSAP saposin A-domain variant causes saposin A deficiency."
diagnosis:
- name: Galactosylceramidase assay pattern with PSAP sequencing
diagnosis_term:
preferred_term: clinical laboratory procedure
term:
id: MAXO:0000006
label: clinical laboratory procedure
description: >-
A Krabbe-like phenotype with deficient galactocerebrosidase activity in leukocytes but
normal activity in cultured fibroblasts implicates an activator (saposin A) defect;
diagnosis is confirmed by PSAP sequencing.
markers: Discordant leukocyte vs fibroblast galactocerebrosidase activity; abnormal galactosylceramide storage.
evidence:
- reference: PMID:15773042
reference_title: "A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "When cultured skin fibroblasts did not show a similar enzyme deficiency"
explanation: "Normal fibroblast enzyme activity despite a Krabbe-like phenotype points to an activator (saposin A) deficiency."
- name: PSAP molecular genetic testing
diagnosis_term:
preferred_term: genetic testing
term:
id: MAXO:0000127
label: genetic testing
description: Confirmatory biallelic PSAP sequencing of the saposin A domain.
evidence:
- reference: PMID:15773042
reference_title: "A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A three base pair deletion was found in the saposin A coding sequence of the prosaposin gene"
explanation: "PSAP sequencing identifies the causative saposin A-domain variant."
differential_diagnoses:
- name: Krabbe disease
description: >-
Classic globoid cell leukodystrophy caused by galactosylceramidase (GALC) enzyme
deficiency, with a clinically near-identical presentation.
disease_term:
preferred_term: Krabbe disease
term:
id: MONDO:0009499
label: Krabbe disease
distinguishing_features:
- Caused by GALC enzyme deficiency (deficient in both leukocytes and fibroblasts), whereas saposin A deficiency spares fibroblast enzyme activity.
evidence:
- reference: PMID:29995202
reference_title: "The Second Case of Saposin A Deficiency and Altered Autophagy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A clinical manifestation almost identical to Krabbe disease is observed when, instead of the galactosylceramidase protein, the saposin A molecule is defective."
explanation: "Saposin A deficiency is clinically near-identical to GALC-deficient Krabbe disease but mechanistically distinct."
treatments:
- name: Supportive Care
description: >-
No saposin-A-specific disease-modifying therapy is established; management is
supportive and symptomatic, as for classic Krabbe disease.
treatment_term:
preferred_term: Supportive Care
term:
id: NCIT:C15747
label: Supportive Care
definitions:
- name: Clinical case definition of saposin A deficiency Krabbe disease
definition_type: CASE_DEFINITION
description: >-
Krabbe disease due to saposin A deficiency is an atypical globoid cell leukodystrophy
defined by biallelic PSAP variants abolishing the saposin A activator, with
galactosylceramide accumulation and demyelination despite a normal GALC gene.
scope: Disease-level case definition for saposin A deficiency Krabbe disease.
evidence:
- reference: PMID:29995202
reference_title: "The Second Case of Saposin A Deficiency and Altered Autophagy."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A clinical manifestation almost identical to Krabbe disease is observed when, instead of the galactosylceramidase protein, the saposin A molecule is defective."
explanation: "Anchors the case definition in saposin A deficiency producing a Krabbe-like disease."
References & Deep Research
Deep Research
11. Disease Information
1.1 Concise overview
Krabbe disease (globoid cell leukodystrophy, GLD) is a lysosomal leukodystrophy characterized by progressive central and peripheral demyelination and neurodegeneration. While most cases are due to primary GALC deficiency, “in very rare cases” a Krabbe phenotype can be caused by lack of active saposin A, a necessary cofactor for GALC activity in vivo. (szymanska2012diagnosticdifficultiesin pages 11-11)
1.2 Key identifiers
- Saposin A deficiency: OMIM #611722 (kose2018thesecondcase pages 6-7)
- Krabbe disease / globoid cell leukodystrophy: OMIM #245200 (kose2018thesecondcase pages 6-7)
- MONDO / Orphanet / ICD-10/ICD-11 / MeSH: not confirmed from the retrieved sources; should be populated from external disease ontologies in a downstream curation step.
1.3 Synonyms / alternative names
- Saposin A deficiency (PSAP saposin A domain deficiency) (kose2018thesecondcase pages 6-7)
- Krabbe-like leukodystrophy due to saposin A deficiency (concept supported by the Krabbe-phenotype description) (kose2018thesecondcase pages 1-3, szymanska2012diagnosticdifficultiesin pages 11-11)
- Globoid cell leukodystrophy (Krabbe disease) phenotype due to saposin A deficiency (szymanska2012diagnosticdifficultiesin pages 11-11)
1.4 Evidence type
Evidence in this report is derived from: - Individual patient case report(s) (human; aggregated only at “n=2 published cases” level in-source) (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 11-12) - Aggregated disease-level sources for classic Krabbe disease (newborn screening policy review; incidence and HSCT risk) (ream2025evidenceandrecommendation pages 1-3) - Model organism studies (mouse; defining mechanism and phenotype timing) (matsuda2001amutationin pages 4-6, matsuda2007thefunctionof pages 2-3)
2. Etiology
2.1 Disease causal factors
Primary cause: germline biallelic pathogenic variants in PSAP affecting the saposin A domain, producing functional saposin A deficiency. The second reported human case carried homozygous PSAP NM_002778.3:c.209T>G (p.Val70Gly). (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 1-3)
Mechanistic cause: saposin A is a non-enzymatic lysosomal activator/stabilizer required for in vivo degradation of galactosylceramide by GALC; saposin A deficiency can therefore impair GalCer catabolism even when the GALC gene is intact, producing a Krabbe phenotype. (kose2018thesecondcase pages 8-9, matsuda2007thefunctionof pages 2-3)
2.2 Risk factors
- Genetic risk: autosomal recessive inheritance is implied by reported homozygous variants (and consanguinity in case reports is common in PSAP-related disorders, though not established here beyond the homozygous state). (kose2018thesecondcase pages 3-6)
- Environmental risk factors: none established in retrieved sources.
