Hermansky-Pudlak Syndrome
Hermansky-Pudlak Syndrome (HPS) is a rare autosomal recessive disorder caused by biallelic pathogenic variants in any of 11 known HPS genes (HPS1–HPS11). The shared mechanistic lesion is defective biogenesis of lysosome-related organelles (LROs), producing three core features: oculocutaneous albinism from defective melanosomes, bleeding diathesis from absent platelet dense granules, and in HPS-1 and HPS-4, progressive pulmonary fibrosis driven by dysfunctional lamellar bodies in alveolar type II cells.
Ask OpenScientist
Ask a research question about Hermansky-Pudlak Syndrome. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (1 reference)
Subtypes
11Pathophysiology
5Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Pathograph
Phenotypes
11Blood 2
Show evidence (1 reference)
Show evidence (1 reference)
Digestive 1
Show evidence (1 reference)
Eye 2
Show evidence (2 references)
Show evidence (1 reference)
Immune 1
Show evidence (1 reference)
Integument 1
Show evidence (2 references)
Respiratory 1
Show evidence (3 references)
Other 3
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
11Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Medical Actions
8Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Clinical Trials
3Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Source YAML
click to showname: Hermansky-Pudlak Syndrome
creation_date: "2026-06-16T00:00:00Z"
category: Genetic
description: >-
Hermansky-Pudlak Syndrome (HPS) is a rare autosomal recessive disorder caused
by biallelic pathogenic variants in any of 11 known HPS genes (HPS1–HPS11).
The shared mechanistic lesion is defective biogenesis of lysosome-related
organelles (LROs), producing three core features: oculocutaneous albinism from
defective melanosomes, bleeding diathesis from absent platelet dense granules,
and in HPS-1 and HPS-4, progressive pulmonary fibrosis driven by dysfunctional
lamellar bodies in alveolar type II cells.
disease_term:
preferred_term: Hermansky-Pudlak syndrome
term:
id: MONDO:0019312
label: Hermansky-Pudlak syndrome
synonyms:
- HPS
- Hermansky-Pudlak syndrome pulmonary fibrosis
- HPS-PF
parents:
- Disorder of lysosome-related organelle biogenesis
- Oculocutaneous albinism
- Inherited platelet function disorder
references:
- reference: PMID:20301464
title: "Hermansky-Pudlak Syndrome."
tags:
- GeneReviews
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:33536261
reference_title: "Hermansky-Pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Hermansky-Pudlak syndrome (HPS), a rare autosomal recessive disorder characterised by abnormal biogenesis of lysosome-related organelles"
explanation: HPS is consistently described as an autosomal recessive disorder of lysosome-related organelle biogenesis.
has_subtypes:
- name: HPS-1
display_name: HPS-1 (HPS1, BLOC-3)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 1
term:
id: MONDO:0008748
label: Hermansky-Pudlak syndrome 1
description: >
Caused by biallelic variants in HPS1, a component of the BLOC-3 complex.
Most common and one of the most severe forms, with oculocutaneous albinism,
bleeding diathesis, and a high lifetime risk of progressive pulmonary
fibrosis and granulomatous colitis. Highly prevalent in northwest Puerto Rico
due to a founder duplication in HPS1.
- name: HPS-2
display_name: HPS-2 (AP3B1, AP-3)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 2
term:
id: MONDO:0011997
label: Hermansky-Pudlak syndrome 2
description: >
Caused by biallelic variants in AP3B1, the beta-3A subunit of the AP-3
adaptor complex. Distinguished by congenital neutropenia and immunodeficiency
with recurrent infections, in addition to albinism and bleeding. Pulmonary
fibrosis can occur and often presents earlier (childhood/young adulthood).
- name: HPS-3
display_name: HPS-3 (HPS3, BLOC-2)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 3
term:
id: MONDO:0013555
label: Hermansky-Pudlak syndrome 3
description: >
Caused by biallelic variants in HPS3, a component of the BLOC-2 complex.
Milder phenotype with subtle hypopigmentation; pulmonary fibrosis is
typically absent.
- name: HPS-4
display_name: HPS-4 (HPS4, BLOC-3)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 4
term:
id: MONDO:0013556
label: Hermansky-Pudlak syndrome 4
description: >
Caused by biallelic variants in HPS4, the second subunit of the BLOC-3
complex (with HPS1). Like HPS-1, it carries a high risk of pulmonary
fibrosis and granulomatous colitis.
- name: HPS-5
display_name: HPS-5 (HPS5, BLOC-2)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 5
term:
id: MONDO:0013557
label: Hermansky-Pudlak syndrome 5
description: >
Caused by biallelic variants in HPS5, a component of the BLOC-2 complex.
Milder form without pulmonary fibrosis; hypopigmentation and nystagmus
may be subtle.
- name: HPS-6
display_name: HPS-6 (HPS6, BLOC-2)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 6
term:
id: MONDO:0013558
label: Hermansky-Pudlak syndrome 6
description: >
Caused by biallelic variants in HPS6, a component of the BLOC-2 complex.
Milder form without pulmonary fibrosis.
- name: HPS-7
display_name: HPS-7 (DTNBP1, BLOC-1)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 7
term:
id: MONDO:0013559
label: Hermansky-Pudlak syndrome 7
description: >
Caused by biallelic variants in DTNBP1 (dysbindin), a component of the
BLOC-1 complex. Rare; albinism and bleeding diathesis.
- name: HPS-8
display_name: HPS-8 (BLOC1S3, BLOC-1)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 8
term:
id: MONDO:0013560
label: Hermansky-Pudlak syndrome 8
description: >
Caused by biallelic variants in BLOC1S3, a component of the BLOC-1 complex.
Rare; albinism and bleeding diathesis.
- name: HPS-9
display_name: HPS-9 (BLOC1S6/PLDN, BLOC-1)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 9
term:
id: MONDO:0013606
label: Hermansky-Pudlak syndrome 9
description: >
Caused by biallelic variants in BLOC1S6 (pallidin, PLDN), a component of
the BLOC-1 complex. Rare.
- name: HPS-10
display_name: HPS-10 (AP3D1, AP-3)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 10
term:
id: MONDO:0014885
label: Hermansky-Pudlak syndrome 10
description: >
Caused by biallelic variants in AP3D1, the delta subunit of the AP-3
adaptor complex. Severe form with neurodevelopmental involvement,
immunodeficiency, and neutropenia in addition to albinism and bleeding.
- name: HPS-11
display_name: HPS-11 (BLOC1S5, BLOC-1)
classification: molecular_complex
subtype_term:
preferred_term: Hermansky-Pudlak syndrome 11
term:
id: MONDO:0030903
label: Hermansky-Pudlak syndrome 11
description: >
Caused by biallelic variants in BLOC1S5, a component of the BLOC-1 complex.
Rare.
pathophysiology:
- name: Defective Biogenesis of Lysosome-Related Organelles
role: Primary
description: >
HPS results from defective biogenesis of lysosome-related organelles (LROs)
such as melanosomes, platelet dense granules, and the lamellar bodies of
alveolar type II cells. The causative genes encode subunits of multi-protein
complexes (the AP-3 adaptor complex and the BLOC-1, BLOC-2, and BLOC-3
complexes) that direct cargo trafficking from early endosomes to maturing
LROs. Loss of any subunit impairs LRO formation, producing the shared
multisystem phenotype.
cell_types:
- preferred_term: melanocyte
term:
id: CL:0000148
label: melanocyte
- preferred_term: platelet
term:
id: CL:0000233
label: platelet
- preferred_term: pulmonary alveolar type 2 cell
term:
id: CL:0002063
label: pulmonary alveolar type 2 cell
biological_processes:
- preferred_term: lysosome organization
term:
id: GO:0007040
label: lysosome organization
modifier: DECREASED
- preferred_term: vesicle-mediated transport
term:
id: GO:0016192
label: vesicle-mediated transport
modifier: DECREASED
evidence:
- reference: PMID:39457053
reference_title: "Pathogenesis and Therapy of Hermansky-Pudlak Syndrome (HPS)-Associated Pulmonary Fibrosis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "the HPS proteins play an essential role in the biogenesis and function of lysosome-related organelles (LROs) in alveolar epithelial type II (AT2) cells"
explanation: HPS proteins are required for biogenesis of lysosome-related organelles, the unifying molecular defect.
- reference: PMID:33536261
reference_title: "Hermansky-Pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "characterised by abnormal biogenesis of lysosome-related organelles, manifests with oculocutaneous albinism and excessive bleeding of variable severity"
explanation: Defective LRO biogenesis is the shared mechanism producing albinism and bleeding across subtypes.
- reference: PMID:35129281
reference_title: "New insights into the pathogenesis of Hermansky-Pudlak syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "HPS protein-associated complexes (HPACs) function in cargo transport, cargo recycling, and cargo removal to maintain LRO homeostasis."
explanation: Defines the role of HPS protein complexes in cargo trafficking that maintains lysosome-related organelle homeostasis.
downstream:
- target: Defective Melanosome Biogenesis
description: Impaired LRO trafficking blocks normal maturation of melanosomes in melanocytes and the retinal pigment epithelium.
causal_link_type: DIRECT
evidence:
- reference: PMID:39457053
reference_title: "Pathogenesis and Therapy of Hermansky-Pudlak Syndrome (HPS)-Associated Pulmonary Fibrosis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "the HPS proteins play an essential role in the biogenesis and function of lysosome-related organelles (LROs)"
explanation: Loss of HPS-complex function impairs biogenesis of melanosomes, a lysosome-related organelle.
- target: Platelet Dense Granule Deficiency
description: Impaired LRO trafficking prevents formation of platelet dense (delta) granules.
causal_link_type: DIRECT
evidence:
- reference: PMID:27529121
reference_title: "Pulmonary Fibrosis in Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "a bleeding diathesis due to platelet dysfunction"
explanation: The platelet defect arises from absent dense granules, a lysosome-related organelle.
