Kallmann Syndrome
Kallmann syndrome is the anosmic form of congenital isolated gonadotropin-releasing hormone (GnRH) deficiency, characterized by hypogonadotropic hypogonadism (absent or incomplete puberty, low sex steroids, infertility) combined with an impaired sense of smell (anosmia or hyposmia). It arises from defective embryonic co-migration of GnRH neurons and olfactory axons from the olfactory placode to the forebrain, with associated failure of olfactory bulb morphogenesis. Kallmann syndrome is genetically heterogeneous and can be inherited in X-linked recessive, autosomal dominant, autosomal recessive, or oligogenic patterns. Causal genes include ANOS1/KAL1 (X-linked), FGFR1/KAL2, FGF8, PROKR2, PROK2, and CHD7, among others. Anosmia distinguishes Kallmann syndrome from normosmic isolated GnRH deficiency, with which it shares a reproductive phenotype.
Ask OpenScientist
Ask a research question about Kallmann Syndrome. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
3Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Subtypes
5Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Pathophysiology
3Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Pathograph
Phenotypes
11Endocrine 2
Show evidence (1 reference)
Show evidence (1 reference)
Genitourinary 4
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Head and Neck 3
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Other 2
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
5Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Medical Actions
2Show evidence (1 reference)
Show evidence (1 reference)
Source YAML
click to showname: Kallmann Syndrome
creation_date: '2026-06-17T12:00:00Z'
category: Mendelian
description: >-
Kallmann syndrome is the anosmic form of congenital isolated gonadotropin-releasing
hormone (GnRH) deficiency, characterized by hypogonadotropic hypogonadism (absent or
incomplete puberty, low sex steroids, infertility) combined with an impaired sense of
smell (anosmia or hyposmia). It arises from defective embryonic co-migration of GnRH
neurons and olfactory axons from the olfactory placode to the forebrain, with associated
failure of olfactory bulb morphogenesis. Kallmann syndrome is genetically heterogeneous
and can be inherited in X-linked recessive, autosomal dominant, autosomal recessive, or
oligogenic patterns. Causal genes include ANOS1/KAL1 (X-linked), FGFR1/KAL2, FGF8,
PROKR2, PROK2, and CHD7, among others. Anosmia distinguishes Kallmann syndrome from
normosmic isolated GnRH deficiency, with which it shares a reproductive phenotype.
disease_term:
preferred_term: Kallmann syndrome
term:
id: MONDO:0018800
label: Kallmann syndrome
parents:
- Hypogonadotropic hypogonadism
- Disorders of puberty
has_subtypes:
- name: ANOS1
display_name: Kallmann syndrome 1 (X-linked, ANOS1/KAL1)
description: >-
X-linked recessive Kallmann syndrome caused by loss-of-function variants in ANOS1
(formerly KAL1), encoding the extracellular matrix adhesion protein anosmin-1. KAL1
mutations tend to cause a more severe reproductive phenotype and are distinctively
associated with unilateral renal agenesis and bimanual synkinesia (mirror movements).
evidence:
- reference: PMID:15605412
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "In this study, unilateral renal agenesis and bimanual synkinesia were exclusively found\nassociated with KAL1mutations, cleft palate and dental agenesia with\nFGFR1mutations."
explanation: A KAL1/FGFR1 cohort distinguishes the KAL1 subtype by renal agenesis and bimanual synkinesia.
- name: FGFR1
display_name: Kallmann syndrome 2 (FGFR1, KAL2)
description: >-
Autosomal dominant Kallmann syndrome caused by loss-of-function variants in FGFR1
(KAL2), with incomplete penetrance and variable expressivity. May include cleft
lip/palate and dental agenesis.
evidence:
- reference: PMID:12627230
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We establish here that loss-of-function mutations in FGFR1 underlie KAL2 whereas a\ngain-of-function mutation in FGFR1 has been shown to cause a form of\ncraniosynostosis."
explanation: The discovery paper establishes FGFR1 loss-of-function as the cause of KAL2.
- name: PROKR2_PROK2
display_name: Kallmann syndrome 3/4 (PROKR2 / PROK2)
description: >-
Kallmann syndrome caused by variants in the prokineticin receptor PROKR2 (KAL3) or its
ligand prokineticin-2, PROK2 (KAL4). PROKR2 variants occur in heterozygous, homozygous,
or compound heterozygous states and frequently contribute to oligogenic inheritance.
evidence:
- reference: PMID:17054399
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "we took a \ncandidate gene strategy and identified ten and four different point mutations in \nthe genes encoding the G protein-coupled prokineticin receptor-2 (PROKR2) and \none of its ligands, prokineticin-2 (PROK2), respectively."
explanation: Establishes PROKR2 and PROK2 as Kallmann syndrome genes.
- name: FGF8
display_name: Kallmann syndrome 6 (FGF8)
description: >-
Kallmann syndrome / GnRH deficiency caused by variants in FGF8, the principal ligand for
FGFR1 in GnRH neuron ontogeny; reduced FGF8 signaling impairs GnRH neuron development.
evidence:
- reference: PMID:18596921
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "While loss-of-function mutations in FGF receptor 1 (FGFR1) cause human GnRH deficiency, to date no specific ligand for FGFR1 has been identified in GnRH neuron ontogeny."
explanation: Establishes the FGF8-FGFR1 ligand-receptor axis underlying this subtype.
- name: CHD7
display_name: Kallmann syndrome 5 (CHD7)
description: >-
Kallmann syndrome caused by heterozygous CHD7 variants, overlapping mechanistically with
CHARGE syndrome. Isolated Kallmann syndrome / IHH can represent a milder allelic variant
of CHARGE syndrome.
evidence:
- reference: PMID:18834967
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We hypothesized that CHD7 would be \ninvolved in the pathogenesis of IHH and KS (IHH/KS) without the CHARGE phenotype \nand that IHH/KS represents a milder allelic variant of CHARGE syndrome."
explanation: Establishes CHD7 as a Kallmann syndrome gene and its allelic relationship to CHARGE.
inheritance:
- name: X-linked recessive
inheritance_term:
preferred_term: X-linked inheritance
term:
id: HP:0001417
label: X-linked inheritance
description: >-
ANOS1 (KAL1) mutations cause X-linked recessive Kallmann syndrome, the first molecularly
defined form. The gender difference in anosmin-1 dosage has been proposed to explain the
higher prevalence of disease in males.
evidence:
- reference: PMID:12627230
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "we suggest that the KAL1 gene product, the\nextracellular matrix protein anosmin-1, is involved in FGF signaling and propose\nthat the gender difference in anosmin-1 dosage (because KAL1 partially escapes X\ninactivation) explains the higher prevalence of the disease in males."
explanation: Supports X-linked inheritance and the male predominance of the KAL1 form.
- name: Autosomal dominant
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
description: >-
FGFR1, FGF8, and CHD7 variants typically cause autosomal dominant Kallmann syndrome with
incomplete penetrance and variable expressivity.
evidence:
- reference: PMID:12627230
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We took advantage of overlapping interstitial deletions at chromosome 8p11-p12\nin two individuals with contiguous gene syndromes and defined an interval of\nroughly 540 kb associated with a dominant form of Kallmann syndrome, KAL2."
explanation: Defines FGFR1/KAL2 as a dominant form of Kallmann syndrome.
- name: Oligogenic inheritance
inheritance_term:
preferred_term: Oligogenic inheritance
term:
id: HP:0010983
label: Oligogenic inheritance
description: >-
A subset of cases reflects oligogenic/digenic inheritance, with variants in two or more
GnRH-deficiency genes combining to produce or modify the phenotype.
evidence:
- reference: PMID:17054399
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "one of the patients heterozygous for a PROKR2 mutation was also carrying a missense \nmutation in KAL1, thus indicating a possible digenic inheritance of the disease \nin this individual."
explanation: Documents digenic PROKR2/KAL1 inheritance, the basis for oligogenic transmission.
pathophysiology:
- name: Defective GnRH neuron and olfactory axon migration
description: >-
During embryogenesis GnRH neurons originate in the olfactory placode and migrate along
olfactory/vomeronasal axons to the hypothalamus. Kallmann syndrome genes disrupt the
molecular guidance, adhesion, and growth-factor signaling required for this co-migration
and for olfactory bulb formation. Anosmin-1 (ANOS1) is an extracellular matrix protein
that binds heparan sulfate proteoglycans and modulates FGFR1 signaling; FGF8 is the
cognate FGFR1 ligand; and prokineticin-2/PROKR2 signaling is also required, so loss of
any of these arrests GnRH neuron migration and impairs olfactory bulb morphogenesis.
cell_types:
- preferred_term: Hypothalamic GnRH neuron
term:
id: CL:0011111
label: hypothalamic gonadotropin-releasing hormone neuron
biological_processes:
- preferred_term: Neuron migration
term:
id: GO:0001764
label: neuron migration
modifier: DECREASED
- preferred_term: Fibroblast growth factor receptor signaling pathway
term:
id: GO:0008543
label: fibroblast growth factor receptor signaling pathway
modifier: DECREASED
evidence:
- reference: PMID:21497178
supports: SUPPORT
evidence_source: OTHER
snippet: "Initial studies suggested a \ncentral role of anosmin-1, in GnRH neuron ontogeny - specifically in GnRH \nneuronal migration from the cribriform plate area into the brain - as well as in \nolfactory bulb development."
explanation: Supports anosmin-1's role in GnRH neuron migration and olfactory bulb development.