2.3 Protective factors / modifiers
No protective variants or environmental protective factors were identified in the retrieved sources.
2.4 Gene–environment interactions
No gene–environment interaction data were identified.
3. Phenotypes (Human)
3.1 Core clinical phenotype (reported 2018 case)
A reported infant (7 months) with saposin A deficiency had a clinical picture described as highly compatible with infantile Krabbe disease, including: - Neurologic regression / loss of milestones (loss of head control, loss of acquired skills) (kose2018thesecondcase pages 1-3) - Seizures (refractory seizures; tonic convulsions) (kose2018thesecondcase pages 1-3) - Hypertonicity and increased deep tendon reflexes (kose2018thesecondcase pages 1-3) - Peripheral neuropathy: severe axonal polyneuropathy (kose2018thesecondcase pages 1-3) - CSF abnormality: elevated CSF protein (135 mg/dL) (kose2018thesecondcase pages 1-3)
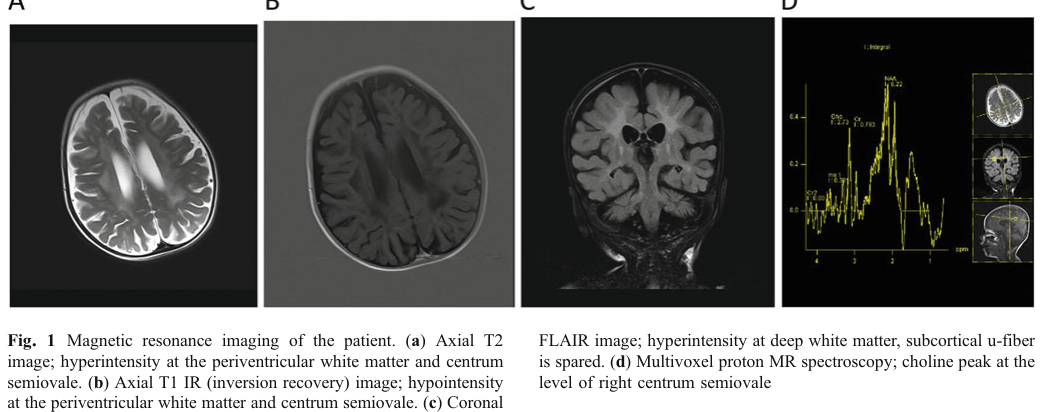
Imaging: MRI showed ventriculomegaly and white matter signal abnormalities, described as compatible with Krabbe disease; optic nerve thickening was also noted. (kose2018thesecondcase pages 1-3, kose2018thesecondcase media 3d158019)
3.2 Phenotype characteristics (limitations)
- Age of onset: infantile in the reported human case; earlier/later-onset spectrum is not well-defined due to extremely small number of published human cases accessible in this corpus. (kose2018thesecondcase pages 1-3, kose2018thesecondcase pages 11-12)
- Progression: progressive neurodegeneration is implied by regression and severe neurologic findings. (kose2018thesecondcase pages 1-3)
- Frequency among affected individuals: not quantifiable in retrieved sources (n≈2 published human cases referenced in-source). (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 11-12)
3.3 Suggested HPO terms (non-exhaustive; mapping based on described features)
- Seizures HP:0001250 (kose2018thesecondcase pages 1-3)
- Developmental regression HP:0002376 (kose2018thesecondcase pages 1-3)
- Hypertonia HP:0001252 (kose2018thesecondcase pages 1-3)
- Hyperreflexia HP:0001347 (kose2018thesecondcase pages 1-3)
- Peripheral neuropathy HP:0009830 / Axonal neuropathy HP:0003477 (kose2018thesecondcase pages 1-3)
- Abnormal brain MRI / White matter abnormalities HP:0002500 / HP:0002669 (kose2018thesecondcase pages 1-3, kose2018thesecondcase media 3d158019)
- Increased cerebrospinal fluid protein HP:0002928 (kose2018thesecondcase pages 1-3)
3.4 Quality-of-life impact
No formal QoL instruments (e.g., PedsQL, PROMIS) were reported in retrieved saposin A deficiency sources; however, the clinical features (seizures, regression, neuropathy) imply severe impairment. (kose2018thesecondcase pages 1-3)
4. Genetic / Molecular Information
4.1 Causal gene
- PSAP (prosaposin), specifically affecting the saposin A domain (kose2018thesecondcase pages 1-3)
4.2 Pathogenic variants (human)
From the retrieved corpus: - PSAP NM_002778.3:c.209T>G (p.Val70Gly), homozygous, associated with infantile Krabbe-like phenotype; GALC gene testing negative in that patient. (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 1-3, kose2018thesecondcase media 3d158019)
Other human saposin A deficiency was referenced as first reported in 2005 (Spiegel et al.), but that primary paper was not retrievable here; variant details are therefore not extractable from the current evidence set. (kose2018thesecondcase pages 12-12, szymanska2012diagnosticdifficultiesin pages 11-11)
4.3 Functional consequence
In the human case report, biochemical findings supported impaired GalCer degradation and lysosomal dysfunction, consistent with loss of function of saposin A cofactor activity. (kose2018thesecondcase pages 8-9)
4.4 Modifier genes / epigenetics / chromosomal abnormalities
No modifier genes, epigenetic mechanisms, or chromosomal abnormalities specific to saposin A deficiency were identified.