- target: Alveolar Type II Cell Lamellar Body Dysfunction
description: Defective LRO biogenesis disrupts lamellar bodies (surfactant-storing LROs) in alveolar type II cells, especially in BLOC-3/AP-3 subtypes.
causal_link_type: DIRECT
evidence:
- reference: PMID:39457053
reference_title: "Pathogenesis and Therapy of Hermansky-Pudlak Syndrome (HPS)-Associated Pulmonary Fibrosis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "HPS-PF is associated with dysfunction of AT2 cells and abnormal immune reactions"
explanation: LRO (lamellar body) dysfunction in AT2 cells initiates the pulmonary-fibrosis cascade.
- name: Defective Melanosome Biogenesis
description: >
Failure to mature melanosomes in melanocytes and the retinal pigment
epithelium reduces melanin production, producing oculocutaneous albinism
with the characteristic ocular features (nystagmus, foveal hypoplasia,
reduced visual acuity).
cell_types:
- preferred_term: melanocyte
term:
id: CL:0000148
label: melanocyte
biological_processes:
- preferred_term: melanosome organization
term:
id: GO:0032438
label: melanosome organization
modifier: DECREASED
evidence:
- reference: PMID:33841163
reference_title: "Hermansky-Pudlak Syndrome and Lung Disease: Pathogenesis and Therapeutics."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "characterized by oculocutaneous albinism (OCA), bleeding diathesis, immunodeficiency, granulomatous colitis, and pulmonary fibrosis"
explanation: Oculocutaneous albinism is a defining feature attributable to defective melanosome biogenesis.
downstream:
- target: Oculocutaneous albinism
description: Reduced melanin in skin, hair, and eyes manifests as hypopigmentation and ocular albinism.
causal_link_type: DIRECT
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Hermansky-Pudlak syndrome (HPS) is characterized by oculocutaneous albinism"
explanation: Defective melanosome biogenesis produces the oculocutaneous albinism phenotype.
- name: Platelet Dense Granule Deficiency
description: >
Absence of platelet dense (delta) granules removes the stored pool of ADP,
ATP, serotonin, and calcium needed for the platelet secretion and second-wave
aggregation response, producing a storage-pool bleeding diathesis. Absent
dense bodies on whole-mount electron microscopy is the diagnostic hallmark.
cell_types:
- preferred_term: platelet
term:
id: CL:0000233
label: platelet
biological_processes:
- preferred_term: platelet dense granule organization
term:
id: GO:0060155
label: platelet dense granule organization
modifier: DECREASED
- preferred_term: platelet degranulation
term:
id: GO:0002576
label: platelet degranulation
modifier: DECREASED
- preferred_term: platelet aggregation
term:
id: GO:0070527
label: platelet aggregation
modifier: DECREASED
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "demonstration of absence of platelet delta granules (dense bodies) on electron microscopy"
explanation: The platelet defect is due to absent dense granules, demonstrable diagnostically by electron microscopy.
downstream:
- target: Bleeding diathesis
description: Impaired dense-granule secretion and aggregation produces variable mucocutaneous and surgical bleeding.
causal_link_type: DIRECT
evidence:
- reference: PMID:27529121
reference_title: "Pulmonary Fibrosis in Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "a bleeding diathesis due to platelet dysfunction"
explanation: Dense-granule deficiency causes the platelet dysfunction underlying the bleeding diathesis.
- name: Alveolar Type II Cell Lamellar Body Dysfunction
description: >
In BLOC-3 (HPS-1, HPS-4) and AP-3 (HPS-2) subtypes, defective lamellar-body
LRO biogenesis causes alveolar type II cell stress and injury. Histopathology
shows vacuolated hyperplastic type II cells with enlarged lamellar bodies and
alveolar macrophages with lipofuscin-like deposits, distinguishing HPS-PF
from idiopathic pulmonary fibrosis.
cell_types:
- preferred_term: pulmonary alveolar type 2 cell
term:
id: CL:0002063
label: pulmonary alveolar type 2 cell
evidence:
- reference: PMID:33536261
reference_title: "Hermansky-Pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Histopathology of HPS pulmonary fibrosis, and not IPF, shows vacuolated hyperplastic type II cells with enlarged lamellar bodies and alveolar macrophages with lipofuscin-like deposits"
explanation: AT2 cell lamellar-body abnormality is the histopathologic correlate of the lung disease.
downstream:
- target: Type 2 Innate Immune Amplification of Fibrosis
description: AT2 cell injury triggers an aberrant type 2 innate immune response that amplifies fibrosis.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- AT2 epithelial injury and CHI3L1 release recruit and activate type 2 innate lymphoid cells.
evidence:
- reference: PMID:39405112
reference_title: "Type 2 innate immunity promotes the development of pulmonary fibrosis in Hermansky-Pudlak syndrome."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "ILC2s were recruited and appeared to contribute to fibrosis development in the Hps1-/- mice"
explanation: Epithelial injury in Hps1-deficient lung drives ILC2 recruitment that contributes to fibrosis.
- name: Type 2 Innate Immune Amplification of Fibrosis
description: >
A CHI3L1-CRTH2 axis recruits and activates type 2 innate lymphoid cells
(ILC2s) in HPS lungs. ILC2s stimulate fibroblast proliferation and
differentiation, partly via amphiregulin-EGFR signaling, amplifying the
fibrotic response after epithelial injury.
cell_types:
- preferred_term: group 2 innate lymphoid cell
term:
id: CL:0001069
label: group 2 innate lymphoid cell
- preferred_term: lung fibroblast
term:
id: CL:0002553
label: fibroblast of lung
biological_processes:
- preferred_term: fibroblast proliferation
term:
id: GO:0048144
label: fibroblast proliferation
modifier: INCREASED
- preferred_term: collagen fibril organization
term:
id: GO:0030199
label: collagen fibril organization
modifier: INCREASED
evidence:
- reference: PMID:39405112
reference_title: "Type 2 innate immunity promotes the development of pulmonary fibrosis in Hermansky-Pudlak syndrome."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: "ILC2s may directly stimulate the proliferation and differentiation of primary lung fibroblasts partially through amphiregulin-EGFR-dependent mechanisms"
explanation: ILC2-fibroblast co-culture experiments show amphiregulin-EGFR-dependent fibroblast activation driving the fibroproliferative response in HPS lung.
downstream:
- target: Pulmonary fibrosis
description: Sustained fibroblast activation and collagen deposition produce progressive restrictive lung disease.
causal_link_type: DIRECT
evidence:
- reference: PMID:39405112
reference_title: "Type 2 innate immunity promotes the development of pulmonary fibrosis in Hermansky-Pudlak syndrome."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "ILC2-mediated, CRTH2-dependent mechanisms might contribute to optimal CHI3L1-induced fibroproliferative repair in HPS-associated pulmonary fibrosis"
explanation: The ILC2/CRTH2/CHI3L1 fibroproliferative response culminates in HPS pulmonary fibrosis.
phenotypes:
- category: Phenotypic
name: Oculocutaneous albinism
description: >
Hypopigmentation of skin, hair, and eyes due to defective melanosome
biogenesis in melanocytes and retinal pigment epithelium. Hair ranges from
white to brown and skin from white to olive, usually lighter than unaffected
family members.
phenotype_term:
preferred_term: Albinism
term:
id: HP:0001022
label: Albinism
frequency: VERY_FREQUENT
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Hermansky-Pudlak syndrome (HPS) is characterized by oculocutaneous albinism, a bleeding diathesis"
explanation: Oculocutaneous albinism is a defining and near-universal feature of HPS.
- reference: PMID:35886065
reference_title: "Clinical Features and Novel Genetic Variants Associated with Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Hermansky-Pudlak syndrome (HPS) is a rare autosomal recessive syndromic form of albinism, characterized by oculocutaneous albinism (OCA) and other systemic complications."
explanation: Independent corroboration that HPS is a syndromic form of oculocutaneous albinism.
- category: Phenotypic
name: Ocular albinism with reduced visual acuity
description: >
Ocular findings include nystagmus, reduced iris and retinal pigment, foveal
hypoplasia with significant reduction in visual acuity (usually 20/50 to
20/400), and strabismus in many individuals.
phenotype_term:
preferred_term: Ocular albinism
term:
id: HP:0001107
label: Ocular albinism
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Ocular findings include nystagmus, reduced iris pigment, reduced retinal pigment, foveal hypoplasia with significant reduction in visual acuity"
explanation: Ocular albinism with foveal hypoplasia and reduced acuity is a core clinical feature.
- category: Phenotypic
name: Nystagmus
description: Involuntary eye movements, part of the ocular albinism phenotype.
phenotype_term:
preferred_term: Nystagmus
term:
id: HP:0000639
label: Nystagmus
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Ocular findings include nystagmus, reduced iris pigment"
explanation: Nystagmus is among the characteristic ocular findings.
- reference: PMID:35886065
reference_title: "Clinical Features and Novel Genetic Variants Associated with Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "photophobia, and nystagmus was detected in all (4/4) patients"
explanation: Nystagmus was present in all genetically confirmed HPS patients in this clinical cohort.
- category: Phenotypic
name: Foveal hypoplasia
description: >
Underdevelopment of the fovea is a characteristic ocular feature of the
albinism in HPS and contributes to reduced visual acuity.
phenotype_term:
preferred_term: Foveal hypoplasia
term:
id: HP:0007750
label: Hypoplasia of the fovea
evidence:
- reference: PMID:35886065
reference_title: "Clinical Features and Novel Genetic Variants Associated with Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Fundus examination revealed fundus hypopigmentation and foveal hypoplasia in all (8/8) eyes."
explanation: Foveal hypoplasia was present in all examined eyes of genetically confirmed HPS patients.
- category: Phenotypic
name: Photophobia
description: >
Light sensitivity arising from iris and retinal hypopigmentation, a common
ocular symptom of the albinism in HPS.
phenotype_term:
preferred_term: Photophobia
term:
id: HP:0000613
label: Photophobia
evidence:
- reference: PMID:35886065
reference_title: "Clinical Features and Novel Genetic Variants Associated with Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "photophobia, and nystagmus was detected in all (4/4) patients"
explanation: Photophobia was present in all genetically confirmed HPS patients in this clinical cohort.