- reference: PMID:12627230
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "we suggest that the KAL1 gene product, the\nextracellular matrix protein anosmin-1, is involved in FGF signaling"
explanation: Links anosmin-1 to the FGFR1 signaling pathway that guides GnRH neuron migration.
downstream:
- target: GnRH deficiency and hypogonadotropic hypogonadism
description: >-
Failure of GnRH neurons to reach the hypothalamus results in deficient hypothalamic
GnRH secretion.
causal_link_type: DIRECT
- target: Impaired olfactory bulb development
description: >-
The same migratory/developmental defect that prevents olfactory axons from reaching
the forebrain also disrupts olfactory bulb formation.
causal_link_type: DIRECT
- name: GnRH deficiency and hypogonadotropic hypogonadism
description: >-
Deficient hypothalamic GnRH leads to inappropriately low pituitary secretion of
gonadotropins (LH and FSH) and consequently low sex steroid levels, producing absent or
incomplete puberty, hypogonadism, and infertility.
cell_types:
- preferred_term: Hypothalamic GnRH neuron
term:
id: CL:0011111
label: hypothalamic gonadotropin-releasing hormone neuron
biological_processes:
- preferred_term: Gonadotropin secretion
term:
id: GO:0032274
label: gonadotropin secretion
modifier: DECREASED
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "Isolated gonadotropin-releasing hormone (GnRH) \ndeficiency (IGD) is characterized by inappropriately low serum concentrations of \nthe gonadotropins LH (luteinizing hormone) and FSH (follicle-stimulating \nhormone) in the presence of low circulating concentrations of sex steroids."
explanation: Defines the core endocrine defect downstream of GnRH deficiency.
- name: Impaired olfactory bulb development
description: >-
Disrupted migration of olfactory axons and failure of olfactory bulb morphogenesis cause
absent or hypoplastic olfactory bulbs, the anatomic basis of the anosmia that defines
Kallmann syndrome.
biological_processes:
- preferred_term: Olfactory bulb development
term:
id: GO:0021772
label: olfactory bulb development
modifier: DECREASED
evidence:
- reference: PMID:17054399
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Kallmann syndrome combines anosmia, related to defective olfactory bulb \nmorphogenesis, and hypogonadism due to gonadotropin-releasing hormone \ndeficiency."
explanation: Attributes anosmia to defective olfactory bulb morphogenesis.
phenotypes:
- name: Hypogonadotropic hypogonadism
description: Low sex steroids with low or inappropriately normal LH and FSH.
frequency: OBLIGATE
phenotype_term:
preferred_term: Hypogonadotropic hypogonadism
term:
id: HP:0000044
label: Hypogonadotropic hypogonadism
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "low serum \ntestosterone or estradiol (hypogonadism) that results from complete or partial \nabsence of GnRH-mediated release of LH and FSH (hypogonadotropic hypogonadism"
explanation: Confirms hypogonadotropic hypogonadism as the cardinal endocrine feature.
- name: Anosmia
description: Absent or impaired sense of smell due to absent/hypoplastic olfactory bulbs; defines Kallmann syndrome.
frequency: OBLIGATE
phenotype_term:
preferred_term: Anosmia
term:
id: HP:0000458
label: Anosmia
evidence:
- reference: PMID:17054399
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Kallmann syndrome combines anosmia, related to defective olfactory bulb \nmorphogenesis, and hypogonadism due to gonadotropin-releasing hormone \ndeficiency."
explanation: Anosmia is a defining feature of Kallmann syndrome.
- name: Delayed puberty
description: Absent or incomplete puberty due to GnRH deficiency.
frequency: VERY_FREQUENT
phenotype_term:
preferred_term: Delayed puberty
term:
id: HP:0000823
label: Delayed puberty
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "Adolescents and adults with IGD have clinical evidence of \nhypogonadism and incomplete sexual maturation on physical examination."
explanation: Confirms absent/incomplete pubertal maturation.
- name: Infertility
description: Inability to conceive due to absent gonadotropin-driven gametogenesis.
frequency: VERY_FREQUENT
phenotype_term:
preferred_term: Infertility
term:
id: HP:0000789
label: Infertility
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "diminished libido, erectile dysfunction, and infertility."
explanation: Infertility is a core consequence of GnRH deficiency.
- name: Micropenis
description: Small penis in infant boys due to fetal gonadotropin deficiency.
frequency: FREQUENT
phenotype_term:
preferred_term: Micropenis
term:
id: HP:0000054
label: Micropenis
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "Infant boys with congenital IGD often have micropenis and \ncryptorchidism."
explanation: Micropenis is a recognized neonatal feature of congenital GnRH deficiency.
- name: Cryptorchidism
description: Undescended testes due to fetal gonadotropin deficiency.
frequency: FREQUENT
phenotype_term:
preferred_term: Cryptorchidism
term:
id: HP:0000028
label: Cryptorchidism
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "Infant boys with congenital IGD often have micropenis and \ncryptorchidism."
explanation: Cryptorchidism is a recognized neonatal feature of congenital GnRH deficiency.
- name: Primary amenorrhea
description: Absence of menstruation in affected females due to estrogen deficiency.
frequency: FREQUENT
phenotype_term:
preferred_term: Primary amenorrhea
term:
id: HP:0000786
label: Primary amenorrhea
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "Adult females have little or no breast development and primary \namenorrhea."
explanation: Primary amenorrhea is the female reproductive manifestation.
- name: Renal agenesis
description: >-
Unilateral renal agenesis, distinctively associated with the X-linked ANOS1/KAL1
subtype, reflecting anosmin-1 expression in the developing kidney.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Unilateral renal agenesis
term:
id: HP:0000122
label: Unilateral renal agenesis
evidence:
- reference: PMID:15605412
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "In this study, unilateral renal agenesis and bimanual synkinesia were exclusively found\nassociated with KAL1mutations"
explanation: Unilateral renal agenesis was exclusively associated with KAL1 mutations in this cohort.
- name: Bimanual synkinesia
description: >-
Mirror movements of the hands, a feature distinctively associated with the X-linked
ANOS1/KAL1 subtype.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Bimanual synkinesia
term:
id: HP:0001335
label: Bimanual synkinesia
evidence:
- reference: PMID:15605412
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "In this study, unilateral renal agenesis and bimanual synkinesia were exclusively found\nassociated with KAL1mutations"
explanation: Bimanual synkinesia was exclusively associated with KAL1 mutations in this cohort.
- name: Orofacial cleft
description: >-
Cleft lip and/or palate, distinctively associated with the FGFR1/KAL2 subtype.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Cleft lip and palate
term:
id: HP:0000202
label: Orofacial cleft
evidence:
- reference: PMID:15605412
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "cleft palate and dental agenesia with\nFGFR1mutations."
explanation: Cleft palate was associated with FGFR1 mutations in this cohort.
- name: Tooth agenesis
description: >-
Dental agenesis (missing teeth), distinctively associated with the FGFR1/KAL2 subtype.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Tooth agenesis
term:
id: HP:0009804
label: Tooth agenesis
evidence:
- reference: PMID:15605412
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "cleft palate and dental agenesia with\nFGFR1mutations."
explanation: Dental agenesis was associated with FGFR1 mutations in this cohort.
genetic:
- name: ANOS1 (KAL1) loss-of-function
association: Causative
gene_term:
preferred_term: ANOS1
term:
id: hgnc:6211
label: ANOS1
notes: >-
Loss-of-function variants in ANOS1 (KAL1) cause X-linked Kallmann syndrome via loss of
anosmin-1, an extracellular matrix protein required for GnRH neuron migration and
olfactory bulb development.
evidence:
- reference: PMID:21497178
supports: SUPPORT
evidence_source: OTHER
snippet: "The gene for X-linked Kallmann's syndrome (KAL-1, encoding anosmin-1) was cloned \nin 1991."
explanation: Establishes KAL1/ANOS1 (anosmin-1) as the X-linked Kallmann syndrome gene.
- name: FGFR1 (KAL2) loss-of-function
association: Causative
gene_term:
preferred_term: FGFR1
term:
id: hgnc:3688
label: FGFR1
notes: >-
Loss-of-function variants in FGFR1 cause autosomal dominant Kallmann syndrome (KAL2).
evidence:
- reference: PMID:12627230
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We establish here that loss-of-function mutations in FGFR1 underlie KAL2 whereas a\ngain-of-function mutation in FGFR1 has been shown to cause a form of\ncraniosynostosis."
explanation: Direct evidence that FGFR1 loss-of-function causes Kallmann syndrome type 2.