5. Environmental Information
No validated environmental, lifestyle, toxic, or infectious contributors were identified in the retrieved sources.
6. Mechanism / Pathophysiology
6.1 Causal chain (current understanding)
- PSAP saposin A-domain deficiency → reduced availability of saposin A cofactor (kose2018thesecondcase pages 1-3)
- Impaired in vivo GALC-mediated degradation of galactosylceramide (saposin A is “essential/indispensable” for this process in vivo in model systems) (matsuda2007thefunctionof pages 2-3, matsuda2007thefunctionof pages 1-2)
- Accumulation of myelin-enriched glycosphingolipids, including GalCer; psychosine accumulation is implicated in Krabbe biology and was increased in saposin A-deficient mice (modest vs twitcher) (matsuda2001amutationin pages 2-4, matsuda2001amutationin pages 6-7)
- Demyelination and neuroinflammation with infiltration of PAS-positive multinucleated macrophages (“globoid cells”) in CNS/PNS (matsuda2001amutationin pages 2-4, matsuda2007thefunctionof pages 2-3)
- Clinical manifestations: progressive neurologic decline, seizures, neuropathy, and white matter disease (human phenotype) (kose2018thesecondcase pages 1-3)
6.2 Autophagy / lysosome dysfunction (human evidence)
Patient fibroblasts showed altered autophagy consistent with impaired autophagic flux: the report describes a twofold increase of LC3 and p62 and impaired autophagosome–lysosome fusion/maturation. (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 11-12)
6.3 Biochemical abnormalities
- In the 2018 human case, fibroblasts showed increased neutral glycosphingolipids including GalCer (3.5-fold), LacCer (1.5-fold), Cer (2-fold), GlcCer (1.4-fold) compared with controls. (kose2018thesecondcase pages 8-9)
- Psychosine could not be measured in that patient due to specimen constraints (“only fibroblasts were available”). (kose2018thesecondcase pages 8-9)
6.4 Suggested ontology terms
GO biological process (examples): - sphingolipid catabolic process (GO) (mechanistic basis supported) (matsuda2007thefunctionof pages 2-3) - lysosomal transport / lysosome organization (GO) (lysosome/autophagy involvement) (kose2018thesecondcase pages 11-12) - autophagy (GO) (kose2018thesecondcase pages 11-12)
GO cellular component: - lysosome (GO:0005764) (supported by LAMP1-positive lysosome increase) (kose2018thesecondcase pages 8-9)
Cell Ontology (CL) (examples): - macrophage / microglia-like phagocytes implicated by globoid cells (histologic globoid macrophages) (matsuda2001amutationin pages 2-4) - oligodendrocyte lineage is implicated by demyelination (general Krabbe biology; directly supported in model pathology) (matsuda2007thefunctionof pages 2-3)
7. Anatomical Structures Affected
7.1 Organ/system level
- Central nervous system (white matter leukodystrophy; MRI changes) (kose2018thesecondcase pages 1-3, kose2018thesecondcase media 3d158019)
- Peripheral nervous system (severe axonal polyneuropathy; in mice, marked PNS demyelination and enlarged peripheral nerves) (kose2018thesecondcase pages 1-3, matsuda2001amutationin pages 4-6)
7.2 Tissue/cell level
- Myelinated tracts / white matter (leukodystrophy and demyelination pathology in models) (matsuda2001amutationin pages 2-4, matsuda2007thefunctionof pages 2-3)
- Macrophage-lineage globoid cells (PAS-positive multinucleated macrophages) (matsuda2001amutationin pages 2-4, matsuda2007thefunctionof pages 2-3)
7.3 Subcellular localization
- Lysosome (lysosomal storage biology; increased LAMP1 signal reported) (kose2018thesecondcase pages 8-9)
7.4 Suggested UBERON terms (examples)
- Brain white matter (UBERON) (kose2018thesecondcase media 3d158019)
- Peripheral nerve (UBERON) (matsuda2001amutationin pages 4-6)
8. Temporal Development
8.1 Human
- Infantile onset with rapid neurologic deterioration by 7 months was reported in the 2018 case. (kose2018thesecondcase pages 1-3)
8.2 Model organism temporal profile (saposin A-deficient mice)
- Weakness onset around ~60 days and lifespan around ~120 days in saposin A-deficient mice, indicating a chronic, later-onset course relative to twitcher (GALC-deficient) mice. (matsuda2007thefunctionof pages 2-3, matsuda2007thefunctionof pages 1-2)
9. Inheritance and Population
9.1 Inheritance
Autosomal recessive inheritance is supported by the reported homozygous PSAP variant in the 2018 case. (kose2018thesecondcase pages 3-6)
9.2 Epidemiology
- Saposin A deficiency specifically: extremely rare; the 2018 report states it is the second reported human case. (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 11-12)
- Krabbe disease overall (mostly GALC-related): a 2025 Pediatrics review reports incidence “0.3–2.6 per 100,000 live births.” (ream2025evidenceandrecommendation pages 1-3)
- A 2023 review cited U.S. estimates of 1 in 310,000 (retrospective analysis) and 1 in 394,000 (New York State NBS). (heller2023preclinicalstudiesin pages 1-2)
Carrier frequency, penetrance, founder effects, and variant geographic distribution for saposin A deficiency were not available in retrieved sources.