- category: Phenotypic
name: Bleeding diathesis
description: >
Prolonged bleeding from platelet dense-granule (delta granule) deficiency,
causing variable bruising, epistaxis, gingival bleeding, postpartum
hemorrhage, and prolonged bleeding with menses or after surgery.
phenotype_term:
preferred_term: Abnormal bleeding
term:
id: HP:0001892
label: Abnormal bleeding
frequency: VERY_FREQUENT
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The bleeding diathesis can result in variable degrees of bruising, epistaxis, gingival bleeding, postpartum hemorrhage"
explanation: The bleeding diathesis is a defining feature with broad mucocutaneous manifestations.
- category: Phenotypic
name: Abnormal platelet function
description: Storage-pool platelet defect with absent dense granules on electron microscopy.
phenotype_term:
preferred_term: Abnormal platelet function
term:
id: HP:0011869
label: Abnormal platelet function
evidence:
- reference: PMID:27529121
reference_title: "Pulmonary Fibrosis in Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "a bleeding diathesis due to platelet dysfunction"
explanation: Platelet dysfunction underlies the bleeding phenotype.
- category: Phenotypic
name: Pulmonary fibrosis
description: >
Progressive restrictive lung disease seen mainly in HPS-1, HPS-2, and HPS-4.
Typically causes symptoms in the early 30s and can progress to death within a
decade. In HPS-1 it is essentially fully penetrant.
phenotype_term:
preferred_term: Pulmonary fibrosis
term:
id: HP:0002206
label: Pulmonary fibrosis
subtypes:
- HPS-1
- HPS-2
- HPS-4
evidence:
- reference: PMID:33841163
reference_title: "Hermansky-Pudlak Syndrome and Lung Disease: Pathogenesis and Therapeutics."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "HPS pulmonary fibrosis (HPS-PF) occurs in 100% of patients with subtype HPS-1 and has a similar presentation to idiopathic pulmonary fibrosis"
explanation: Pulmonary fibrosis is a major complication, fully penetrant in HPS-1.
- reference: PMID:33536261
reference_title: "Hermansky-Pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Pulmonary fibrosis is highly prevalent in three out of 10 genetic types of HPS (HPS-1, HPS-2 and HPS-4)"
explanation: Confirms the subtype distribution of pulmonary fibrosis.
- reference: PMID:35129281
reference_title: "New insights into the pathogenesis of Hermansky-Pudlak syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "typically manifesting with oculocutaneous albinism or ocular albinism, bleeding tendency, and in some cases with pulmonary fibrosis, inflammatory bowel disease or immunodeficiency, neuropsychological disorders."
explanation: Pulmonary fibrosis is listed among the variable multisystem complications of HPS.
- category: Phenotypic
name: Granulomatous colitis
description: >
Inflammatory bowel disease resembling Crohn disease; severe in about 15% of
affected individuals.
phenotype_term:
preferred_term: Colitis
term:
id: HP:0002583
label: Colitis
frequency: OCCASIONAL
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Granulomatous colitis is severe in about 15% of affected individuals"
explanation: Granulomatous colitis affects a clinically significant minority of patients.
- category: Phenotypic
name: Immunodeficiency
description: >
Immunodeficiency and immune defects occur primarily in individuals with
pathogenic variants in AP3B1 (HPS-2) and AP3D1 (HPS-10).
phenotype_term:
preferred_term: Immunodeficiency
term:
id: HP:0002721
label: Immunodeficiency
subtypes:
- HPS-2
- HPS-10
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Neutropenia and/or immune defects occur primarily in individuals with pathogenic variants in AP3B1 and AP3D1"
explanation: Immunodeficiency is concentrated in the AP-3 subtypes (HPS-2 and HPS-10).
- category: Phenotypic
name: Neutropenia
description: Congenital/cyclic neutropenia in AP-3 subtypes, contributing to recurrent infections.
phenotype_term:
preferred_term: Decreased total neutrophil count
term:
id: HP:0001875
label: Decreased total neutrophil count
subtypes:
- HPS-2
- HPS-10
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Neutropenia and/or immune defects occur primarily in individuals with pathogenic variants in AP3B1 and AP3D1"
explanation: Neutropenia is a feature of the AP-3 subtypes.
genetic:
- name: HPS1
gene_term:
preferred_term: HPS1
term:
id: hgnc:5163
label: HPS1
association: Causative
subtype: HPS-1
notes: >
BLOC-3 complex component. A recurrent 16-bp duplication in exon 15
(c.1472_1487dup) is a founder variant in northwest Puerto Rico. HPS-1 is
fully penetrant for pulmonary fibrosis.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Identification of biallelic pathogenic variants in AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, or HPS6 confirms the diagnosis"
explanation: Biallelic HPS1 variants are diagnostic of HPS-1.
- name: AP3B1
gene_term:
preferred_term: AP3B1
term:
id: hgnc:566
label: AP3B1
association: Causative
subtype: HPS-2
notes: Beta-3A subunit of the AP-3 adaptor complex. Associated with neutropenia and immunodeficiency.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Neutropenia and/or immune defects occur primarily in individuals with pathogenic variants in AP3B1 and AP3D1"
explanation: Biallelic AP3B1 variants cause HPS-2 with immune involvement.
- name: HPS3
gene_term:
preferred_term: HPS3
term:
id: hgnc:15597
label: HPS3
association: Causative
subtype: HPS-3
notes: BLOC-2 complex component. Milder phenotype; hypopigmentation may not be clinically evident.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Identification of biallelic pathogenic variants in AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, or HPS6 confirms the diagnosis"
explanation: Biallelic HPS3 variants are diagnostic of HPS-3.
- name: HPS4
gene_term:
preferred_term: HPS4
term:
id: hgnc:15844
label: HPS4
association: Causative
subtype: HPS-4
notes: BLOC-3 complex component (with HPS1). High risk of pulmonary fibrosis and granulomatous colitis.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Identification of biallelic pathogenic variants in AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, or HPS6 confirms the diagnosis"
explanation: Biallelic HPS4 variants are diagnostic of HPS-4.
- name: HPS5
gene_term:
preferred_term: HPS5
term:
id: hgnc:17022
label: HPS5
association: Causative
subtype: HPS-5
notes: BLOC-2 complex component. Milder form; pulmonary fibrosis typically absent.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "In families with HPS3-, HPS5-, or HPS6-related HPS (milder types of HPS in which hypopigmentation and nystagmus may not be clinically evident)"
explanation: HPS5-related HPS is a milder BLOC-2 form.
- name: HPS6
gene_term:
preferred_term: HPS6
term:
id: hgnc:18817
label: HPS6
association: Causative
subtype: HPS-6
notes: BLOC-2 complex component. Milder form; pulmonary fibrosis typically absent.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "In families with HPS3-, HPS5-, or HPS6-related HPS (milder types of HPS in which hypopigmentation and nystagmus may not be clinically evident)"
explanation: HPS6-related HPS is a milder BLOC-2 form.
- name: DTNBP1
gene_term:
preferred_term: DTNBP1
term:
id: hgnc:17328
label: DTNBP1
association: Causative
subtype: HPS-7
notes: Dysbindin; BLOC-1 complex component.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Identification of biallelic pathogenic variants in AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, or HPS6 confirms the diagnosis"
explanation: Biallelic DTNBP1 variants cause HPS-7.
- name: BLOC1S3
gene_term:
preferred_term: BLOC1S3
term:
id: hgnc:20914
label: BLOC1S3
association: Causative
subtype: HPS-8
notes: BLOC-1 complex component.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Identification of biallelic pathogenic variants in AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, or HPS6 confirms the diagnosis"
explanation: Biallelic BLOC1S3 variants cause HPS-8.
- name: BLOC1S6
gene_term:
preferred_term: BLOC1S6
term:
id: hgnc:8549
label: BLOC1S6
association: Causative
subtype: HPS-9
notes: Pallidin (PLDN); BLOC-1 complex component.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Identification of biallelic pathogenic variants in AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, or HPS6 confirms the diagnosis"
explanation: Biallelic BLOC1S6 variants cause HPS-9.
- name: AP3D1
gene_term:
preferred_term: AP3D1
term:
id: hgnc:568
label: AP3D1
association: Causative
subtype: HPS-10
notes: Delta subunit of the AP-3 adaptor complex. Severe form with neurodevelopmental involvement, immunodeficiency, and neutropenia.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Neutropenia and/or immune defects occur primarily in individuals with pathogenic variants in AP3B1 and AP3D1"
explanation: Biallelic AP3D1 variants cause HPS-10 with immune involvement.
- name: BLOC1S5
gene_term:
preferred_term: BLOC1S5
term:
id: hgnc:18561
label: BLOC1S5
association: Causative
subtype: HPS-11
notes: BLOC-1 complex component.
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Identification of biallelic pathogenic variants in AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, or HPS6 confirms the diagnosis"
explanation: Biallelic BLOC1S5 variants cause HPS-11.
treatments:
- name: Lung transplantation
description: >
Lung transplantation is the only potentially life-prolonging treatment for
end-stage HPS pulmonary fibrosis.
treatment_term:
preferred_term: whole lung transplantation
term:
id: MAXO:0010038
label: whole lung transplantation
therapeutic_modality: SURGERY
target_phenotypes:
- preferred_term: Pulmonary fibrosis
term:
id: HP:0002206
label: Pulmonary fibrosis
evidence:
- reference: PMID:27529121
reference_title: "Pulmonary Fibrosis in Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "lung transplantation remains the only potentially life-prolonging treatment"
explanation: Lung transplantation is the principal life-prolonging intervention for HPS-PF.
- name: Antifibrotic therapy (pirfenidone, nintedanib)
description: >
Pirfenidone and nintedanib are antifibrotic drugs approved for idiopathic
pulmonary fibrosis but not specifically approved for HPS pulmonary fibrosis;
they have prompted trials and off-label consideration. A randomized
NIH/NHGRI pirfenidone trial in HPS-PF was stopped for futility at interim
analysis.
treatment_term:
preferred_term: Pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
therapeutic_agent:
- preferred_term: pirfenidone
term:
id: CHEBI:32016
label: pirfenidone
- preferred_term: nintedanib
term:
id: CHEBI:85164
label: nintedanib
therapeutic_modality: SMALL_MOLECULE
target_phenotypes:
- preferred_term: Pulmonary fibrosis
term:
id: HP:0002206
label: Pulmonary fibrosis
evidence:
- reference: PMID:27529121
reference_title: "Pulmonary Fibrosis in Hermansky-Pudlak Syndrome."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "the approval of two new antifibrotic drugs, pirfenidone and nintedanib, has prompted new interest in identifying drugs capable of reversing or halting the progression of HPS-PF"
explanation: Antifibrotics are under investigation for HPS-PF but are not established disease-modifying therapy.