- name: PROKR2 / PROK2 variants
association: Causative
gene_term:
preferred_term: PROKR2
term:
id: hgnc:15836
label: PROKR2
notes: >-
Variants in the prokineticin receptor PROKR2 (KAL3) and its ligand PROK2 (KAL4) cause
Kallmann syndrome; insufficient prokineticin signaling through PROKR2 impairs olfactory
system and reproductive axis development.
evidence:
- reference: PMID:17054399
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "These findings reveal that insufficient \nprokineticin-signaling through PROKR2 leads to abnormal development of the \nolfactory system and reproductive axis in man."
explanation: Establishes the PROKR2/PROK2 prokineticin pathway in Kallmann syndrome pathogenesis.
- name: FGF8 variants
association: Causative
gene_term:
preferred_term: FGF8
term:
id: hgnc:3686
label: FGF8
notes: >-
Decreased FGF8 signaling causes GnRH deficiency in humans and mice; FGF8 is the
principal FGFR1 ligand in GnRH neuron ontogeny.
evidence:
- reference: PMID:18596921
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "While loss-of-function mutations in FGF receptor 1 (FGFR1) cause human GnRH deficiency, to date no specific ligand for FGFR1 has been identified in GnRH neuron ontogeny."
explanation: Establishes FGF8 as the FGFR1 ligand whose deficiency causes GnRH deficiency.
- name: CHD7 variants
association: Causative
gene_term:
preferred_term: CHD7
term:
id: hgnc:20626
label: CHD7
notes: >-
Heterozygous CHD7 variants cause Kallmann syndrome / IHH; isolated disease can represent
a milder allelic variant of CHARGE syndrome.
evidence:
- reference: PMID:18834967
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Seven heterozygous mutations, two splice and \nfive missense, which were absent in > or = 180 controls, were identified in \nthree sporadic KS and four sporadic normosmic IHH patients."
explanation: CHD7 mutations were identified in sporadic Kallmann syndrome patients.
diagnosis:
- name: Biochemical confirmation of hypogonadotropic hypogonadism
description: >-
Diagnosis requires biochemical testing showing low serum testosterone or estradiol with
low or inappropriately normal LH and FSH, in the setting of otherwise normal anterior
pituitary anatomy and function and after excluding secondary causes.
diagnosis_term:
preferred_term: circulating hormone measurement
term:
id: MAXO:0035005
label: circulating hormone measurement
results: Low sex steroids with low or inappropriately normal LH and FSH support isolated GnRH deficiency.
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "using biochemical testing that reveals low serum \ntestosterone or estradiol (hypogonadism) that results from complete or partial \nabsence of GnRH-mediated release of LH and FSH"
explanation: GeneReviews defines the biochemical, exclusion-based diagnostic pattern.
treatments:
- name: Sex steroid replacement therapy
description: >-
Testosterone (or hCG injections) in males or estrogen and progestin in females to induce
and maintain secondary sexual characteristics.
treatment_term:
preferred_term: Pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "To induce and maintain secondary sex \ncharacteristics, gradually increasing doses of testosterone or human chorionic \ngonadotropin (hCG) injections in males or estrogen and progestin in females"
explanation: GeneReviews recommends sex steroid replacement to induce and maintain secondary sex characteristics.
- name: Fertility-inducing gonadotropin or pulsatile GnRH therapy
description: >-
Combined gonadotropin therapy or pulsatile GnRH therapy to stimulate spermatogenesis or
folliculogenesis when fertility is desired.
treatment_term:
preferred_term: Pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
evidence:
- reference: PMID:20301509
supports: SUPPORT
evidence_source: OTHER
snippet: "to \nstimulate spermatogenesis or folliculogenesis, either combined gonadotropin \ntherapy"
explanation: GeneReviews describes gonadotropin/pulsatile GnRH therapy to restore fertility.
references:
- reference: PMID:20301509
title: "Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency."
tags:
- GeneReviews
References & Deep Research
References
1Deep Research
11. Disease Information
1.1 Overview (current understanding)
KS is a rare congenital disorder characterized by hypogonadotropic hypogonadism plus impaired sense of smell, with broad phenotypic heterogeneity (reproductive and non-reproductive anomalies). (laitinen2011incidencephenotypicfeatures pages 1-2, zak2024kallmannsyndromecausessymptoms pages 1-5)
Key mechanistic definition: KS “results from disturbed intrauterine migration of gonadotropin-releasing hormone (GnRH) neurons from the olfactory placode to the hypothalamus.” (laitinen2011incidencephenotypicfeatures pages 1-2)
1.2 Key identifiers (available in retrieved sources)
- OMIM (MIM): KS is explicitly listed as MIM#147950. (laitinen2011incidencephenotypicfeatures pages 1-2)
- Related OMIM: hypogonadotropic hypogonadism is cited as MIM#146110 in the same source. (laitinen2011incidencephenotypicfeatures pages 1-2)
- ICD-10 (as used in Finnish discharge register ascertainment): hypogonadotropic hypogonadism E23.04 (and ICD-9 253.4). (laitinen2011incidencephenotypicfeatures pages 1-2)
Not found in the retrieved evidence snippets (therefore cannot be asserted here with citations): Orphanet ORPHA code, MeSH ID, MONDO ID, and ICD-11 code for KS specifically. (laitinen2011incidencephenotypicfeatures pages 1-2, meczekalski2013kallmannsyndromein pages 1-2)
1.3 Synonyms and alternative names
A women-focused KS review explicitly lists alternative names/synonyms: “de Morsier syndrome”, “dysplasia olfactogenitalis”, and “familial hypogonadism with anosmia”. (meczekalski2013kallmannsyndromein pages 1-2)
1.4 Evidence provenance
Most information in this report is derived from aggregated disease-level resources: population-based epidemiology (Finland), cohort sequencing studies, and clinical genetics reviews—not from EHR-only sources. (laitinen2011incidencephenotypicfeatures pages 1-2, kałuzna2021defectsingnrh pages 1-2, sayed2023paneltestingfor pages 1-2)
2. Etiology
2.1 Disease causal factors
Primary cause: genetic defects affecting GnRH neuron development/migration and/or hypothalamic–pituitary signaling, producing congenital GnRH deficiency and associated olfactory defects. (laitinen2011incidencephenotypicfeatures pages 1-2, kałuzna2021defectsingnrh pages 1-2)
2.2 Risk factors
- Genetic: pathogenic/likely pathogenic variants in multiple genes (see Section 4). KS is “genetically heterogeneous,” with multiple inheritance modes (X-linked, autosomal dominant/recessive, and oligogenic). (laitinen2011incidencephenotypicfeatures pages 1-2, sayed2023paneltestingfor pages 1-2)
- Non-genetic/environmental: no specific environmental risk factors were identified in the retrieved evidence; KS is generally treated as primarily genetic/developmental in the cited sources. (laitinen2011incidencephenotypicfeatures pages 1-2, sayed2023paneltestingfor pages 1-2)
2.3 Protective factors
No protective genetic or environmental factors were identified in the retrieved evidence. (laitinen2011incidencephenotypicfeatures pages 1-2, sayed2023paneltestingfor pages 1-2)
2.4 Gene–environment interactions
No specific gene–environment interaction evidence was identified in the retrieved texts. (laitinen2011incidencephenotypicfeatures pages 1-2, sayed2023paneltestingfor pages 1-2)
3. Phenotypes
KS has a wide phenotype spectrum including reproductive, olfactory, congenital anomaly, neurologic, and psychosocial manifestations. (laitinen2011incidencephenotypicfeatures pages 1-2, zak2024kallmannsyndromecausessymptoms pages 1-5)
| Phenotype (plain) | Suggested HPO term(s) | Typical timing/onset | Notes on frequency | Key evidence (include abstract quotes if present) | Key references/URL |
|---|---|---|---|---|---|
| Hypogonadotropic hypogonadism | HP:0000044 Hypogonadotropic hypogonadism | Congenital; usually recognized in infancy (mini-puberty) or adolescence | Core/defining feature; effectively universal in KS by definition | KS is defined as congenital HH with olfactory dysfunction; Laitinen: KS is “comprised of congenital hypogonadotropic hypogonadism (HH) and anosmia” (laitinen2011incidencephenotypicfeatures pages 1-2) | Laitinen 2011 https://doi.org/10.1186/1750-1172-6-41; Żak 2024 review (zak2024kallmannsyndromecausessymptoms pages 1-5) |
| Delayed or absent puberty | HP:0000823 Delayed puberty; HP:0008197 Absent puberty | Adolescence | Very common/core presentation; often the reason for diagnosis | Żak 2024 notes absent or incomplete pubertal development in adolescence; Meczekalski 2013 describes “absence of spontaneous puberty” and arrested sexual maturation in female KS (zak2024kallmannsyndromecausessymptoms pages 1-5, meczekalski2013kallmannsyndromein pages 3-4) | Meczekalski 2013 https://doi.org/10.3109/09513590.2012.752459; Żak 2024 review (zak2024kallmannsyndromecausessymptoms pages 1-5) |