10. Diagnostics
10.1 Clinical testing (biochemical)
Key diagnostic pitfall: saposin A deficiency can present as Krabbe disease while having atypical enzymology/genetics for GALC. - In the 2018 saposin A deficiency case, GALC activity was reduced (dried blood; leukocytes) but described as higher than expected for classic Krabbe, and other lysosomal enzymes were normal. (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 8-9) - Fibroblast lipid studies demonstrated GalCer and related glycosphingolipid accumulation. (kose2018thesecondcase pages 8-9)
Newborn screening context (Krabbe overall): - NBS uses low GALC activity in dried blood spots, with second-tier psychosine testing to improve specificity. (ream2025evidenceandrecommendation pages 1-3) - The 2024 expedited evidence review evaluated/implemented a referral strategy based on psychosine ≥10 nM. (kemperUnknownyearexpeditedevidencebasedreview pages 1-4)
10.2 Imaging
MRI white matter abnormalities consistent with leukodystrophy were reported in the 2018 saposin A deficiency case. (kose2018thesecondcase pages 1-3, kose2018thesecondcase media 3d158019)
10.3 Genetic testing strategy
For a Krabbe phenotype with inconclusive GALC findings, the 2018 case supports: - initial GALC testing (enzyme + gene) followed by - exome sequencing and confirmatory Sanger sequencing to identify PSAP saposin A-domain variants. (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 1-3)
10.4 Differential diagnosis
- Classic GALC-related Krabbe disease (globoid cell leukodystrophy) remains the main differential. (kose2018thesecondcase pages 6-7, szymanska2012diagnosticdifficultiesin pages 11-11)
- Saposin A deficiency should be considered when Krabbe phenotype is present but standard testing is not definitive. (kose2018thesecondcase pages 11-12)
10.5 Suggested LOINC-style test concepts (since specific LOINC IDs not retrieved)
- GALC enzyme activity (dried blood spot; leukocytes) (kose2018thesecondcase pages 3-6)
- Psychosine (galactosylsphingosine) in dried blood spot / plasma (Krabbe NBS) (kemperUnknownyearexpeditedevidencebasedreview pages 1-4)
- PSAP sequencing (targeted or exome) (kose2018thesecondcase pages 3-6)
11. Outcomes / Prognosis
11.1 Saposin A deficiency (human)
No longitudinal survival outcomes or treatment response were available in the retrieved saposin A deficiency case excerpts.
11.2 Krabbe disease (overall; policy/review evidence)
The 2025 Pediatrics review summarizes infantile Krabbe disease as untreated leading to “death in early childhood.” (Direct quote from abstract-style summary) (ream2025evidenceandrecommendation pages 1-3)
12. Treatment
12.1 Disease-modifying therapy (Krabbe overall)
- HSCT: The 2025 Pediatrics review states: “Hematopoietic stem cell transplantation (HSCT) for IKD approximately 1 month after birth can improve long-term survival but has about a 10% risk of mortality within 100 days.” (direct quote) (ream2025evidenceandrecommendation pages 1-3)
- Newborn screening enabling early HSCT: The 2024 expedited evidence review modeled that with a psychosine ≥10 nM referral threshold, ~11.3 infants/year (range 5.6–20.2) would be diagnosed with infantile Krabbe disease in a 3.65M-birth cohort, with ~1.0 (0.3–1.2) HSCT deaths within 100 days among those transplanted. (kemperUnknownyearexpeditedevidencebasedreview pages 21-24)
12.2 Applicability to saposin A deficiency
No direct evidence in the retrieved human saposin A deficiency case report excerpts documents HSCT or gene therapy use in saposin A deficiency patients. Therefore, extrapolation from GALC-Krabbe therapeutic literature should be done cautiously.
12.3 Suggested MAXO terms (examples)
- Hematopoietic stem cell transplantation (MAXO) (Krabbe overall) (ream2025evidenceandrecommendation pages 1-3)
- Genetic testing (MAXO) / Whole exome sequencing (MAXO) (kose2018thesecondcase pages 3-6)
13. Prevention
13.1 Secondary prevention (screening)
- Newborn screening for Krabbe (overall) is based on dried blood spot GALC activity with psychosine second-tier testing; in 2024, infantile Krabbe disease was added to the U.S. Recommended Uniform Screening Panel (RUSP) as summarized in the 2025 Pediatrics review. (ream2025evidenceandrecommendation pages 1-3)
Primary prevention (environmental) is not applicable based on current evidence.
14. Other Species / Natural Disease
No naturally occurring saposin A deficiency “Krabbe due to saposin A deficiency” was identified in non-human species from the retrieved sources (separate from experimental models).