- name: Desmopressin (DDAVP)
description: >
DDAVP (desmopressin acetate) is used to support hemostasis for procedures
such as wisdom tooth extraction and other invasive procedures in HPS.
treatment_term:
preferred_term: Pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
therapeutic_agent:
- preferred_term: desmopressin
term:
id: CHEBI:4450
label: desmopressin
therapeutic_modality: PEPTIDE
target_phenotypes:
- preferred_term: Abnormal bleeding
term:
id: HP:0001892
label: Abnormal bleeding
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "DDAVP® (desmopressin acetate) for wisdom tooth extraction and invasive procedures"
explanation: DDAVP is part of bleeding management for procedures in HPS.
- name: Platelet transfusion
description: >
Platelet or red blood cell transfusions are used for surgery or protracted
bleeding; HLA-matched single-donor platelets are used as needed.
treatment_term:
preferred_term: platelet transfusion
term:
id: MAXO:0001490
label: platelet transfusion
target_phenotypes:
- preferred_term: Abnormal bleeding
term:
id: HP:0001892
label: Abnormal bleeding
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "platelet or red blood cell transfusions for surgery or protracted bleeding; HLA-matched single-donor platelets as needed"
explanation: Transfusion supports hemostasis during bleeding or surgery.
- name: Corticosteroids for granulomatous colitis
description: >
Granulomatous colitis in HPS is managed with corticosteroids as a first-line
anti-inflammatory agent.
treatment_term:
preferred_term: Pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
therapeutic_agent:
- preferred_term: corticosteroid
term:
id: CHEBI:50858
label: corticosteroid
therapeutic_modality: SMALL_MOLECULE
target_phenotypes:
- preferred_term: Colitis
term:
id: HP:0002583
label: Colitis
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "steroids, other anti-inflammatory agents, and/or Remicade® for granulomatous colitis"
explanation: GeneReviews lists corticosteroids among anti-inflammatory agents for HPS-associated granulomatous colitis.
- name: Infliximab for granulomatous colitis
description: >
Infliximab (Remicade), an anti-TNF monoclonal antibody, is used for
HPS-associated granulomatous colitis refractory to other anti-inflammatory
agents.
treatment_term:
preferred_term: Pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
therapeutic_agent:
- preferred_term: infliximab
term:
id: NCIT:C1789
label: Infliximab
therapeutic_modality: MONOCLONAL_ANTIBODY
target_phenotypes:
- preferred_term: Colitis
term:
id: HP:0002583
label: Colitis
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "steroids, other anti-inflammatory agents, and/or Remicade® for granulomatous colitis"
explanation: GeneReviews lists Remicade (infliximab) as an anti-TNF option for HPS-associated granulomatous colitis.
- name: Genetic counseling
description: >
HPS is autosomal recessive; carrier testing, prenatal testing, and
preimplantation genetic testing are possible once familial variants are
identified. Cascade testing is appropriate in milder HPS-3/HPS-5/HPS-6 types.
treatment_term:
preferred_term: Genetic Counseling
term:
id: NCIT:C15240
label: Genetic Counseling
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "carrier testing for at-risk family members, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible"
explanation: Genetic counseling and testing are part of standard management.
- name: Bleeding-risk avoidance (NSAIDs and aspirin)
description: >
Over-the-counter nonsteroidal anti-inflammatory products, aspirin-containing
products, and other anticoagulants should be avoided unless medically
indicated because they further impair the already-defective platelet
function. Tobacco and lung-injurious inhalants should also be avoided.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
evidence:
- reference: PMID:20301464
reference_title: "Hermansky-Pudlak Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Over-the-counter nonsteroidal anti-inflammatory products, aspirin-containing products, and other anticoagulants unless medically indicated"
explanation: GeneReviews lists NSAIDs/aspirin among agents to avoid given the bleeding diathesis.
clinical_trials:
- name: NCT00001596
phase: PHASE_II
status: COMPLETED
description: >
Randomized, double-blind trial of oral pirfenidone for the pulmonary
fibrosis of Hermansky-Pudlak syndrome; an interim analysis stopped the study
for futility.
target_phenotypes:
- preferred_term: Pulmonary fibrosis
term:
id: HP:0002206
label: Pulmonary fibrosis
evidence:
- reference: clinicaltrials:NCT00001596
supports: SUPPORT
snippet: "The drug pirfenidone blocks the biochemical process of inflammation and has been reported to slow or reverse pulmonary fibrosis in animal systems."
explanation: NIH/NHGRI placebo-controlled pirfenidone trial in HPS pulmonary fibrosis.
- name: NCT00001456
status: RECRUITING
description: >
NHGRI natural history study, Clinical and Basic Investigations Into
Hermansky-Pudlak Syndrome, including mutation analysis and longitudinal
phenotyping.
evidence:
- reference: clinicaltrials:NCT00001456
supports: SUPPORT
snippet: "Hermansky-Pudlak Syndrome (HPS) is an inherited disease which results in decreased pigmentation (oculocutaneous albinism), bleeding problems due to a platelet abnormality (platelet storage pool defect)"
explanation: Long-running NIH natural history study of HPS, including mutation analysis.
- name: NCT04193592
description: >
Efficacy and Safety of Pirfenidone Treatment in HPS interstitial lung
disease.
target_phenotypes:
- preferred_term: Pulmonary fibrosis
term:
id: HP:0002206
label: Pulmonary fibrosis
evidence:
- reference: clinicaltrials:NCT04193592
supports: SUPPORT
snippet: "This research study will explore the safety and efficacy of the drug, pirfenidone, in patients with a diagnosis of Hermansky-Pudlak Syndrome (HPS) who have an associated interstitial lung disease (ILD)"
explanation: Trial of pirfenidone for HPS-related interstitial lung disease.
References & Deep Research
References
1Deep Research
11. Disease Information
1.1 Concise overview (current understanding)
Hermansky–Pudlak syndrome (HPS) is a group of rare, autosomal recessive disorders characterized by oculocutaneous albinism, platelet dysfunction with a bleeding diathesis (classically due to platelet dense-granule/delta-granule deficiency), and variable systemic involvement including pulmonary fibrosis, granulomatous colitis, and immunodeficiency depending on subtype. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, vicary2016pulmonaryfibrosisin pages 1-2, velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2)
1.2 Key identifiers
Evidence retrieved in this session robustly supports MONDO identifiers (from OpenTargets), but did not contain explicit OMIM/Orphanet/ICD/MeSH IDs.
| Concept | Identifier system | ID | Notes | Key supporting citation |
|---|---|---|---|---|
| Hermansky-Pudlak syndrome | MONDO | MONDO_0019312 | Main disease entry in OpenTargets evidence; associated with 12 targets including HPS1, HPS4, AP3B1, HPS3, HPS5, HPS6, DTNBP1, BLOC1S3, BLOC1S5, BLOC1S6, AP3D1. | (OpenTargets Search: Hermansky-Pudlak syndrome) |
| Hermansky-Pudlak syndrome with pulmonary fibrosis | MONDO | MONDO_0016501 | Subgroup in OpenTargets evidence linked to pulmonary-fibrosis-associated targets HPS4, HPS1, AP3B1. Terminology aligns with HPS pulmonary fibrosis / HPS-PF. | (OpenTargets Search: Hermansky-Pudlak syndrome) |
| Hermansky-Pudlak syndrome without pulmonary fibrosis | MONDO | MONDO_0016502 | Subgroup in OpenTargets evidence linked to non-PF-associated targets such as HPS3, HPS5, HPS6. | (OpenTargets Search: Hermansky-Pudlak syndrome) |
| Hermansky-Pudlak syndrome 10 | MONDO | MONDO_0014885 | Subtype entry in OpenTargets evidence associated with AP3D1. | (OpenTargets Search: Hermansky-Pudlak syndrome) |
| Hermansky-Pudlak syndrome 11 | MONDO | MONDO_0030903 | Subtype entry in OpenTargets evidence associated with BLOC1S5. | (OpenTargets Search: Hermansky-Pudlak syndrome) |
| Hermansky-Pudlak syndrome | Preferred disease name / synonym | HPS | Major shorthand abbreviation used across reviews and trials. | (hu2024pathogenesisandtherapy pages 1-2, velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2, NCT00001456 chunk 1) |
| Hermansky–Pudlak syndrome | Preferred disease name / synonym | — | Standard full disease name; described as a rare autosomal recessive disorder. | (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, vicary2016pulmonaryfibrosisin pages 1-2) |
| HPS pulmonary fibrosis | Disease feature / synonym | HPS-PF | Common term for pulmonary fibrosis occurring in HPS, especially HPS-1, HPS-2, and HPS-4. | (velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2, vicary2016pulmonaryfibrosisin pages 1-2, hu2024pathogenesisandtherapy pages 1-2) |
| HPS-associated pulmonary fibrosis | Disease feature / synonym | HPS-PF (descriptive synonym) | Used in recent literature for the fibrotic lung manifestation; interchangeable with HPS pulmonary fibrosis in context. | (hu2024pathogenesisandtherapy pages 1-2, hu2024pathogenesisandtherapy pages 2-3) |
| Inheritance | Inheritance pattern | Autosomal recessive | Consistently described across reviews and natural history sources. | (hu2024pathogenesisandtherapy pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, vicary2016pulmonaryfibrosisin pages 1-2) |
| OMIM identifier | OMIM | Not found in gathered evidence | Requested identifier system, but no specific OMIM disease ID was retrieved in available evidence. | (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4, vicary2016pulmonaryfibrosisin pages 1-2) |
| Orphanet identifier | Orphanet | Not found in gathered evidence | Requested identifier system, but no specific Orphanet ID was retrieved in available evidence. | (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, hu2024pathogenesisandtherapy pages 1-2) |
| ICD-10 / ICD-11 identifier | ICD | Not found in gathered evidence | Requested identifier system, but no specific ICD code was retrieved in available evidence. | (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, hu2024pathogenesisandtherapy pages 1-2) |
| MeSH identifier | MeSH | Not found in gathered evidence | Requested identifier system, but no specific MeSH ID was retrieved in available evidence. | (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, hu2024pathogenesisandtherapy pages 1-2) |
Table: This table summarizes the disease naming and identifier information for Hermansky-Pudlak syndrome using only evidence gathered in the session. It is useful for quickly mapping MONDO terms, common synonyms, inheritance, and identifier gaps that still need confirmation from external databases.