| Anosmia / hyposmia | HP:0000458 Anosmia; HP:0004409 Hyposmia | Congenital, though often recognized in childhood/adolescence | Core/defining feature; required to distinguish KS from normosmic CHH | Sayed 2023 lists anosmia as a clinical “red flag”; Laitinen used UPSIT with anosmia defined as “<5th percentile for age”; Żak 2024 and Liu 2022 define KS by HH plus hyposmia/anosmia (laitinen2011incidencephenotypicfeatures pages 1-2, sayed2023paneltestingfor pages 1-2, liu2022advancesingenetic pages 1-2) | Sayed 2023 https://doi.org/10.1038/s41431-022-01261-0; Laitinen 2011 https://doi.org/10.1186/1750-1172-6-41 |
| Infertility / subfertility | HP:0000789 Infertility | Usually recognized in adulthood | Very common if untreated; treatment-responsive in many patients | Sayed 2023 states CHH/KS causes “reduced potential for fertility” and that fertility can be restored in “approximately 75% of men and women”; Żak 2024 notes infertility is common in adults with KS (sayed2023paneltestingfor pages 1-2, zak2024kallmannsyndromecausessymptoms pages 1-5) | Sayed 2023 https://doi.org/10.1038/s41431-022-01261-0; Żak 2024 review (zak2024kallmannsyndromecausessymptoms pages 1-5) |
| Cryptorchidism | HP:0000028 Cryptorchidism | Neonatal/infancy in males | Important early clue in boys; qualitative frequency high enough to be a classic red flag; one Chinese IHH cohort reported 35% overall among male patients, not KS-specific (zak2024kallmannsyndromecausessymptomsa pages 1-5) | Sayed 2023 includes cryptorchidism among KS/CHH “red flag” features; Żak 2024 lists neonatal male presentation with cryptorchidism; Liu 2022 highlights neonatal male signs including cryptorchidism (sayed2023paneltestingfor pages 1-2, zak2024kallmannsyndromecausessymptoms pages 1-5, liu2022advancesingenetic pages 1-2) | Sayed 2023 https://doi.org/10.1038/s41431-022-01261-0; Żak 2024 review (zak2024kallmannsyndromecausessymptoms pages 1-5) |
| Micropenis | HP:0000054 Micropenis | Neonatal/infancy in males | Important early clue; qualitative classic sign of congenital GnRH deficiency | Sayed 2023 lists micropenis as a clinical red flag; Liu 2022 notes neonatal male signs such as “cryptorchidism and micropenis (stretched penile length <2.5 cm)”; Żak 2024 also notes micropenis in neonatal males (sayed2023paneltestingfor pages 1-2, liu2022advancesingenetic pages 1-2, zak2024kallmannsyndromecausessymptoms pages 1-5) | Sayed 2023 https://doi.org/10.1038/s41431-022-01261-0; Liu 2022 https://doi.org/10.1007/s43032-021-00638-8 |
| Renal agenesis (often unilateral) | HP:0000122 Renal agenesis; HP:0010957 Unilateral renal agenesis | Congenital | Non-reproductive associated anomaly; frequency variable and gene-dependent | Żak 2024 lists “Unilateral renal agenesis”; Laitinen 2011 and Liu 2022 include renal agenesis among associated non-reproductive features; Sayed 2023 lists renal agenesis among red flags (zak2024kallmannsyndromecausessymptoms pages 5-8, laitinen2011incidencephenotypicfeatures pages 1-2, sayed2023paneltestingfor pages 1-2, liu2022advancesingenetic pages 1-2) | Laitinen 2011 https://doi.org/10.1186/1750-1172-6-41; Sayed 2023 https://doi.org/10.1038/s41431-022-01261-0 |
| Cleft lip and/or palate | HP:0000202 Cleft palate; HP:0000204 Cleft upper lip | Congenital | Non-reproductive associated anomaly; variable | Laitinen 2011 lists “cleft lip/palate”; Żak 2024 lists “cleft palate and lip”; Sayed 2023 includes midline defects such as cleft palate (laitinen2011incidencephenotypicfeatures pages 1-2, zak2024kallmannsyndromecausessymptoms pages 5-8, sayed2023paneltestingfor pages 1-2) | Laitinen 2011 https://doi.org/10.1186/1750-1172-6-41; Żak 2024 review (zak2024kallmannsyndromecausessymptoms pages 5-8) |
| Dental agenesis / hypodontia | HP:0009804 Tooth agenesis; HP:0000674 Hypodontia | Congenital; often recognized in childhood/adolescence | Variable but well-established associated feature | Laitinen 2011 lists “dental agenesis”; Żak 2024 lists “hypodontia”; Liu 2022 includes “dental agenesis” in the broad phenotype; FGFR1-associated dental anomalies are repeatedly cited (laitinen2011incidencephenotypicfeatures pages 1-2, zak2024kallmannsyndromecausessymptoms pages 5-8, liu2022advancesingenetic pages 1-2) | Laitinen 2011 https://doi.org/10.1186/1750-1172-6-41; Liu 2022 https://doi.org/10.1007/s43032-021-00638-8 |
| Synkinesis / mirror movements | HP:0003128 Mirror movements | Childhood onward; often longstanding | Classic associated neurologic sign; variable, gene-enriched | Laitinen 2011 lists “mirror movements”; Sayed 2023 lists “synkinesis (mirror movements)” as a red flag; Liu 2022 includes “mirror movements” in the phenotypic spectrum (laitinen2011incidencephenotypicfeatures pages 1-2, sayed2023paneltestingfor pages 1-2, liu2022advancesingenetic pages 1-2) | Laitinen 2011 https://doi.org/10.1186/1750-1172-6-41; Sayed 2023 https://doi.org/10.1038/s41431-022-01261-0 |
| Hearing impairment | HP:0000365 Hearing impairment | Congenital or early-life; may be recognized later | Variable associated feature | Laitinen 2011 lists “hearing impairment”; Żak 2024 notes “central hearing impairment”; Liu 2022 lists “hearing loss” among broader phenotypes; He 2023 lists hearing loss among non-reproductive features (laitinen2011incidencephenotypicfeatures pages 1-2, zak2024kallmannsyndromecausessymptoms pages 5-8, liu2022advancesingenetic pages 1-2, he2023clinicalmanifestationsgenetic pages 1-2) | Laitinen 2011 https://doi.org/10.1186/1750-1172-6-41; He 2023 https://doi.org/10.2147/ijgm.s430904 |
| Eye movement abnormalities / ataxia | HP:0000640 Oculomotor apraxia/abnormality of eye movement; HP:0001251 Ataxia | Childhood onward | Variable neurologic manifestations; not universal | Liu 2022 lists “eye movement abnormalities”; Żak 2024 lists “ataxia”; Laitinen 2011 includes associated anomalies and later reviews emphasize cerebellar/oculomotor involvement in some patients (liu2022advancesingenetic pages 1-2, zak2024kallmannsyndromecausessymptoms pages 5-8, laitinen2011incidencephenotypicfeatures pages 1-2) | Liu 2022 https://doi.org/10.1007/s43032-021-00638-8; Żak 2024 review (zak2024kallmannsyndromecausessymptoms pages 5-8) |
| Psychological impact / reduced quality of life | HP:0012735 Emotional lability or use broader term: HP:0000716 Depression; HP:0000739 Anxiety | Often emerges in adolescence/adulthood | Qualitatively important; related to delayed puberty, infertility, body image, chronic disease burden | Żak 2024 notes “significant psychological morbidity” and elevated BDI/BAI/ASEX scores in cited literature; the review emphasizes psychosocial burden and the need for psychological support (zak2024kallmannsyndromecausessymptomsa pages 5-8, zak2024kallmannsyndromecausessymptomsa pages 8-13) | Żak 2024 review (zak2024kallmannsyndromecausessymptomsa pages 5-8, zak2024kallmannsyndromecausessymptomsa pages 8-13) |
| Low gonadotropins and inhibin B | HP:0011968 Decreased circulating luteinizing hormone level; HP:0011969 Decreased circulating follicle stimulating hormone level; HP:0034343 Decreased circulating inhibin B level | Detectable in infancy (mini-puberty) and at diagnostic evaluation later | Characteristic laboratory abnormality; central to diagnosis | Żak 2024: “Patients of both sexes with KS exhibit very low plasma levels of gonadotropins, including FSH, LH and inhibin B”; He 2023 describes “low or inappropriately normal serum levels of luteinizing hormone (LH), follicle-stimulating hormone (FSH)” with low sex steroids (zak2024kallmannsyndromecausessymptomsa pages 5-8, he2023clinicalmanifestationsgenetic pages 1-2, zak2024kallmannsyndromecausessymptoms pages 5-8) | Żak 2024 review (zak2024kallmannsyndromecausessymptomsa pages 5-8); He 2023 https://doi.org/10.2147/ijgm.s430904 |
| Primary amenorrhea / absent breast development in affected females | HP:0000786 Primary amenorrhea; HP:0000066 Hypogonadism; HP:0000824 Delayed menarche | Adolescence | Common female presentation but female cases are under-recognized | Żak 2024 notes underdeveloped breasts and primary amenorrhea in girls; Meczekalski 2013 discusses incomplete secondary sexual characteristics in women with KS (zak2024kallmannsyndromecausessymptoms pages 1-5, meczekalski2013kallmannsyndromein pages 3-4) | Meczekalski 2013 https://doi.org/10.3109/09513590.2012.752459; Żak 2024 review (zak2024kallmannsyndromecausessymptoms pages 1-5) |
| Osteopenia / osteoporosis / fracture risk | HP:0000939 Osteoporosis; HP:0002758 Osteopenia | Usually chronic, emerging in adolescence/adulthood if untreated | Secondary complication of untreated hypogonadism; clinically important | Meczekalski 2013 states “Untreated KS patients have increased risk of osteoporosis” and “higher incidence of osteopenia or osteoporosis and have a greater fracture risk”; Żak 2024 notes risk of early osteoporotic fractures (meczekalski2013kallmannsyndromein pages 3-4, zak2024kallmannsyndromecausessymptomsa pages 1-5) | Meczekalski 2013 https://doi.org/10.3109/09513590.2012.752459; Żak 2024 review (zak2024kallmannsyndromecausessymptomsa pages 1-5) |
Table: This table summarizes the core reproductive, olfactory, neurologic, congenital, psychological, and laboratory phenotypes reported in Kallmann syndrome, with suggested HPO mappings and evidence-linked notes. It is useful for structured disease knowledge-base curation and phenotype annotation.