15. Model Organisms
15.1 Mouse models (saposin A deficiency)
Mouse models provide strong mechanistic support that saposin A deficiency can cause globoid cell leukodystrophy: - Saposin A-deficient mice show Krabbe-like demyelination with PAS-positive multinucleated macrophage/globoid cells in CNS/PNS, weakness at ~60 days, and lifespan ~120 days. (matsuda2007thefunctionof pages 2-3, matsuda2007thefunctionof pages 1-2) - In a saposin A-domain mutant mouse model, mean survival was ~122±17 days versus twitcher ~48±5 days, and brain GALC activity was ~half of wild type, consistent with saposin A acting as an essential activator/stabilizer for GALC in vivo. (matsuda2001amutationin pages 4-6)
15.2 Comparative pathology (saposin A vs twitcher)
A comparative clinico-pathological study used demyelination markers (Luxol fast blue loss; PAS-positive macrophages) and compared terminal-stage saposin A-deficient mice at PND 180 vs twitcher at PND 50, emphasizing the later course in saposin A deficiency relative to primary GALC deficiency. (yagi2004comparativeclinicopathologicalstudy pages 1-3)
15.3 Zebrafish (combined saposin deficiency; broader PSAP biology)
A CRISPR-Cas9 zebrafish psap knockout model recapitulated major LSD pathologies including impaired locomotion and severe myelin loss and identified acid sphingomyelinase modulation as a potential therapeutic direction for sphingolipidoses (not specific to isolated saposin A deficiency). (kose2018thesecondcase media bc75f129)
Summary Table (human + models)
| Cohort/model | Age at onset / stage | Key phenotypes | Imaging / pathology | Biochemical findings | Genetic variant / genotype | Rarity / notes |
|---|---|---|---|---|---|---|
| Human case 1: first reported saposin A deficiency presenting as Krabbe disease | Infantile onset; exact onset not available in retrieved context | Krabbe-like / globoid cell leukodystrophy phenotype in an infant (szymanska2012diagnosticdifficultiesin pages 11-11) | Not available in retrieved context | Saposin A deficiency reported as cause of Krabbe phenotype; detailed GALC/psychosine values not available in retrieved context (szymanska2012diagnosticdifficultiesin pages 11-11) | Mutation in saposin A coding region of PSAP; exact HGVS not available in retrieved context (kose2018thesecondcase pages 12-12, szymanska2012diagnosticdifficultiesin pages 11-11) | First human report; establishes that saposin A deficiency can phenocopy Krabbe disease (kose2018thesecondcase pages 12-12, szymanska2012diagnosticdifficultiesin pages 11-11) |
| Human case 2: JIMD Reports 2018 proband | Normal early infancy, then deterioration by 7 months; infantile presentation (kose2018thesecondcase pages 1-3) | Refractory seizures, loss of milestones/head control, feeding difficulty, hypertonicity, increased deep tendon reflexes, severe axonal polyneuropathy, elevated CSF protein 135 mg/dL; phenotype highly compatible with infantile Krabbe disease (kose2018thesecondcase pages 1-3) | Brain MRI: bilateral ventricular enlargement, periventricular/centrum semiovale white matter hyperintensities, optic nerve thickening; MRI compatible with Krabbe disease (kose2018thesecondcase pages 1-3, kose2018thesecondcase media 3d158019) | GALC activity low in dried blood and reduced in leukocytes, but higher than expected for classic Krabbe; other lysosomal enzymes normal. Fibroblasts: GalCer 3.5-fold, LacCer 1.5-fold, Cer 2-fold, GlcCer 1.4-fold vs controls; increased LAMP1-positive lysosomes. Psychosine could not be assessed because only fibroblasts were available (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 8-9, kose2018thesecondcase pages 6-7, kose2018thesecondcase media 3d158019) | PSAP NM_002778.3:c.209T>G (p.Val70Gly), homozygous, in saposin A domain; GALC gene negative (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 1-3, kose2018thesecondcase media 3d158019) | Second known human case; authors emphasize extreme rarity and recommend considering PSAP when Krabbe phenotype is present but GALC testing is inconclusive (kose2018thesecondcase pages 3-6, kose2018thesecondcase pages 11-12) |
| Mouse model: saposin A domain mutant / saposin A-deficient (C106F; often denoted SAP-A−/− or A−/−) | Subtle weakness/sluggishness around 60 days to 2.5 months; hind-leg atrophy/paralysis and weight plateau by ~3 months (matsuda2007thefunctionof pages 2-3, matsuda2001amutationin pages 1-2) | Chronic, milder Krabbe-like disease with progressive neuromotor decline; occasional seizures/hyperactivity (matsuda2001amutationin pages 2-4, matsuda2001amutationin pages 1-2) | Demyelination in CNS and PNS; PAS-positive multinucleated macrophages / globoid-like cells around vessels; enlarged peripheral nerves; pathology detectable by ~30 days in some studies (matsuda2001amutationin pages 2-4, matsuda2007thefunctionof pages 1-2, matsuda2001amutationin pages 6-7, yagi2004comparativeclinicopathologicalstudy pages 1-3) | Brain GALC activity ~0.74 ± 0.15 vs 1.41 ± 0.23 nmol/h/mg in wild type; slight brain GalCer increase, marked kidney GalCer increase; brain psychosine ~2–3× normal / approximately doubled by 2 months (matsuda2001amutationin pages 4-6, matsuda2001amutationin pages 2-4, matsuda2001amutationin pages 6-7) | Targeted Psap saposin A-domain C106F mutation disrupting conserved disulfide bond (matsuda2001amutationin pages 1-2, matsuda2001amutationin pages 2-4) | Demonstrates saposin A is indispensable for in vivo GALC-mediated GalCer degradation and that saposin A deficiency is an alternative cause of globoid cell leukodystrophy (matsuda2007thefunctionof pages 2-3, matsuda2007thefunctionof pages 1-2) |