1.3 Common synonyms / alternative names
Common names include Hermansky–Pudlak syndrome, HPS, and pulmonary-fibrosis-specific terms such as HPS pulmonary fibrosis (HPS-PF) or HPS-associated pulmonary fibrosis. (velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2, hu2024pathogenesisandtherapy pages 1-2)
1.4 Evidence provenance (patient-level vs aggregated)
The evidence base in this run is dominated by aggregated disease-level sources (reviews in 2021 and 2024; clinical trial registry entries) plus some cohort-based and translational studies, including human lung tissue immunostaining and mouse models. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, hu2024pathogenesisandtherapy pages 1-2, sorkhdini2024type2innate pages 2-4, NCT00001596 chunk 1)
2. Etiology
2.1 Disease causal factors
Primary cause: biallelic pathogenic variants in genes encoding components of the AP-3 adaptor complex and BLOC (biogenesis of lysosome-related organelles complex) pathways, leading to defective biogenesis/trafficking of lysosome-related organelles (LROs). LROs implicated include melanosomes (albinism), platelet dense/alpha granules (bleeding), and in the lung lamellar bodies in alveolar type II (AT2) cells (pulmonary fibrosis). (hu2024pathogenesisandtherapy pages 2-3, hu2024pathogenesisandtherapy pages 1-2)
2.2 Risk factors
Genetic risk factors: Subtype strongly influences major complications. Pulmonary fibrosis is concentrated in specific genotypes (HPS-1/HPS-2/HPS-4). (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4, hu2024pathogenesisandtherapy pages 1-2)
Founder effects / population risk: Northwest Puerto Rico shows a strong founder effect for HPS-1; one review reports regional prevalence about ~1/1800 and carrier frequency ~1/22 for the recurrent HPS1 exon 15 duplication c.1472_1487dup16-bp. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4)
Age as a risk factor (for pulmonary fibrosis): HPS pulmonary fibrosis often manifests earlier than idiopathic pulmonary fibrosis, frequently around ages 30–40 in HPS-1 according to a major pulmonary-fibrosis-focused review. (vicary2016pulmonaryfibrosisin pages 1-2)
2.3 Protective factors
No protective genetic or environmental factors were identified in the retrieved evidence set.
2.4 Gene–environment interactions
Pulmonary fibrosis can be modeled by bleomycin challenge in mice with Hps1 deficiency, indicating that environmental or injury triggers can interact with genetic susceptibility to amplify fibrotic responses. (sorkhdini2024type2innate pages 2-4, sorkhdini2024type2innate pages 8-10)
3. Phenotypes (clinical features)
3.1 Core phenotypes (human)
- Oculocutaneous albinism (hypopigmentation, ocular albinism features; poor vision reported in multiple descriptions). (vicary2016pulmonaryfibrosisin pages 1-2, hu2024pathogenesisandtherapy pages 1-2)
-
Suggested HPO terms: HP:0001022 (Albinism), HP:0000539 (Abnormality of the fundus), HP:0000568 (Nystagmus) (general suggestions; HPO IDs not explicitly present in evidence).
-
Bleeding diathesis / platelet function defect due to platelet storage pool/dense-granule defect. A key diagnostic feature is demonstrable by EM: “δ-granules are absent in platelets ... imaged using whole-mount transmission electron microscopy.” (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4)
-
Suggested HPO terms: HP:0001892 (Bleeding diathesis), HP:0001873 (Thrombocytopenia) (context-dependent), HP:0001928 (Abnormality of coagulation).
-
Pulmonary fibrosis (HPS-PF)
- Concentrated in HPS-1, HPS-2, and HPS-4; several reviews describe HPS-1 as highly penetrant for PF, including statements of 100% PF in HPS-1. (vicary2016pulmonaryfibrosisin pages 1-2, hu2024pathogenesisandtherapy pages 1-2)
- Onset: often middle-aged adults for HPS-1/HPS-4; earlier in HPS-2 (children/young adults). (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, hu2024pathogenesisandtherapy pages 1-2)
-
Suggested HPO terms: HP:0002099 (Pulmonary fibrosis), HP:0002093 (Respiratory insufficiency), HP:0002875 (Progressive respiratory failure).
-
Granulomatous colitis / inflammatory bowel disease phenotype
- One major review reports granulomatous colitis in ~15% of HPS patients. (velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2)
-
Suggested HPO terms: HP:0002037 (Inflammatory bowel disease), HP:0002570 (Colitis), HP:0100602 (Granulomatous inflammation).
-
Immunodeficiency/immune dysfunction (subset-dependent)
- Immunodeficiency is emphasized particularly in AP-3–related disease (e.g., HPS-2), and immune dysregulation is increasingly implicated in HPS pulmonary fibrosis mechanisms. (velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2, hu2024pathogenesisandtherapy pages 1-2)
- Suggested HPO terms: HP:0002721 (Immunodeficiency), HP:0011107 (Recurrent infections).
3.2 Quality-of-life impact
Pulmonary fibrosis progression leads to worsening dyspnea, hypoxemia, and eventually respiratory failure, imposing substantial functional limitation; this is repeatedly emphasized in PF-focused reviews. (vicary2016pulmonaryfibrosisin pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
4. Genetic / Molecular Information
4.1 Causal genes and subtype architecture
Recent synthesis (2024) lists 11 HPS genes and organizes them into four trafficking complexes. (hu2024pathogenesisandtherapy pages 1-2, hu2024pathogenesisandtherapy pages 2-3)
| Complex | HPS subtype(s) / gene aliases in evidence | Component genes/subunits listed in evidence | Main disease associations highlighted in retrieved evidence | Key evidence |
|---|---|---|---|---|
| BLOC-1 | HPS-7 = BLOC1S8; HPS-8 = BLOC1S3; HPS-9 = BLOC1S6; HPS-11 = BLOC1S5 | BLOC1S1, BLOC1S2, BLOC1S3/HPS8, BLOC1S4, BLOC1S5/HPS11, BLOC1S6/HPS9, BLOC1S7, BLOC1S8/HPS7 | Causes HPS through lysosome-related organelle (LRO) trafficking defects; core manifestations across HPS include oculocutaneous albinism and platelet dense-granule deficiency/bleeding diathesis. Pulmonary fibrosis is not the major association emphasized for BLOC-1 subtypes in the retrieved reviews. | (hu2024pathogenesisandtherapy pages 2-3, velazquezdiaz2021hermanskypudlaksyndromeand pages 2-3, hu2024pathogenesisandtherapy pages 1-2) |
| BLOC-2 | HPS-3, HPS-5, HPS-6 | HPS3, HPS5, HPS6 | Associated with classic HPS manifestations (albinism and bleeding); overall HPS review notes granulomatous colitis in ~15% of patients, and non-PF subgroup associations are enriched for HPS3/HPS5/HPS6 in OpenTargets evidence. Pulmonary fibrosis is generally not the principal association highlighted for these subtypes in the retrieved reviews. | (velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2, OpenTargets Search: Hermansky-Pudlak syndrome, hu2024pathogenesisandtherapy pages 2-3) |
| BLOC-3 | HPS-1, HPS-4 | HPS1, HPS4 | Major pulmonary-fibrosis-associated complex in HPS. Reviews state HPS-PF is especially linked to HPS-1 and HPS-4, typically beginning in adulthood (often 30–50 years); HPS-1 is described as highly penetrant for PF, with some reviews stating 100% of HPS-1 patients develop HPS-PF. BLOC-3 also acts as a Rab32/38 guanine nucleotide exchange factor relevant to melanosome/LRO cargo trafficking. | (velazquezdiaz2021hermanskypudlaksyndromeand pages 2-3, yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4, hu2024pathogenesisandtherapy pages 1-2) |
| AP-3 | HPS-2 = AP3B1; HPS-10 = AP3D1 | AP3B1 (β3A; HPS2), AP3D1 (δ; HPS10), μ3, σ3 | AP-3 disease is linked to HPS with albinism and platelet dysfunction; HPS-2 is specifically associated with pulmonary fibrosis/interstitial lung disease, often earlier in life, and AP-3 defects are also linked to immunodeficiency/immune dysfunction in retrieved reviews. | (hu2024pathogenesisandtherapy pages 2-3, velazquezdiaz2021hermanskypudlaksyndromeand pages 2-3, yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, hu2024pathogenesisandtherapy pages 1-2) |
| Cross-complex clinical summary | HPS overall | BLOC-1, BLOC-2, BLOC-3, AP-3 pathways affecting LRO biogenesis | Across HPS subtypes, the recurring phenotype triad is oculocutaneous albinism, platelet dense-granule defect with bleeding diathesis, and variable systemic disease. Pulmonary fibrosis is concentrated in HPS1/HPS2/HPS4; immunodeficiency is most emphasized for AP-3 disease; granulomatous colitis affects about 15% overall in one major review. | (velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, hu2024pathogenesisandtherapy pages 1-2) |
Table: This table summarizes the major molecular complexes underlying Hermansky-Pudlak syndrome, the subtype-defining genes captured in the retrieved evidence, and the main genotype-associated clinical features. It is useful for linking HPS subtypes to pathobiology and phenotype patterns such as pulmonary fibrosis, immunodeficiency, and colitis.