3.1 Phenotypic spectrum and timing
- Neonatal/infancy (males): cryptorchidism and micropenis may be early “red flags” for CHH/KS. (sayed2023paneltestingfor pages 1-2, liu2022advancesingenetic pages 1-2)
- Adolescence: absent or incomplete puberty and delayed sexual maturation are common triggers for diagnosis. (zak2024kallmannsyndromecausessymptomsa pages 1-5, meczekalski2013kallmannsyndromein pages 3-4)
- Adulthood: infertility/subfertility becomes prominent if untreated; chronic hypogonadism contributes to low bone mineral density and fracture risk. (meczekalski2013kallmannsyndromein pages 3-4, zak2024kallmannsyndromecausessymptoms pages 1-5)
3.2 Frequency data and cohort statistics (available)
- Finnish epidemiology/ascertainment: KS minimal incidence 1:48,000 overall (sex-stratified in Section 9). (laitinen2011incidencephenotypicfeatures pages 1-2)
- Genetic diagnostic yield in a KS cohort (Poland, panel sequencing): P/LP variants detected in 43.5% (20/46) and oligogenic P/LP defects in 26% (12/46). (kałuzna2021defectsingnrh pages 1-2)
For specific non-reproductive phenotype frequencies (e.g., renal agenesis rate, mirror movements rate), the retrieved evidence identifies these features but does not provide consistent percentages in the snippets available; therefore only qualitative assertions are provided. (laitinen2011incidencephenotypicfeatures pages 1-2, zak2024kallmannsyndromecausessymptoms pages 5-8)
4. Genetic / Molecular Information
4.1 Causal genes (representative, non-exhaustive)
Multiple genes are implicated, including ANOS1 (KAL1), FGFR1, FGF8, PROK2, PROKR2, CHD7, WDR11, SOX10, SEMA3A, among others; genetic heterogeneity and incomplete penetrance are emphasized across sources. (laitinen2011incidencephenotypicfeatures pages 1-2, sayed2023paneltestingfor pages 1-2, he2023clinicalmanifestationsgenetic pages 1-2, zak2024kallmannsyndromecausessymptoms pages 1-5)
| Gene (HGNC symbol) | Typical inheritance in KS | Biological role/mechanism (short) | Evidence (include abstract quote if present) | Key references (with DOI/URL) |
|---|---|---|---|---|
| ANOS1 (formerly KAL1) | X-linked recessive; can contribute to oligogenic KS | Encodes anosmin-1; involved in olfactory axon/GnRH neuron development and migration from olfactory placode to hypothalamus | Review evidence lists KAL1/ANOS1 among core KS genes and classifies it as X-linked; KS mechanistically “results from disturbed intrauterine migration of gonadotropin-releasing hormone (GnRH) neurons from the olfactory placode to the hypothalamus” (zak2024kallmannsyndromecausessymptoms pages 1-5, laitinen2011incidencephenotypicfeatures pages 1-2). Laitinen et al. found KAL1 mutations in 3 men in the Finnish cohort (laitinen2011incidencephenotypicfeatures pages 1-2). | Laitinen 2011, Orphanet J Rare Dis — https://doi.org/10.1186/1750-1172-6-41; Żak 2024 review (zak2024kallmannsyndromecausessymptoms pages 1-5, laitinen2011incidencephenotypicfeatures pages 1-2) |
| FGFR1 | Usually autosomal dominant; incomplete penetrance; also seen in oligogenic disease | FGF receptor controlling olfactory bulb development, GnRH neuron ontogeny/migration, craniofacial/dental development | Laitinen: “a monoallelic mutation in FGFR1 underlies approximately 10% of KS cases” and mutations were found in all 5 women vs. 4/25 men in their cohort (laitinen2011incidencephenotypicfeatures pages 1-2). Chu 2023: “autosomal dominant (FGFR1, FGF8, and CHD7...)” and identified novel FGFR1 variants including pathogenic frameshift/CNV lesions (laitinen2011incidencephenotypicfeatures pages 1-2). | Laitinen 2011 — https://doi.org/10.1186/1750-1172-6-41; Chu 2023 — https://doi.org/10.1186/s12958-023-01074-w |
| FGF8 | Usually autosomal dominant; sometimes incomplete penetrance/oligogenic contribution | Ligand for FGFR1 pathway; critical for embryonic olfactory/GnRH neuronal development | Core KS reviews consistently include FGF8 in dominant KS genetics and within the neurodevelopmental class of genes linked to anosmic CHH/KS (zak2024kallmannsyndromecausessymptoms pages 1-5, stamou2018kallmannsyndromephenotype pages 1-3, zak2024kallmannsyndromecausessymptoms pages 5-8). The FGFR1/FGF8 axis is repeatedly associated with craniofacial and dental phenotypes in KS (zak2024kallmannsyndromecausessymptoms pages 5-8). | Chu 2023 — https://doi.org/10.1186/s12958-023-01074-w; Stamou 2018 — https://doi.org/10.1016/j.metabol.2017.10.012 |
| PROK2 | Usually autosomal recessive; may participate in digenic/oligogenic KS | Ligand in prokineticin signaling required for olfactory bulb morphogenesis and GnRH neuron migration/guidance | Gene lists from Finnish and later reviews include PROK2 among canonical KS genes with AR inheritance patterns and possible oligogenicity (laitinen2011incidencephenotypicfeatures pages 1-2, liu2022advancesingenetic pages 1-2). Sayed 2023 emphasizes CHH/KS complexity with “di- and oligogenic, as well as classic monogenic, inheritance and incomplete penetrance” (sayed2023paneltestingfor pages 1-2). | Laitinen 2011 — https://doi.org/10.1186/1750-1172-6-41; Sayed 2023 — https://doi.org/10.1038/s41431-022-01261-0 |
| PROKR2 | Often autosomal recessive or dominant with reduced penetrance; frequent digenic/oligogenic contributor | GPCR for PROK2; regulates olfactory bulb formation and GnRH neuron migration; variants can also affect broader neuroendocrine phenotypes | Martinez-Mayer 2023 abstract: “Mice lacking Prokr2 have been shown to present abnormal olfactory bulb formation as well as defects in GnRH neuron migration. Patients carrying mutations in PROKR2 typically present hypogonadotropic hypogonadism, anosmia/hyposmia or Kallmann Syndrome” (zak2024kallmannsyndromecausessymptomsa pages 1-5). He 2023 found PROKR2 heterozygous variants in both KS and nIHH; Kałużna 2021 reported oligogenic P/LP defects in 26% of KS patients (he2023clinicalmanifestationsgenetic pages 1-2, kałuzna2021defectsingnrh pages 1-2). | Martinez-Mayer 2023 — https://doi.org/10.3389/fendo.2023.1132787; He 2023 — https://doi.org/10.2147/ijgm.s430904; Kałużna 2021 — https://doi.org/10.3390/genes12060868 |
| CHD7 | Usually autosomal dominant; variable expressivity; incomplete penetrance; oligogenic cases reported | Chromatin remodeler implicated in neural crest/olfactory/GnRH development; overlaps KS–CHARGE spectrum | Laitinen notes some KS patients show CHARGE-like features even without CHD7 mutation, supporting pathway overlap (laitinen2011incidencephenotypicfeatures pages 1-2). He 2023 detected CHD7 variants in both KS and nIHH; Sayed 2023 highlights incomplete penetrance and oligogenic inheritance in CHH panels (he2023clinicalmanifestationsgenetic pages 1-2, sayed2023paneltestingfor pages 1-2). | Laitinen 2011 — https://doi.org/10.1186/1750-1172-6-41; He 2023 — https://doi.org/10.2147/ijgm.s430904; Sayed 2023 — https://doi.org/10.1038/s41431-022-01261-0 |
| WDR11 | Usually autosomal dominant or oligogenic contributor; incomplete penetrance reported | Developmental regulator affecting GnRH neuronal development and hypothalamic-pituitary signaling | Included in canonical KS gene sets from Laitinen and later NGS studies (laitinen2011incidencephenotypicfeatures pages 1-2, he2023clinicalmanifestationsgenetic pages 1-2). Kałużna 2021 showed that genes affecting “GnRH neuron migration/development and hypothalamic-pituitary signaling” contribute to clinical variability in KS, supporting WDR11 as a pathway gene (kałuzna2021defectsingnrh pages 1-2). | Laitinen 2011 — https://doi.org/10.1186/1750-1172-6-41; He 2023 — https://doi.org/10.2147/ijgm.s430904; Kałużna 2021 — https://doi.org/10.3390/genes12060868 |
| SOX10 | Usually autosomal dominant; can be syndromic; reduced penetrance/variable expressivity | Neural crest transcription factor; links KS with hearing/pigmentary phenotypes and olfactory/GnRH developmental defects | He 2023: “a novel likely pathogenic variant in the SOX10 (c.429–1G>C) was considered to cause the KS phenotype” (he2023clinicalmanifestationsgenetic pages 1-2). Żak 2024 also lists SOX10 among implicated autosomal dominant genes and notes hearing/pigmentary manifestations in KS (zak2024kallmannsyndromecausessymptomsa pages 1-5). | He 2023 — https://doi.org/10.2147/ijgm.s430904; Żak 2024 review (zak2024kallmannsyndromecausessymptomsa pages 1-5) |