| Mouse comparator: twitcher (classic GALC-deficient Krabbe model) | Earlier onset than saposin A-deficient mice; terminal stage around PND 50 in comparative pathology studies (yagi2004comparativeclinicopathologicalstudy pages 1-3) | Severe, rapidly progressive Krabbe phenotype (matsuda2007thefunctionof pages 1-2, yagi2004comparativeclinicopathologicalstudy pages 1-3) | More severe demyelination and globoid cell pathology than saposin A-deficient mice at earlier ages (matsuda2007thefunctionof pages 1-2, yagi2004comparativeclinicopathologicalstudy pages 1-3) | Much greater psychosine accumulation than saposin A-deficient mice; terminal twitcher mice show markedly elevated psychosine, with saposin A-deficient mice having only modest increases (matsuda2001amutationin pages 2-4, matsuda2001amutationin pages 6-7) | Galc-deficient twitcher genotype (matsuda2007thefunctionof pages 1-2, yagi2004comparativeclinicopathologicalstudy pages 1-3) | Benchmark canonical Krabbe model used to show saposin A deficiency causes a milder, later-onset but mechanistically related leukodystrophy (matsuda2001amutationin pages 4-6, matsuda2007thefunctionof pages 1-2, yagi2004comparativeclinicopathologicalstudy pages 1-3) |
| Mouse comparator: saposin A-deficient vs twitcher lifespan | SAP-A−/− mean survival ~122 ± 17 days vs twitcher ~48 ± 5 days (matsuda2001amutationin pages 4-6) | SAP-A−/− chronic course vs twitcher fulminant course (matsuda2001amutationin pages 4-6, matsuda2007thefunctionof pages 1-2) | SAP-A−/− terminal-stage pathology compared at PND 180 vs twitcher at PND 50 (yagi2004comparativeclinicopathologicalstudy pages 1-3) | SAP-A−/− retains partial GALC-related function / compensation, whereas twitcher lacks primary GALC activity (matsuda2001amutationin pages 4-6, matsuda2007thefunctionof pages 2-3) | Comparative model evidence rather than a separate genotype row (matsuda2001amutationin pages 4-6, yagi2004comparativeclinicopathologicalstudy pages 1-3) | Useful for interpreting why human saposin A deficiency may show Krabbe phenotype despite noncanonical GALC findings (kose2018thesecondcase pages 3-6, matsuda2001amutationin pages 4-6) |
Table: This table summarizes the reported human saposin A deficiency cases with Krabbe-like presentation and the main saposin A animal models used to define disease mechanism. It is useful for comparing clinical rarity, diagnostic findings, and the mechanistic contrast between PSAP/saposin A deficiency and classic GALC-deficient Krabbe disease.
Key gaps / limitations in the current evidence set
- The primary first human saposin A deficiency report (Spiegel et al., 2005) and the psychosine-focused saposin A differential report (Calderwood et al., 2020) were referenced but not retrievable in this tool session; variant-level details and psychosine findings from those sources could not be extracted here. (kose2018thesecondcase pages 12-12, ream2025evidenceandrecommendation pages 8-8)
- MONDO/Orphanet/ICD/MeSH identifiers for “Krabbe disease due to saposin A deficiency” were not available in the retrieved sources.
- No dedicated clinical trials or treatment outcome series for saposin A deficiency (PSAP saposin A domain) were identified in the retrieved corpus; most recent (2023–2025) advances pertain to GALC-Krabbe screening and treatment policy rather than saposin A deficiency specifically. (kemperUnknownyearexpeditedevidencebasedreview pages 21-24, ream2025evidenceandrecommendation pages 1-3)
URLs and publication dates (from retrieved sources)
- Kose et al. JIMD Reports (Jul 2018). https://doi.org/10.1007/8904_2018_114 (kose2018thesecondcase pages 3-6)
- Matsuda et al. Human Molecular Genetics (May 2001). https://doi.org/10.1093/hmg/10.11.1191 (matsuda2001amutationin pages 1-2)
- Matsuda et al. Journal of Neurochemistry (Nov 2007). https://doi.org/10.1111/j.1471-4159.2007.04709.x (matsuda2007thefunctionof pages 2-3)
- Yagi et al. Journal of Neuropathology & Experimental Neurology (Jul 2004). https://doi.org/10.1093/jnen/63.7.721 (yagi2004comparativeclinicopathologicalstudy pages 1-3)
- Ream et al. Pediatrics (Mar/Apr 2025). https://doi.org/10.1542/peds.2024-069152 (ream2025evidenceandrecommendation pages 1-3)
- Kemper et al. HRSA report “Expedited Evidence-Based Review…” (Feb 1, 2024; PDF URL referenced in-source). (kemperUnknownyearexpeditedevidencebasedreview pages 1-4)
References
-
(kose2018thesecondcase pages 1-3): Melis Kose, Secil Akyildiz Demir, Gulcin Akinci, Cenk Eraslan, Unsal Yilmaz, Serdar Ceylaner, Eser Sozmen Yildirim, and Volkan Seyrantepe. The second case of saposin a deficiency and altered autophagy. JIMD reports, 44:43-54, Jul 2018. URL: https://doi.org/10.1007/8904_2018_114, doi:10.1007/8904_2018_114. This article has 11 citations and is from a peer-reviewed journal.