4.2 Pathogenic variant types (representative examples in retrieved evidence)
- Puerto Rico founder HPS-1 variant: c.1472_1487dup16-bp (exon 15), recurrent in NW Puerto Rico, with reported prevalence and carrier frequency. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4)
- Reviews also note many HPS1 variants (e.g., “67 HPS1 variants”). (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4)
Variant classes across HPS genes include frameshift/nonsense/deletion and splice-disrupting variants (general statement consistent with subtypes and founder events in evidence). (vicary2016pulmonaryfibrosisin pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4)
4.3 Functional consequences
Mechanistically, HPS is framed as an LRO biogenesis/trafficking disorder affecting: - Melanosomes → hypopigmentation/albinism - Platelet dense granules → impaired secretion/aggregation → bleeding - AT2 lamellar bodies → surfactant organelle dysfunction → epithelial stress/injury → fibrosis (hu2024pathogenesisandtherapy pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
4.4 Modifier genes / epigenetic information
A 2024 review highlights multiple epigenetic regulators relevant to fibrosis broadly but notes that “studies related to the epigenetics of HPS-PF have not been reported” (as of that review). (hu2024pathogenesisandtherapy pages 15-16)
5. Environmental Information
No clear environmental exposures were identified as primary drivers in the retrieved evidence; however, experimental lung-injury models (bleomycin) and age-associated immune activation support a role for injury/aging context in disease expression. (sorkhdini2024type2innate pages 8-10, hu2024pathogenesisandtherapy pages 1-2)
6. Mechanism / Pathophysiology
6.1 Core mechanistic concept: lysosome-related organelle (LRO) dysfunction
HPS proteins form trafficking complexes required for LRO maturation and cargo delivery, implicating a membrane trafficking / endosomal-lysosomal pathway disorder. A 2024 review explicitly lists LROs relevant to clinical phenotypes, including “LBs of AT2 cells, melanosomes..., alpha and dense granules in platelets,” among others. (hu2024pathogenesisandtherapy pages 1-2)
Suggested GO biological process terms (examples): - GO:0006897 (Endocytosis) - GO:0005764 (Lysosome; cellular component) - GO:0031410 (Lysosomal transport) - GO:0032541 (Assembly of protein-containing complex)
6.2 Pulmonary fibrosis mechanisms (prioritize 2023–2024)
6.2.1 Type 2 innate immune axis (CHI3L1–CRTH2–ILC2) as an amplifying fibrotic mechanism (2024)
A 2024 JCI Insight paper provides mechanistic evidence that type 2 innate lymphoid cells (ILC2s) and a CHI3L1–CRTH2 signaling axis promote fibrosis in HPS models and are present in human HPSPF lungs. (sorkhdini2024type2innate pages 2-4)
Key chain (as supported in the evidence): 1) Hps1 deficiency → exaggerated fibrotic response after injury (bleomycin) → increased recruitment/activation of ILC2s. (sorkhdini2024type2innate pages 2-4) 2) CHI3L1 interacts with CRTH2, and CRTH2 contributes to ILC2 accumulation; CRTH2 inhibition reduces collagen and ILC2 accumulation. (sorkhdini2024type2innate pages 2-4, sorkhdini2024type2innate pages 4-6) 3) ILC2s produce profibrotic mediators; direct quote: “IL-5, IL-13, and AREG were significantly increased in sorted ILC2s.” (sorkhdini2024type2innate pages 12-13) 4) ILC2s stimulate fibroblast proliferation/differentiation at least partly through amphiregulin/EGFR signaling; direct quote: “the AREG/EGFR pathway was partially responsible for increased fibroblast proliferation and differentiation.” (sorkhdini2024type2innate pages 12-13)
Suggested CL (cell type) terms: - ILC2: CL:0000934 (innate lymphoid cell) with subtype annotation (ILC2; specific CL ID not provided in evidence) - Lung fibroblast: CL:0000057 (fibroblast) - AT2 cell: CL:0002062 (alveolar type II pneumocyte)
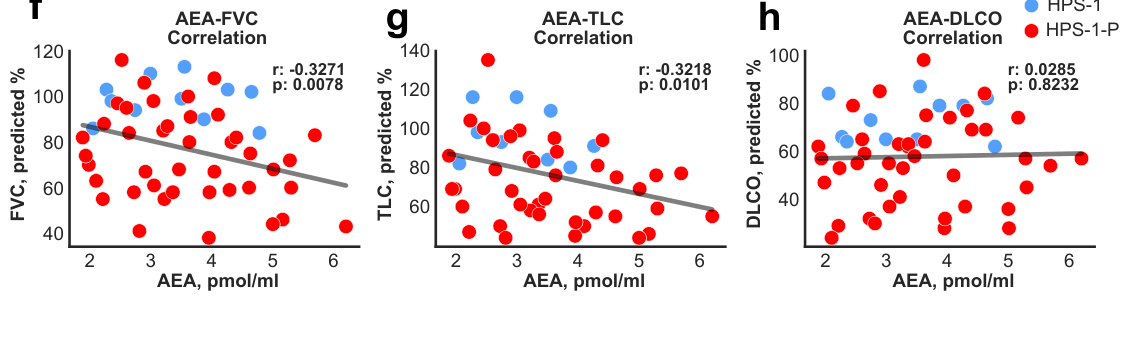
6.2.2 Biomarker-linked mechanistic signal: endocannabinoid anandamide (AEA) (2024 preprint)
A 2024 medRxiv study reports serum anandamide (AEA) is increased in HPS-1 (with or without PF) and not elevated in HPS-3 or IPF comparator groups, with negative correlations with pulmonary function and longitudinal rise during subclinical PF evolution. (cinar2024anandamideisan pages 1-6, cinar2024anandamideisan pages 21-24)
Quantitative/statistical highlights reported in the text/figures include correlation coefficients such as r = −0.3271 (p = 0.0078) and stronger negative correlations in subgroups (e.g., r = −0.6180 (p = 0.0037)), consistent with higher AEA tracking worse physiology. (cinar2024anandamideisan pages 21-24)
Evidence from retrieved figures supports the association between circulating AEA and impaired lung function, and individual trajectories over time. (cinar2024anandamideisan media 7046981e, cinar2024anandamideisan media 48b57ed7, cinar2024anandamideisan media 3c6bdc33, cinar2024anandamideisan media 7d3a8a16)
Translation to therapy hypothesis in that work: in an HPSPF mouse model, a peripheral CB1R/iNOS antagonist (zevaquenabant / MRI-1867) reduced elevated AEA and attenuated fibrosis. (cinar2024anandamideisan pages 1-6)
6.3 Tissue damage mechanisms in HPS-PF
PF-focused reviews describe characteristic histopathology distinct from idiopathic pulmonary fibrosis, including vacuolated hyperplastic type II cells with enlarged lamellar bodies and macrophages with lipofuscin-like deposits, consistent with an AT2/LRO-centered pathobiology. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
7. Anatomical Structures Affected
7.1 Organ/system level
- Eye/skin/hair pigmentation system (albinism): melanosome dysfunction (hu2024pathogenesisandtherapy pages 1-2)
- Hematologic/vascular system: platelet dense-granule dysfunction → bleeding (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4, vicary2016pulmonaryfibrosisin pages 1-2)
- Respiratory system (lung interstitium/alveolar region): progressive pulmonary fibrosis (hu2024pathogenesisandtherapy pages 1-2, vicary2016pulmonaryfibrosisin pages 1-2)
- Gastrointestinal tract: granulomatous colitis/IBD phenotype in a subset (velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2)
7.2 Tissue/cell level (suggested ontologies)
- Lung alveolar epithelium (UBERON:0002048 lung; alveolar region): AT2 cells (CL:0002062) with lamellar bodies as key subcellular compartment (GO:0031904, secretory granule/lamellar body-related annotations; not explicitly provided in evidence). (hu2024pathogenesisandtherapy pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
- Platelets (CL:0000233 platelet): dense granules/delta granules (GO cellular component). (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4)
- Melanocytes (CL:0000148 melanocyte): melanosomes. (hu2024pathogenesisandtherapy pages 1-2)
8. Temporal Development
8.1 Onset
- Congenital/early-life manifestations: albinism and platelet-function defect generally allow early recognition. (vicary2016pulmonaryfibrosisin pages 1-2)
- Pulmonary fibrosis: typically middle-aged onset in HPS-1 and HPS-4, and earlier for HPS-2. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, hu2024pathogenesisandtherapy pages 1-2)
8.2 Progression
HPS-PF is progressive and often fatal; one review emphasizes short time from onset to respiratory failure (“approximately 3 years” post-onset to respiratory failure) and severe prognosis, though this estimate varies by source and patient subset. (velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2)
9. Inheritance and Population
9.1 Epidemiology (key statistics)
- Global prevalence estimates of HPS: ~1–9 per million. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4, hu2024pathogenesisandtherapy pages 1-2)
- Puerto Rico founder effect (HPS-1): prevalence ~1/1800 and carrier frequency ~1/22 in NW Puerto Rico reported in a major PF review. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4)
- Another pulmonary-fibrosis-focused review reports that “approximately 50% of all cases globally” are diagnosed in Puerto Rico. (vicary2016pulmonaryfibrosisin pages 1-2)
9.2 Inheritance
Autosomal recessive inheritance is consistently reported. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, hu2024pathogenesisandtherapy pages 1-2)
9.3 Penetrance / expressivity
Pulmonary fibrosis penetrance is subtype-dependent; HPS-1 is repeatedly described as highly penetrant and sometimes reported as “100%,” while other subtypes have milder disease courses. (vicary2016pulmonaryfibrosisin pages 1-2, hu2024pathogenesisandtherapy pages 1-2)
10. Diagnostics
10.1 Key clinical/functional tests
- Pulmonary function tests (FVC, TLC, DLCO) and HRCT are central for evaluating HPS-PF progression and serve as endpoints in clinical trials. (NCT00001596 chunk 1, NCT00001596 chunk 2)
10.2 Platelet testing (hallmark diagnostic)
A key confirmatory test for platelet dense-granule deficiency is whole-mount TEM: “δ-granules are absent in platelets ... imaged using whole-mount transmission electron microscopy.” (yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4)
10.3 Genetic testing
Genotyping is emphasized as clinically relevant because lung-disease risk is concentrated in specific subtypes (HPS-1, -2, -4). (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
10.4 Differential diagnosis
Not systematically extractable from the retrieved evidence; however, PF reviews repeatedly position HPS-PF as overlapping clinically with idiopathic pulmonary fibrosis while differing in age-of-onset and histopathology. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