| SEMA3A | Likely autosomal dominant susceptibility/modifier gene; often oligogenic | Axon guidance cue influencing olfactory/GnRH neuron pathfinding | Żak 2024 lists SEMA3A among genes “under investigation” in KS (zak2024kallmannsyndromecausessymptoms pages 1-5). He 2023 includes SEMA3A among common IHH/KS genes in the NGS era (he2023clinicalmanifestationsgenetic pages 1-2). Kałużna 2021 places KS genes within the broader class of migration/guidance genes; Sayed 2023 highlights panel-based diagnosis amid oligogenicity/incomplete penetrance (kałuzna2021defectsingnrh pages 1-2, sayed2023paneltestingfor pages 1-2). | He 2023 — https://doi.org/10.2147/ijgm.s430904; Sayed 2023 — https://doi.org/10.1038/s41431-022-01261-0 |
| RMST (lncRNA) | Structural-disruption/LOF mechanism reported in isolated case; inheritance not yet established as classic Mendelian pattern | Long noncoding RNA regulating neural crest/GnRH ontogeny; affects downstream developmental genes | Stamou 2020 abstract: “A novel deletion in RMST implicates the loss of function of a lncRNA as a unique cause of KS and suggests it plays a critical role in the ontogeny of GnRH neurons and puberty” (zak2024kallmannsyndromecausessymptomsa pages 1-5). In patient-derived cells, RMST reduction was associated with abnormal neural crest morphology and altered expression of SOX2, PAX3, CHD7, TUBB3, MKRN3 (zak2024kallmannsyndromecausessymptomsa pages 1-5). | Stamou 2020 — https://doi.org/10.1210/clinem/dgz011 |
| Cross-gene architecture note | Monogenic, digenic, and oligogenic inheritance; incomplete penetrance common | KS is genetically heterogeneous; neurodevelopmental and hypothalamic-pituitary pathway defects converge on GnRH deficiency plus olfactory dysfunction | Kałużna 2021 abstract: “The prevalence of oligogenic P/LP defects in selected genes among KS patients was 26% (12/46)” and P/LP variants were found in 43.5% of the cohort (kałuzna2021defectsingnrh pages 1-2). Sayed 2023: CHH genetics includes “di- and oligogenic, as well as classic monogenic, inheritance and incomplete penetrance” (sayed2023paneltestingfor pages 1-2). Liu 2022: “Approximately 40% of KS patients have one or several rare sequence variants that have been identified” (liu2022advancesingenetic pages 1-2). | Kałużna 2021 — https://doi.org/10.3390/genes12060868; Sayed 2023 — https://doi.org/10.1038/s41431-022-01261-0; Liu 2022 — https://doi.org/10.1007/s43032-021-00638-8 |
Table: This table summarizes the main genes implicated in Kallmann syndrome, their typical inheritance patterns, and the developmental mechanisms linking them to GnRH deficiency and olfactory dysfunction. It is useful for rapid comparison of core KS genes while highlighting oligogenicity and incomplete penetrance.
4.2 Pathogenic variant types and classification practices
- A 2023 KS mutation study reported identification of novel ANOS1 variants including splice-altering mutations validated by functional splicing assay, and FGFR1 variants including frameshift and pathogenic CNV deletions, interpreted under ACMG/ClinGen standards. (zak2024kallmannsyndromecausessymptomsa pages 1-5)
- A 2023 sporadic IHH/KS cohort explicitly classified variants according to ACMG-AMP guidelines and reported a novel likely pathogenic SOX10 splice variant (c.429–1G>C) in a KS patient. (he2023clinicalmanifestationsgenetic pages 1-2)
4.3 Oligogenicity and diagnostic yield (recent emphasis)
- A KS cohort sequencing study found “The prevalence of oligogenic P/LP defects…was 26% (12/46)” and P/LP variants in “43.5%.” (kałuzna2021defectsingnrh pages 1-2)
- A CHH genetic testing perspective emphasizes that CHH/KS genetics includes “di- and oligogenic, as well as classic monogenic, inheritance and incomplete penetrance,” and notes a curated 14-gene panel adopted in the UK NHS Genomic Medicine Service for CHH molecular diagnosis. (sayed2023paneltestingfor pages 1-2)
4.4 Epigenetic information
Direct epigenetic mechanisms (methylation/histone marks) were not identified in the retrieved evidence; however, chromatin remodeling genes (e.g., CHD7) can contribute to KS/CHH phenotypes. (sayed2023paneltestingfor pages 1-2, he2023clinicalmanifestationsgenetic pages 1-2)
4.5 Chromosomal/structural abnormalities
A structural defect affecting a long noncoding RNA was described: a KS patient with a balanced translocation implicating RMST. The abstract concludes: “A novel deletion in RMST implicates the loss of function of a lncRNA as a unique cause of KS and suggests it plays a critical role in the ontogeny of GnRH neurons and puberty.” (zak2024kallmannsyndromecausessymptomsa pages 1-5)
5. Environmental Information
No specific toxins, lifestyle exposures, or infectious agents were identified as contributors in the retrieved KS-focused evidence; the disorder is predominantly presented as genetic/developmental. (laitinen2011incidencephenotypicfeatures pages 1-2, sayed2023paneltestingfor pages 1-2)
6. Mechanism / Pathophysiology
6.1 Causal chain (current consensus in retrieved sources)
- Upstream developmental defect: genetic disruptions in pathways controlling olfactory system development and GnRH neuron ontogeny/migration. (laitinen2011incidencephenotypicfeatures pages 1-2, kałuzna2021defectsingnrh pages 1-2)
- Key developmental event: GnRH neurons normally migrate from the olfactory placode/nasal region to the hypothalamus; KS arises when this is disturbed. (laitinen2011incidencephenotypicfeatures pages 1-2, kałuzna2021defectsingnrh pages 1-2)
- Downstream endocrine consequence: impaired GnRH pulsatility → low/inappropriately normal LH/FSH → hypogonadism, absent puberty, infertility. (he2023clinicalmanifestationsgenetic pages 1-2, zak2024kallmannsyndromecausessymptomsa pages 5-8)
- Parallel olfactory phenotype: olfactory bulb/tract hypoplasia or agenesis may accompany the GnRH neuronal defect (e.g., MRI findings). (zak2024kallmannsyndromecausessymptomsa pages 5-8, zak2024kallmannsyndromecausessymptoms pages 5-8)
6.2 Molecular/cellular processes and example pathway anchors
- GnRH neuronal migration and axon guidance: A KS genetics study describes GnRH neurons originating in the nasal region and migrating toward the brain, consistent with shared developmental origins of the olfactory and reproductive axes. (kałuzna2021defectsingnrh pages 1-2)
- PROKR2 biology: Review-level mechanistic evidence links PROKR2 loss to abnormal olfactory bulb formation and GnRH neuron migration; the abstract states: “Mice lacking Prokr2 have been shown to present abnormal olfactory bulb formation as well as defects in GnRH neuron migration. Patients carrying mutations in PROKR2 typically present hypogonadotropic hypogonadism, anosmia/hyposmia or Kallmann Syndrome.” (zak2024kallmannsyndromecausessymptomsa pages 1-5)
6.3 Suggested ontology terms
- GO biological processes (suggested):
- GnRH neuron migration/development (e.g., neuron migration; axon guidance; olfactory bulb development; regulation of GnRH secretion)
- Cell types (CL, suggested):
- Gonadotropin-releasing hormone neuron; olfactory ensheathing cell (supporting GnRH axon pathfinding); neural crest cell (as implicated in RMST/iPSC-NCC modeling) (kałuzna2021defectsingnrh pages 1-2, zak2024kallmannsyndromecausessymptomsa pages 1-5)
7. Anatomical Structures Affected
7.1 Organ and system level
- Hypothalamic–pituitary–gonadal axis (endocrine/reproductive) leading to hypogonadism and infertility. (he2023clinicalmanifestationsgenetic pages 1-2, sayed2023paneltestingfor pages 1-2)
- Olfactory system (olfactory bulbs/tracts), often with structural or functional deficits. (zak2024kallmannsyndromecausessymptomsa pages 5-8, zak2024kallmannsyndromecausessymptoms pages 5-8)
7.2 Tissue and cell level (suggested)
- Neuroendocrine neurons: GnRH neurons (hypothalamic). (laitinen2011incidencephenotypicfeatures pages 1-2)
- Olfactory pathway-associated cells: olfactory-related guidance/support (e.g., olfactory ensheathing cells, based on migration framework). (kałuzna2021defectsingnrh pages 1-2)
7.3 Subcellular level
No consistent subcellular compartment pathology (e.g., mitochondria/ER) is specified in the retrieved evidence.