-
(kose2018thesecondcase pages 11-12): Melis Kose, Secil Akyildiz Demir, Gulcin Akinci, Cenk Eraslan, Unsal Yilmaz, Serdar Ceylaner, Eser Sozmen Yildirim, and Volkan Seyrantepe. The second case of saposin a deficiency and altered autophagy. JIMD reports, 44:43-54, Jul 2018. URL: https://doi.org/10.1007/8904_2018_114, doi:10.1007/8904_2018_114. This article has 11 citations and is from a peer-reviewed journal.
-
(szymanska2012diagnosticdifficultiesin pages 11-11): Krystyna Szymańska, Agnieszka Ługowska, Milena Laure-Kamionowska, Monika Bekiesińska-Figatowska, Dorota Gieruszczak-Białek, Małgorzata Musielak, Sabrina Eichler, Anne-Katrin Giese, and Arndt Rolfs. Diagnostic difficulties in krabbe disease: a report of two cases and review of literature. Folia neuropathologica, 50 4:346-56, Aug 2012. URL: https://doi.org/10.5114/fn.2012.32364, doi:10.5114/fn.2012.32364. This article has 31 citations and is from a peer-reviewed journal.
-
(kose2018thesecondcase pages 6-7): Melis Kose, Secil Akyildiz Demir, Gulcin Akinci, Cenk Eraslan, Unsal Yilmaz, Serdar Ceylaner, Eser Sozmen Yildirim, and Volkan Seyrantepe. The second case of saposin a deficiency and altered autophagy. JIMD reports, 44:43-54, Jul 2018. URL: https://doi.org/10.1007/8904_2018_114, doi:10.1007/8904_2018_114. This article has 11 citations and is from a peer-reviewed journal.
-
(kose2018thesecondcase pages 3-6): Melis Kose, Secil Akyildiz Demir, Gulcin Akinci, Cenk Eraslan, Unsal Yilmaz, Serdar Ceylaner, Eser Sozmen Yildirim, and Volkan Seyrantepe. The second case of saposin a deficiency and altered autophagy. JIMD reports, 44:43-54, Jul 2018. URL: https://doi.org/10.1007/8904_2018_114, doi:10.1007/8904_2018_114. This article has 11 citations and is from a peer-reviewed journal.
-
(ream2025evidenceandrecommendation pages 1-3): Margie A. Ream, Wendy K. K. Lam, Scott D. Grosse, Jelili Ojodu, Elizabeth Jones, Lisa A. Prosser, Angela M. Rose, Anne Marie Comeau, Susan Tanksley, Katie P. DiCostanzo, and Alex R. Kemper. Evidence and recommendation for infantile krabbe disease newborn screening. Pediatrics, Mar 2025. URL: https://doi.org/10.1542/peds.2024-069152, doi:10.1542/peds.2024-069152. This article has 7 citations and is from a highest quality peer-reviewed journal.
-
(matsuda2001amutationin pages 4-6): J. Matsuda, M. Vanier, Y. Saito, J. Tohyama, Kinuko Suzuki, and Kunihiko Suzuki. A mutation in the saposin a domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Human molecular genetics, 10 11:1191-9, May 2001. URL: https://doi.org/10.1093/hmg/10.11.1191, doi:10.1093/hmg/10.11.1191. This article has 173 citations and is from a domain leading peer-reviewed journal.
-
(matsuda2007thefunctionof pages 2-3): Junko Matsuda, Azusa Yoneshige, and Kunihiko Suzuki. The function of sphingolipids in the nervous system: lessons learnt from mouse models of specific sphingolipid activator protein deficiencies. Journal of Neurochemistry, 103:32-38, Nov 2007. URL: https://doi.org/10.1111/j.1471-4159.2007.04709.x, doi:10.1111/j.1471-4159.2007.04709.x. This article has 45 citations and is from a domain leading peer-reviewed journal.
-
(kose2018thesecondcase pages 8-9): Melis Kose, Secil Akyildiz Demir, Gulcin Akinci, Cenk Eraslan, Unsal Yilmaz, Serdar Ceylaner, Eser Sozmen Yildirim, and Volkan Seyrantepe. The second case of saposin a deficiency and altered autophagy. JIMD reports, 44:43-54, Jul 2018. URL: https://doi.org/10.1007/8904_2018_114, doi:10.1007/8904_2018_114. This article has 11 citations and is from a peer-reviewed journal.
-
(kose2018thesecondcase media 3d158019): Melis Kose, Secil Akyildiz Demir, Gulcin Akinci, Cenk Eraslan, Unsal Yilmaz, Serdar Ceylaner, Eser Sozmen Yildirim, and Volkan Seyrantepe. The second case of saposin a deficiency and altered autophagy. JIMD reports, 44:43-54, Jul 2018. URL: https://doi.org/10.1007/8904_2018_114, doi:10.1007/8904_2018_114. This article has 11 citations and is from a peer-reviewed journal.