11. Outcome / Prognosis
Pulmonary fibrosis is a leading driver of morbidity and mortality in HPS genotypes that develop lung disease. Reviews describe poor prognosis once clinically apparent fibrosis develops. (hu2024pathogenesisandtherapy pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
A pulmonary-fibrosis-focused review reports reduced life expectancy (“40–50 years”) in a Puerto Rico-focused context and emphasizes early onset of PF (30–40 years). (vicary2016pulmonaryfibrosisin pages 1-2)
12. Treatment
12.1 Current management and real-world implementation
-
Lung transplantation: consistently described as the only clearly life-prolonging option for end-stage HPS-PF and is highlighted as the main effective option in reviews. (hu2024pathogenesisandtherapy pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
-
Antifibrotics (pirfenidone, nintedanib): approved for idiopathic pulmonary fibrosis but not specifically approved for HPS-PF in PF reviews; nevertheless, they motivate trials and off-label consideration. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, vicary2016pulmonaryfibrosisin pages 1-2)
12.2 Clinical trials landscape (ClinicalTrials.gov)
Pirfenidone RCT in HPS pulmonary fibrosis - Trial: Oral Pirfenidone for the Pulmonary Fibrosis of Hermansky-Pudlak Syndrome (NCT00001596; Phase 2; randomized, quadruple-masked; 2:1 allocation; 801 mg TID). (NCT00001596 chunk 1) - Dates: Start 2005-09, primary completion 2009-09, completion 2016-05-09; results posted around 2012-01-26; interim analysis after 30 enrollments stopped for futility. (NCT00001596 chunk 1) - Endpoints: change in FVC at 36 months (primary), with TLC, DLCOa, and 6MWT as secondary endpoints. (NCT00001596 chunk 1, NCT00001596 chunk 2)
Natural history study - Trial: Clinical and Basic Investigations Into Hermansky-Pudlak Syndrome (NCT00001456; NHGRI; start 1995-11-06; recruiting as of update posted 2026-06-16; up to 600). (NCT00001456 chunk 1)
Colitis treatment (withdrawn trial, but informative algorithm) - Trial: Medical Treatment of Colitis in Patients With Hermansky-Pudlak Syndrome (NCT00514982; Phase 2; open-label step-up IBD regimen; start 2007-08-07; withdrawn; completion 2011-03-08). (NCT00514982 chunk 1) - Step-up treatments include mesalamine → corticosteroids → infliximab + 6-mercaptopurine → adalimumab → tacrolimus. (NCT00514982 chunk 1)
Additional pirfenidone study - Trial: Efficacy and Safety of Pirfenidone Treatment in HPS-ILD (NCT04193592; 2019; pirfenidone). Available evidence in this run contains eligibility/exclusion criteria and sites but not full endpoints/dates. (NCT04193592 chunk 2)
12.3 Experimental / advanced therapeutics (gene therapy / gene editing)
PF-focused reviews summarize proof-of-concept correction approaches, including lentiviral HPS1 correction in patient-derived cells and CRISPR/Cas9 correction of a recurrent HPS1 duplication in B-lymphoblastoid cells, supporting feasibility of gene-based therapies (though not yet established clinically). (yokoyama2021hermansky–pudlaksyndromepulmonary pages 10-11)
12.4 Suggested MAXO terms (examples)
- MAXO:0000747 (lung transplantation) (conceptual mapping)
- MAXO:0001296 (antifibrotic therapy) (conceptual mapping)
- MAXO:0001001 (genetic testing) (conceptual mapping)
(MAXO IDs are suggested; specific MAXO identifiers were not present in the retrieved evidence text.)
13. Prevention
No primary prevention is possible for a monogenic autosomal recessive condition beyond reproductive-risk management; however, the NIH natural history study explicitly includes mutation analysis and family involvement, supporting cascade testing and genetic counseling as standard preventive strategies. (NCT00001456 chunk 1)
Suggested prevention-related actions (conceptual): genetic counseling, carrier testing in high-prevalence populations, and early pulmonary monitoring in HPS-1/HPS-4/HPS-2 genotypes. (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4)
14. Other Species / Natural Disease
No naturally occurring veterinary disease or OMIA-mapped animal syndrome was retrieved in the evidence set for this run.
15. Model Organisms
15.1 Mouse models and translational systems
- Hps1−/− mice challenged with bleomycin are used to model exaggerated HPS fibrotic responses and to interrogate immune–fibroblast crosstalk mechanisms (ILC2s; CHI3L1–CRTH2). (sorkhdini2024type2innate pages 2-4)
- An HPSPF mouse model (“pale ear”) is used in the 2024 biomarker preprint to link serum anandamide dynamics to fibrosis onset and therapeutic response (MRI-1867). (cinar2024anandamideisan pages 1-6)
15.2 Model applications and limitations
These models are particularly well-suited to dissect upstream inflammatory and epithelial stress pathways (e.g., type 2 innate immunity, EGFR signaling) but may not fully capture the chronic multidecade human natural history. (sorkhdini2024type2innate pages 2-4, hu2024pathogenesisandtherapy pages 1-2)
Recent developments and expert analysis (2023–2024 priority)
1) Mechanistic advance (2024): The identification of a CHI3L1–CRTH2–ILC2 axis with amphiregulin/EGFR-mediated fibroblast activation provides a concrete, targetable immunologic pathway for HPSPF beyond generic “epithelial injury” paradigms. (sorkhdini2024type2innate pages 2-4, sorkhdini2024type2innate pages 12-13)
2) Biomarker development (2024): Serum anandamide (AEA) is proposed as an early blood biomarker of HPSPF with longitudinal rise in subclinical disease and negative correlations with PFTs; if validated in peer-reviewed form and larger cohorts, this could support earlier trial enrollment and pharmacodynamic readouts. (cinar2024anandamideisan pages 1-6, cinar2024anandamideisan pages 21-24, cinar2024anandamideisan media 7046981e)
3) Clinical translation gap: Contemporary reviews continue to emphasize that no established disease-modifying therapy exists for HPSPF and that transplantation remains central, underscoring unmet need and the importance of mechanistically guided trials. (hu2024pathogenesisandtherapy pages 1-2, yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
Evidence gaps in this run (important for knowledge-base completeness)
- Specific OMIM, Orphanet, ICD-10/ICD-11, and MeSH identifiers were not contained in the retrieved sources and should be filled from authoritative databases directly. (OpenTargets Search: Hermansky-Pudlak syndrome)
- Phenotype frequency statistics beyond PF penetrance in HPS-1 and colitis (~15%) were limited in the retrieved evidence. (velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2, vicary2016pulmonaryfibrosisin pages 1-2)
- Many key 2023–2024 clinical genetics cohorts and systematic reviews were listed as “unobtainable” in tool output, limiting the ability to provide updated variant-frequency and global prevalence estimates from population screening studies.
Key source URLs and publication dates (selected)
- Hu X. et al. Int J Mol Sci. 2024-10. “Pathogenesis and Therapy of HPS-Associated Pulmonary Fibrosis.” https://doi.org/10.3390/ijms252011270 (hu2024pathogenesisandtherapy pages 1-2)
- Sorkhdini P. et al. JCI Insight. 2024-11. “Type 2 innate immunity promotes the development of pulmonary fibrosis in HPS.” https://doi.org/10.1172/jci.insight.178381 (sorkhdini2024type2innate pages 2-4)
- Cinar R. et al. medRxiv preprint. 2024-05. “Anandamide is an Early Blood Biomarker of HPS Pulmonary Fibrosis.” https://doi.org/10.1101/2024.05.16.24307300 (cinar2024anandamideisan pages 1-6)
- Yokoyama T., Gochuico BR. Eur Respir Rev. 2021-02. https://doi.org/10.1183/16000617.0193-2020 (yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2)
- ClinicalTrials.gov: NCT00001596 (pirfenidone RCT), first posted (per record) 2000s, start 2005-09. https://clinicaltrials.gov/study/NCT00001596 (NCT00001596 chunk 1)
References
-
(yokoyama2021hermansky–pudlaksyndromepulmonary pages 1-2): Tadafumi Yokoyama and Bernadette R. Gochuico. Hermansky–pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease. European Respiratory Review, 30:200193, Feb 2021. URL: https://doi.org/10.1183/16000617.0193-2020, doi:10.1183/16000617.0193-2020. This article has 61 citations and is from a peer-reviewed journal.
-
(vicary2016pulmonaryfibrosisin pages 1-2): Glenn W. Vicary, Yeidyly Vergne, Alberto Santiago-Cornier, Lisa R. Young, and Jesse Roman. Pulmonary fibrosis in hermansky–pudlak syndrome. Annals of the American Thoracic Society, 13(10):1839-1846, Oct 2016. URL: https://doi.org/10.1513/annalsats.201603-186fr, doi:10.1513/annalsats.201603-186fr. This article has 160 citations and is from a domain leading peer-reviewed journal.
-
(velazquezdiaz2021hermanskypudlaksyndromeand pages 1-2): Pamela Velázquez-Díaz, Erika Nakajima, Parand Sorkhdini, Ashley Hernandez-Gutierrez, Adam Eberle, Dongqin Yang, and Yang Zhou. Hermansky-pudlak syndrome and lung disease: pathogenesis and therapeutics. Frontiers in Pharmacology, Mar 2021. URL: https://doi.org/10.3389/fphar.2021.644671, doi:10.3389/fphar.2021.644671. This article has 43 citations.
-
(OpenTargets Search: Hermansky-Pudlak syndrome): Open Targets Query (Hermansky-Pudlak syndrome, 19 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(hu2024pathogenesisandtherapy pages 1-2): Xiao Hu, Zhixiao Wei, Yumeng Wu, Manhan Zhao, Liming Zhou, and Qiong Lin. Pathogenesis and therapy of hermansky–pudlak syndrome (hps)-associated pulmonary fibrosis. International Journal of Molecular Sciences, 25:11270, Oct 2024. URL: https://doi.org/10.3390/ijms252011270, doi:10.3390/ijms252011270. This article has 4 citations.