8. Temporal Development
- Onset: congenital, but diagnosis commonly occurs in adolescence; neonatal male signs may enable earlier recognition. (zak2024kallmannsyndromecausessymptomsa pages 1-5, sayed2023paneltestingfor pages 1-2)
- Course: chronic lifelong without treatment; phenotypic severity ranges from severe HH with absent puberty to partial puberty, and “reversal of hypogonadotropism later in life” is possible in some cases. (laitinen2011incidencephenotypicfeatures pages 1-2)
- Reversal: a recent review states spontaneous recovery occurs in “approximately 10–20%,” though details are not in the snippet. (zak2024kallmannsyndromecausessymptoms pages 5-8)
9. Inheritance and Population
9.1 Epidemiology (recently cited benchmark)
A Finnish population-based ascertainment study reports: “The minimal incidence estimate of KS in Finland was 1:48 000, with clear difference between males (1:30 000) and females (1:125 000).” (laitinen2011incidencephenotypicfeatures pages 1-2)
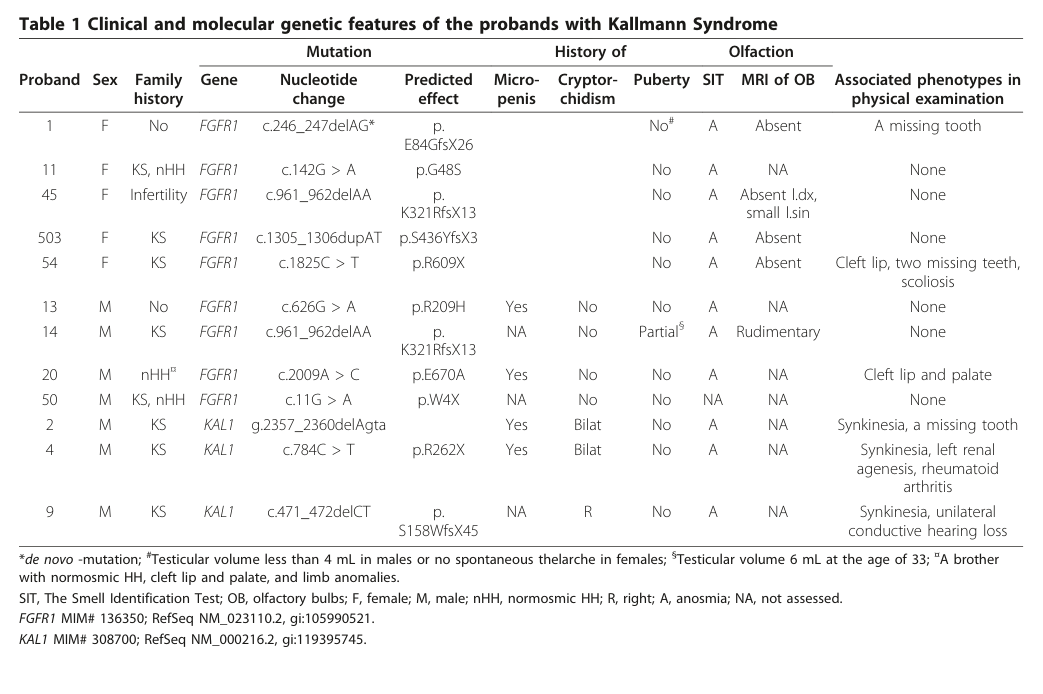
A key visual summary of clinical features and genotypes appears in Table 1 of this Finnish paper (cropped). (laitinen2011incidencephenotypicfeatures media a022cd77)
9.2 Inheritance patterns
- Family patterns include X-linked recessive, autosomal dominant, autosomal recessive, and oligogenic inheritance. (laitinen2011incidencephenotypicfeatures pages 1-2)
- Clinical genetics perspective underscores incomplete penetrance and the need for careful interpretation of genetic results in panels. (sayed2023paneltestingfor pages 1-2)
10. Diagnostics
10.1 Clinical and laboratory testing
- Hormones: KS commonly shows very low gonadotropins; one review states: “Patients of both sexes with KS exhibit very low plasma levels of gonadotropins, including FSH, LH and inhibin B.” (zak2024kallmannsyndromecausessymptomsa pages 5-8)
- Mini-puberty window (infants): highlighted as a diagnostic opportunity (1–3 months) to assess congenital GnRH deficiency. (zak2024kallmannsyndromecausessymptomsa pages 5-8)
10.2 Olfactory testing and imaging
- Formal olfactory assessment: UPSIT thresholds were used in Finnish ascertainment (anosmia <5th percentile for age). (laitinen2011incidencephenotypicfeatures pages 1-2)
- MRI: reviews emphasize MRI findings such as unilateral/bilateral agenesis of olfactory bulbs/tracts in KS patients, supporting diagnosis and phenotyping. (zak2024kallmannsyndromecausessymptomsa pages 5-8, zak2024kallmannsyndromecausessymptoms pages 5-8)
10.3 Genetic testing (real-world implementation)
- Modern practice increasingly uses NGS-based panels/WES due to genetic heterogeneity. (he2023clinicalmanifestationsgenetic pages 1-2, sayed2023paneltestingfor pages 1-2)
- In the UK NHS Genomic Medicine Service, a curated 14-gene CHH diagnostic panel has been adopted (review report). (sayed2023paneltestingfor pages 1-2)
10.4 Differential diagnosis
Reviews highlight differentiation from constitutional delay of growth and puberty (CDGP), CHARGE syndrome, tumors causing acquired HH, and functional hypogonadotropic hypogonadism. (zak2024kallmannsyndromecausessymptomsa pages 5-8, zak2024kallmannsyndromecausessymptoms pages 5-8)
11. Outcome / Prognosis
- Fertility: CHH/KS is considered treatable for fertility; one authoritative review states fertility “can be restored in approximately 75% of men and women” with appropriate therapy. (sayed2023paneltestingfor pages 1-2)
- Bone health: untreated KS is associated with osteoporosis/osteopenia and increased fracture risk (secondary to chronic hypogonadism). (meczekalski2013kallmannsyndromein pages 3-4)
- Psychosocial burden: reviews note substantial psychological morbidity and emphasize psychological support as part of management. (zak2024kallmannsyndromecausessymptomsa pages 5-8, zak2024kallmannsyndromecausessymptomsa pages 8-13)
Mortality/life expectancy statistics specific to KS were not identified in the retrieved evidence snippets.