-
(kose2018thesecondcase pages 12-12): Melis Kose, Secil Akyildiz Demir, Gulcin Akinci, Cenk Eraslan, Unsal Yilmaz, Serdar Ceylaner, Eser Sozmen Yildirim, and Volkan Seyrantepe. The second case of saposin a deficiency and altered autophagy. JIMD reports, 44:43-54, Jul 2018. URL: https://doi.org/10.1007/8904_2018_114, doi:10.1007/8904_2018_114. This article has 11 citations and is from a peer-reviewed journal.
-
(matsuda2007thefunctionof pages 1-2): Junko Matsuda, Azusa Yoneshige, and Kunihiko Suzuki. The function of sphingolipids in the nervous system: lessons learnt from mouse models of specific sphingolipid activator protein deficiencies. Journal of Neurochemistry, 103:32-38, Nov 2007. URL: https://doi.org/10.1111/j.1471-4159.2007.04709.x, doi:10.1111/j.1471-4159.2007.04709.x. This article has 45 citations and is from a domain leading peer-reviewed journal.
-
(matsuda2001amutationin pages 2-4): J. Matsuda, M. Vanier, Y. Saito, J. Tohyama, Kinuko Suzuki, and Kunihiko Suzuki. A mutation in the saposin a domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Human molecular genetics, 10 11:1191-9, May 2001. URL: https://doi.org/10.1093/hmg/10.11.1191, doi:10.1093/hmg/10.11.1191. This article has 173 citations and is from a domain leading peer-reviewed journal.
-
(matsuda2001amutationin pages 6-7): J. Matsuda, M. Vanier, Y. Saito, J. Tohyama, Kinuko Suzuki, and Kunihiko Suzuki. A mutation in the saposin a domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Human molecular genetics, 10 11:1191-9, May 2001. URL: https://doi.org/10.1093/hmg/10.11.1191, doi:10.1093/hmg/10.11.1191. This article has 173 citations and is from a domain leading peer-reviewed journal.
-
(heller2023preclinicalstudiesin pages 1-2): Gregory Heller, Allison M. Bradbury, Mark S. Sands, and Ernesto R. Bongarzone. Preclinical studies in krabbe disease: a model for the investigation of novel combination therapies for lysosomal storage diseases. Molecular Therapy, 31:7-23, Jan 2023. URL: https://doi.org/10.1016/j.ymthe.2022.09.017, doi:10.1016/j.ymthe.2022.09.017. This article has 15 citations and is from a highest quality peer-reviewed journal.
-
(kemperUnknownyearexpeditedevidencebasedreview pages 1-4): AR Kemper, KK Lam, M Ream, and K DiCostanzo. Expedited evidence-based review of newborn screening for krabbe disease final report: february 1, 2024. Unknown journal, Unknown year.
-
(kemperUnknownyearexpeditedevidencebasedreview pages 21-24): AR Kemper, KK Lam, M Ream, and K DiCostanzo. Expedited evidence-based review of newborn screening for krabbe disease final report: february 1, 2024. Unknown journal, Unknown year.
-
(yagi2004comparativeclinicopathologicalstudy pages 1-3): Takashi Yagi, Junko Matsuda, Shoichi Takikita, Ikuko Mohri, Kunihiko Suzuki, and Kinuko Suzuki. Comparative clinico-pathological study of saposin-a-deficient (sap-a−/−) and twitcher mice. Journal of Neuropathology & Experimental Neurology, 63:721-734, Jul 2004. URL: https://doi.org/10.1093/jnen/63.7.721, doi:10.1093/jnen/63.7.721. This article has 13 citations and is from a peer-reviewed journal.
-
(kose2018thesecondcase media bc75f129): Melis Kose, Secil Akyildiz Demir, Gulcin Akinci, Cenk Eraslan, Unsal Yilmaz, Serdar Ceylaner, Eser Sozmen Yildirim, and Volkan Seyrantepe. The second case of saposin a deficiency and altered autophagy. JIMD reports, 44:43-54, Jul 2018. URL: https://doi.org/10.1007/8904_2018_114, doi:10.1007/8904_2018_114. This article has 11 citations and is from a peer-reviewed journal.
-
(matsuda2001amutationin pages 1-2): J. Matsuda, M. Vanier, Y. Saito, J. Tohyama, Kinuko Suzuki, and Kunihiko Suzuki. A mutation in the saposin a domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Human molecular genetics, 10 11:1191-9, May 2001. URL: https://doi.org/10.1093/hmg/10.11.1191, doi:10.1093/hmg/10.11.1191. This article has 173 citations and is from a domain leading peer-reviewed journal.
-
(ream2025evidenceandrecommendation pages 8-8): Margie A. Ream, Wendy K. K. Lam, Scott D. Grosse, Jelili Ojodu, Elizabeth Jones, Lisa A. Prosser, Angela M. Rose, Anne Marie Comeau, Susan Tanksley, Katie P. DiCostanzo, and Alex R. Kemper. Evidence and recommendation for infantile krabbe disease newborn screening. Pediatrics, Mar 2025. URL: https://doi.org/10.1542/peds.2024-069152, doi:10.1542/peds.2024-069152. This article has 7 citations and is from a highest quality peer-reviewed journal.