-
(NCT00001456 chunk 1): Clinical and Basic Investigations Into Hermansky-Pudlak Syndrome. National Human Genome Research Institute (NHGRI). 1995. ClinicalTrials.gov Identifier: NCT00001456
-
(hu2024pathogenesisandtherapy pages 2-3): Xiao Hu, Zhixiao Wei, Yumeng Wu, Manhan Zhao, Liming Zhou, and Qiong Lin. Pathogenesis and therapy of hermansky–pudlak syndrome (hps)-associated pulmonary fibrosis. International Journal of Molecular Sciences, 25:11270, Oct 2024. URL: https://doi.org/10.3390/ijms252011270, doi:10.3390/ijms252011270. This article has 4 citations.
-
(yokoyama2021hermansky–pudlaksyndromepulmonary pages 2-4): Tadafumi Yokoyama and Bernadette R. Gochuico. Hermansky–pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease. European Respiratory Review, 30:200193, Feb 2021. URL: https://doi.org/10.1183/16000617.0193-2020, doi:10.1183/16000617.0193-2020. This article has 61 citations and is from a peer-reviewed journal.
-
(sorkhdini2024type2innate pages 2-4): Parand Sorkhdini, Kiran Klubock-Shukla, Selena Sheth, Dongqin Yang, Alina Xiaoyu Yang, Carmelissa Norbrun, Wendy J. Introne, Bernadette R. Gochuico, and Yang Zhou. Type 2 innate immunity promotes the development of pulmonary fibrosis in hermansky-pudlak syndrome. JCI Insight, Nov 2024. URL: https://doi.org/10.1172/jci.insight.178381, doi:10.1172/jci.insight.178381. This article has 13 citations and is from a domain leading peer-reviewed journal.
-
(NCT00001596 chunk 1): William Gahl, M.D.. Oral Pirfenidone for the Pulmonary Fibrosis of Hermansky-Pudlak Syndrome. William Gahl, M.D.. 2005. ClinicalTrials.gov Identifier: NCT00001596
-
(sorkhdini2024type2innate pages 8-10): Parand Sorkhdini, Kiran Klubock-Shukla, Selena Sheth, Dongqin Yang, Alina Xiaoyu Yang, Carmelissa Norbrun, Wendy J. Introne, Bernadette R. Gochuico, and Yang Zhou. Type 2 innate immunity promotes the development of pulmonary fibrosis in hermansky-pudlak syndrome. JCI Insight, Nov 2024. URL: https://doi.org/10.1172/jci.insight.178381, doi:10.1172/jci.insight.178381. This article has 13 citations and is from a domain leading peer-reviewed journal.
-
(velazquezdiaz2021hermanskypudlaksyndromeand pages 2-3): Pamela Velázquez-Díaz, Erika Nakajima, Parand Sorkhdini, Ashley Hernandez-Gutierrez, Adam Eberle, Dongqin Yang, and Yang Zhou. Hermansky-pudlak syndrome and lung disease: pathogenesis and therapeutics. Frontiers in Pharmacology, Mar 2021. URL: https://doi.org/10.3389/fphar.2021.644671, doi:10.3389/fphar.2021.644671. This article has 43 citations.
-
(hu2024pathogenesisandtherapy pages 15-16): Xiao Hu, Zhixiao Wei, Yumeng Wu, Manhan Zhao, Liming Zhou, and Qiong Lin. Pathogenesis and therapy of hermansky–pudlak syndrome (hps)-associated pulmonary fibrosis. International Journal of Molecular Sciences, 25:11270, Oct 2024. URL: https://doi.org/10.3390/ijms252011270, doi:10.3390/ijms252011270. This article has 4 citations.
-
(sorkhdini2024type2innate pages 4-6): Parand Sorkhdini, Kiran Klubock-Shukla, Selena Sheth, Dongqin Yang, Alina Xiaoyu Yang, Carmelissa Norbrun, Wendy J. Introne, Bernadette R. Gochuico, and Yang Zhou. Type 2 innate immunity promotes the development of pulmonary fibrosis in hermansky-pudlak syndrome. JCI Insight, Nov 2024. URL: https://doi.org/10.1172/jci.insight.178381, doi:10.1172/jci.insight.178381. This article has 13 citations and is from a domain leading peer-reviewed journal.
-
(sorkhdini2024type2innate pages 12-13): Parand Sorkhdini, Kiran Klubock-Shukla, Selena Sheth, Dongqin Yang, Alina Xiaoyu Yang, Carmelissa Norbrun, Wendy J. Introne, Bernadette R. Gochuico, and Yang Zhou. Type 2 innate immunity promotes the development of pulmonary fibrosis in hermansky-pudlak syndrome. JCI Insight, Nov 2024. URL: https://doi.org/10.1172/jci.insight.178381, doi:10.1172/jci.insight.178381. This article has 13 citations and is from a domain leading peer-reviewed journal.
-
(cinar2024anandamideisan pages 1-6): Resat Cinar, Abhishek Basu, Muhammad Arif, Joshua K. Park, Charles N. Zawatsky, Ben Long G. Zuo, Mei Xing G. Zuo, Kevin J. O’Brien, Molly Behan, Wendy Introne, Malliga R. Iyer, William A. Gahl, May Christine V. Malicdan, and Bernadette R. Gochuico. Anandamide is an early blood biomarker of hermansky-pudlak syndrome pulmonary fibrosis. medRxiv, May 2024. URL: https://doi.org/10.1101/2024.05.16.24307300, doi:10.1101/2024.05.16.24307300. This article has 3 citations.
-
(cinar2024anandamideisan pages 21-24): Resat Cinar, Abhishek Basu, Muhammad Arif, Joshua K. Park, Charles N. Zawatsky, Ben Long G. Zuo, Mei Xing G. Zuo, Kevin J. O’Brien, Molly Behan, Wendy Introne, Malliga R. Iyer, William A. Gahl, May Christine V. Malicdan, and Bernadette R. Gochuico. Anandamide is an early blood biomarker of hermansky-pudlak syndrome pulmonary fibrosis. medRxiv, May 2024. URL: https://doi.org/10.1101/2024.05.16.24307300, doi:10.1101/2024.05.16.24307300. This article has 3 citations.
-
(cinar2024anandamideisan media 7046981e): Resat Cinar, Abhishek Basu, Muhammad Arif, Joshua K. Park, Charles N. Zawatsky, Ben Long G. Zuo, Mei Xing G. Zuo, Kevin J. O’Brien, Molly Behan, Wendy Introne, Malliga R. Iyer, William A. Gahl, May Christine V. Malicdan, and Bernadette R. Gochuico. Anandamide is an early blood biomarker of hermansky-pudlak syndrome pulmonary fibrosis. medRxiv, May 2024. URL: https://doi.org/10.1101/2024.05.16.24307300, doi:10.1101/2024.05.16.24307300. This article has 3 citations.
-
(cinar2024anandamideisan media 48b57ed7): Resat Cinar, Abhishek Basu, Muhammad Arif, Joshua K. Park, Charles N. Zawatsky, Ben Long G. Zuo, Mei Xing G. Zuo, Kevin J. O’Brien, Molly Behan, Wendy Introne, Malliga R. Iyer, William A. Gahl, May Christine V. Malicdan, and Bernadette R. Gochuico. Anandamide is an early blood biomarker of hermansky-pudlak syndrome pulmonary fibrosis. medRxiv, May 2024. URL: https://doi.org/10.1101/2024.05.16.24307300, doi:10.1101/2024.05.16.24307300. This article has 3 citations.
-
(cinar2024anandamideisan media 3c6bdc33): Resat Cinar, Abhishek Basu, Muhammad Arif, Joshua K. Park, Charles N. Zawatsky, Ben Long G. Zuo, Mei Xing G. Zuo, Kevin J. O’Brien, Molly Behan, Wendy Introne, Malliga R. Iyer, William A. Gahl, May Christine V. Malicdan, and Bernadette R. Gochuico. Anandamide is an early blood biomarker of hermansky-pudlak syndrome pulmonary fibrosis. medRxiv, May 2024. URL: https://doi.org/10.1101/2024.05.16.24307300, doi:10.1101/2024.05.16.24307300. This article has 3 citations.
-
(cinar2024anandamideisan media 7d3a8a16): Resat Cinar, Abhishek Basu, Muhammad Arif, Joshua K. Park, Charles N. Zawatsky, Ben Long G. Zuo, Mei Xing G. Zuo, Kevin J. O’Brien, Molly Behan, Wendy Introne, Malliga R. Iyer, William A. Gahl, May Christine V. Malicdan, and Bernadette R. Gochuico. Anandamide is an early blood biomarker of hermansky-pudlak syndrome pulmonary fibrosis. medRxiv, May 2024. URL: https://doi.org/10.1101/2024.05.16.24307300, doi:10.1101/2024.05.16.24307300. This article has 3 citations.
-
(NCT00001596 chunk 2): William Gahl, M.D.. Oral Pirfenidone for the Pulmonary Fibrosis of Hermansky-Pudlak Syndrome. William Gahl, M.D.. 2005. ClinicalTrials.gov Identifier: NCT00001596
-
(NCT00514982 chunk 1): Medical Treatment of Colitis in Patients With Hermansky-Pudlak Syndrome. National Institute of Allergy and Infectious Diseases (NIAID). 2007. ClinicalTrials.gov Identifier: NCT00514982
-
(NCT04193592 chunk 2): Jesse Roman. Efficacy and Safety of Pirfenidone Treatment in HPS-ILD. Jesse Roman. 2019. ClinicalTrials.gov Identifier: NCT04193592
-
(yokoyama2021hermansky–pudlaksyndromepulmonary pages 10-11): Tadafumi Yokoyama and Bernadette R. Gochuico. Hermansky–pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease. European Respiratory Review, 30:200193, Feb 2021. URL: https://doi.org/10.1183/16000617.0193-2020, doi:10.1183/16000617.0193-2020. This article has 61 citations and is from a peer-reviewed journal.