12. Treatment
12.1 Hormone replacement (puberty induction / maintenance)
- Males: testosterone replacement is used to induce/maintain virilization and secondary sexual characteristics. (zak2024kallmannsyndromecausessymptoms pages 5-8, sayed2023paneltestingfor pages 1-2)
- Females: 17β-estradiol with later addition of progesterone (or estrogen–progestin regimens) to induce/maintain feminization and protect endometrium. (zak2024kallmannsyndromecausessymptoms pages 5-8, meczekalski2013kallmannsyndromein pages 3-4)
12.2 Fertility induction
- Pulsatile GnRH or gonadotropin therapy (e.g., hCG/FSH-based regimens) are used for fertility induction in adults (both sexes). (zak2024kallmannsyndromecausessymptoms pages 5-8)
- Treatment success for fertility restoration is summarized at population level as ~75% in the CHH panel-testing review. (sayed2023paneltestingfor pages 1-2)
12.3 Surgical/interventional and supportive care
- Cryptorchidism repair is recommended early in infancy (review suggests 6–12 months). (zak2024kallmannsyndromecausessymptoms pages 5-8)
- Psychological support is repeatedly recommended given psychosocial impacts. (zak2024kallmannsyndromecausessymptomsa pages 1-5, zak2024kallmannsyndromecausessymptomsa pages 5-8)
12.4 Experimental/advanced therapeutics
No KS-specific gene therapy or RNA therapy clinical trials were identified in retrieved evidence.
12.5 Suggested MAXO terms (examples)
- Hormone replacement therapy (sex steroid replacement)
- Gonadotropin therapy
- Pulsatile GnRH therapy
- Orchidopexy (cryptorchidism repair)
13. Prevention
Primary prevention is not currently established for KS (congenital genetic/developmental). However, reproductive genetic options are discussed in a genetic diagnosis review, including prenatal diagnosis and preimplantation genetic testing (PGT) to reduce transmission risk in affected families. (liu2022advancesingenetic pages 1-2)
14. Other Species / Natural Disease
No naturally occurring veterinary analogs were identified in the retrieved evidence.
15. Model Organisms
Mouse models are used to support mechanism. For example, Prokr2 loss in mice is associated with “abnormal olfactory bulb formation” and “defects in GnRH neuron migration,” paralleling the human KS phenotype spectrum. (zak2024kallmannsyndromecausessymptomsa pages 1-5)
Recent developments and latest research emphasis (2023–2024)
- Clinical deployment of curated genetic panels (UK NHS): A 2023 European Journal of Human Genetics review reports a curated 14-gene panel for CHH diagnosis within the NHS Genomic Medicine Service and highlights pitfalls/benefits in real-world endocrine diagnostics, stressing oligogenicity and incomplete penetrance. Publication date: Dec 2023; URL: https://doi.org/10.1038/s41431-022-01261-0 (sayed2023paneltestingfor pages 1-2)
- Oligogenicity quantified in KS cohort sequencing: A KS cohort NGS study reports 26% oligogenic P/LP defects and 43.5% P/LP detection (Genes, 2021; still widely relevant as quantification). (kałuzna2021defectsingnrh pages 1-2)
- 2024 literature synthesis: A 2024 KS review summarizes contemporary diagnostic workup (including mini-puberty, olfactory testing, MRI), modern genetics, and treatment approaches including reported reversal (10–20%). (zak2024kallmannsyndromecausessymptomsa pages 1-5, zak2024kallmannsyndromecausessymptoms pages 5-8)
Key visual evidence
A cropped Table 1 from the Finland epidemiology/genetics study summarizes clinical and molecular genetic features across KS probands (supporting phenotypic heterogeneity and genotype-phenotype mapping). (laitinen2011incidencephenotypicfeatures media a022cd77)
Limitations of this evidence package
- Standard ontology identifiers (MONDO, Orphanet ORPHA, MeSH ID, ICD-11 for KS) were not present in the retrieved full-text snippets and therefore are not asserted here.
- Several clinically important phenotype frequencies (e.g., renal agenesis %, mirror movements %) are identified qualitatively but were not numerically extractable from the available snippets.
References
-
(laitinen2011incidencephenotypicfeatures pages 1-2): Eeva-Maria Laitinen, Kirsi Vaaralahti, Johanna Tommiska, Elina Eklund, Mari Tervaniemi, Leena Valanne, and Taneli Raivio. Incidence, phenotypic features and molecular genetics of kallmann syndrome in finland. Orphanet Journal of Rare Diseases, 6:41-41, Jun 2011. URL: https://doi.org/10.1186/1750-1172-6-41, doi:10.1186/1750-1172-6-41. This article has 244 citations and is from a peer-reviewed journal.
-
(zak2024kallmannsyndromecausessymptoms pages 1-5): N Żak. Kallmann syndrome-causes, symptoms, treatment-review of literature. Unknown journal, 2024.
-
(meczekalski2013kallmannsyndromein pages 3-4): Blazej Meczekalski, Agnieszka Podfigurna-Stopa, Roman Smolarczyk, Krzysztof Katulski, and Andrea R. Genazzani. Kallmann syndrome in women: from genes to diagnosis and treatment. Gynecological Endocrinology, 29:296-300, Mar 2013. URL: https://doi.org/10.3109/09513590.2012.752459, doi:10.3109/09513590.2012.752459. This article has 46 citations and is from a peer-reviewed journal.
-
(zak2024kallmannsyndromecausessymptomsa pages 1-5): N Żak. Kallmann syndrome-causes, symptoms, treatment-review of literature. Unknown journal, 2024.
-
(meczekalski2013kallmannsyndromein pages 1-2): Blazej Meczekalski, Agnieszka Podfigurna-Stopa, Roman Smolarczyk, Krzysztof Katulski, and Andrea R. Genazzani. Kallmann syndrome in women: from genes to diagnosis and treatment. Gynecological Endocrinology, 29:296-300, Mar 2013. URL: https://doi.org/10.3109/09513590.2012.752459, doi:10.3109/09513590.2012.752459. This article has 46 citations and is from a peer-reviewed journal.
-
(kałuzna2021defectsingnrh pages 1-2): Małgorzata Kałużna, Bartłomiej Budny, Michał Rabijewski, Jarosław Kałużny, Agnieszka Dubiel, Małgorzata Trofimiuk-Müldner, Elżbieta Wrotkowska, Alicja Hubalewska-Dydejczyk, Marek Ruchała, and Katarzyna Ziemnicka. Defects in gnrh neuron migration/development and hypothalamic-pituitary signaling impact clinical variability of kallmann syndrome. Genes, 12:868, Jun 2021. URL: https://doi.org/10.3390/genes12060868, doi:10.3390/genes12060868. This article has 19 citations.
-
(sayed2023paneltestingfor pages 1-2): Yasmin Al Sayed and Sasha R. Howard. Panel testing for the molecular genetic diagnosis of congenital hypogonadotropic hypogonadism – a clinical perspective. European Journal of Human Genetics, 31:387-394, Dec 2023. URL: https://doi.org/10.1038/s41431-022-01261-0, doi:10.1038/s41431-022-01261-0. This article has 35 citations and is from a domain leading peer-reviewed journal.
-
(liu2022advancesingenetic pages 1-2): Yujun Liu and Xu Zhi. Advances in genetic diagnosis of kallmann syndrome and genetic interruption. Reproductive Sciences, 29:1697-1709, Jul 2022. URL: https://doi.org/10.1007/s43032-021-00638-8, doi:10.1007/s43032-021-00638-8. This article has 42 citations and is from a peer-reviewed journal.
-
(zak2024kallmannsyndromecausessymptoms pages 5-8): N Żak. Kallmann syndrome-causes, symptoms, treatment-review of literature. Unknown journal, 2024.
-
(he2023clinicalmanifestationsgenetic pages 1-2): Dongye He, Hailing Sun, Mei Zhang, Yanying Li, Fupeng Liu, Yanhong Zhang, Mingming He, and Bo Ban. Clinical manifestations, genetic variants and therapeutic evaluation in sporadic chinese patients with idiopathic hypogonadotropic hypogonadism. International Journal of General Medicine, 16:4429-4439, Sep 2023. URL: https://doi.org/10.2147/ijgm.s430904, doi:10.2147/ijgm.s430904. This article has 1 citations.
-
(zak2024kallmannsyndromecausessymptomsa pages 5-8): N Żak. Kallmann syndrome-causes, symptoms, treatment-review of literature. Unknown journal, 2024.
-
(zak2024kallmannsyndromecausessymptomsa pages 8-13): N Żak. Kallmann syndrome-causes, symptoms, treatment-review of literature. Unknown journal, 2024.
-
(stamou2018kallmannsyndromephenotype pages 1-3): Maria I. Stamou and Neoklis A. Georgopoulos. Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Sep 2018. URL: https://doi.org/10.1016/j.metabol.2017.10.012, doi:10.1016/j.metabol.2017.10.012. This article has 222 citations.
-
(laitinen2011incidencephenotypicfeatures media a022cd77): Eeva-Maria Laitinen, Kirsi Vaaralahti, Johanna Tommiska, Elina Eklund, Mari Tervaniemi, Leena Valanne, and Taneli Raivio. Incidence, phenotypic features and molecular genetics of kallmann syndrome in finland. Orphanet Journal of Rare Diseases, 6:41-41, Jun 2011. URL: https://doi.org/10.1186/1750-1172-6-41, doi:10.1186/1750-1172-6-41. This article has 244 citations and is from a peer-reviewed journal.