Boucher-Neuhauser Syndrome
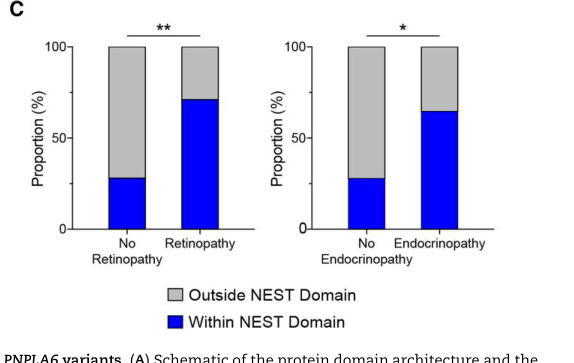
Boucher-Neuhauser syndrome (BNS) is a rare autosomal recessive neurodegenerative disorder caused by biallelic pathogenic variants in PNPLA6, which encodes neuropathy target esterase (NTE). It is classically defined by the triad of cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy, and is now recognized as one cluster on a continuous PNPLA6-disorder spectrum that also includes Gordon Holmes syndrome, Oliver-McFarlane syndrome, Laurence-Moon syndrome, and spastic paraplegia type 39. NTE is an endoplasmic-reticulum-associated phospholipase/esterase required for phospholipid (phosphatidylcholine/lysophospholipid) homeostasis; loss of NTE activity causes degeneration of cerebellar, retinal, and hypothalamic-pituitary neurons. Residual NTE activity correlates inversely with the presence of retinopathy and endocrinopathy.
Ask OpenScientist
Ask a research question about Boucher-Neuhauser Syndrome. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (2 references)
Pathophysiology
2Show evidence (2 references)
Show evidence (3 references)
Pathograph
Phenotypes
10Endocrine 1
Show evidence (2 references)
Eye 3
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Musculoskeletal 1
Show evidence (1 reference)
Nervous System 3
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Other 2
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
1Show evidence (3 references)
Medical Actions
3Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Source YAML
click to showname: Boucher-Neuhauser Syndrome
creation_date: "2026-06-04T12:00:00Z"
category: Mendelian
synonyms:
- BNHS
- Boucher-Neuhäuser syndrome
- Spinocerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy
- Ataxia-hypogonadism-choroidal dystrophy syndrome
description: >-
Boucher-Neuhauser syndrome (BNS) is a rare autosomal recessive
neurodegenerative disorder caused by biallelic pathogenic variants in PNPLA6,
which encodes neuropathy target esterase (NTE). It is classically defined by
the triad of cerebellar ataxia, hypogonadotropic hypogonadism, and
chorioretinal dystrophy, and is now recognized as one cluster on a continuous
PNPLA6-disorder spectrum that also includes Gordon Holmes syndrome,
Oliver-McFarlane syndrome, Laurence-Moon syndrome, and spastic paraplegia
type 39. NTE is an endoplasmic-reticulum-associated phospholipase/esterase
required for phospholipid (phosphatidylcholine/lysophospholipid) homeostasis;
loss of NTE activity causes degeneration of cerebellar, retinal, and
hypothalamic-pituitary neurons. Residual NTE activity correlates inversely
with the presence of retinopathy and endocrinopathy.
disease_term:
preferred_term: Boucher-Neuhauser syndrome
term:
id: MONDO:0008980
label: ataxia-hypogonadism-choroidal dystrophy syndrome
parents:
- Hereditary Ataxia
- Congenital Hypogonadotropic Hypogonadism
references:
- reference: PMID:25299038
title: "PNPLA6 Disorders."
tags:
- GeneReviews
inheritance:
- name: Autosomal recessive inheritance
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
description: >-
BNS is caused by biallelic (homozygous or compound heterozygous) pathogenic
variants in PNPLA6 and is inherited in an autosomal recessive manner.
evidence:
- reference: PMID:38683245
reference_title: "Two case reports of a novel missense mutation in the PNPLA6 gene in two siblings with chorioretinal dystrophy, hypogonadotropic hypogonadism, and cerebellar ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Boucher Neuhäuser Syndrome (BNS) is a rare disease with autosomal recessive inheritance defined by the classical triad; early-onset ataxia, hypogonadism and chorioretinal dystrophy.
explanation: >-
Directly states the autosomal recessive inheritance pattern and the
defining clinical triad.
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
PNPLA6 disorders are inherited in an autosomal recessive manner.
explanation: >-
GeneReviews confirms autosomal recessive inheritance for the PNPLA6
disorder spectrum, which includes BNS.
pathophysiology:
- name: Loss of neuropathy target esterase (NTE) activity
description: >-
PNPLA6 encodes neuropathy target esterase (NTE), a patatin-like serine
hydrolase on the cytoplasmic face of the endoplasmic reticulum with
phospholipase/lysophospholipase activity. NTE deacylates glycerophospholipids
and is central to phosphatidylcholine homeostasis. Biallelic pathogenic
PNPLA6 variants, which cluster in the C-terminal phospholipase/esterase
(patatin-like) domain, reduce NTE esterase activity, the principal
loss-of-function mechanism underlying BNS.
genes:
- preferred_term: PNPLA6
term:
id: hgnc:16268
label: PNPLA6
biological_processes:
- preferred_term: phospholipid metabolic process
term:
id: GO:0006644
label: phospholipid metabolic process
modifier: DECREASED
- preferred_term: phosphatidylcholine metabolic process
term:
id: GO:0046470
label: phosphatidylcholine metabolic process
modifier: ABNORMAL
molecular_functions:
- preferred_term: glycerophospholipase (NTE esterase) activity
term:
id: GO:0004620
label: glycerophospholipase activity
modifier: DECREASED
- preferred_term: carboxylic ester hydrolase activity
term:
id: GO:0052689
label: carboxylic ester hydrolase activity
modifier: DECREASED
cellular_components:
- preferred_term: endoplasmic reticulum membrane
term:
id: GO:0005789
label: endoplasmic reticulum membrane
evidence:
- reference: PMID:37732399
reference_title: "PNPLA6 disorders: what's in a name?"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
PNPLA6 encodes NTE, an enzyme involved in maintaining phospholipid homeostasis and trafficking in the nervous system.
explanation: >-
Establishes NTE's role in phospholipid homeostasis whose disruption is the
molecular basis of BNS.
- reference: PMID:38735647
reference_title: "Neuropathy target esterase activity defines phenotypes among PNPLA6 disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Measuring esterase activity of 46 disease-associated and 20 common variants observed across PNPLA6-associated clinical diagnoses unambiguously reclassified 36 variants as pathogenic and 10 variants as likely pathogenic
explanation: >-
Demonstrates that disease-associated PNPLA6 variants reduce NTE esterase
activity, supporting loss of enzymatic function as the mechanism.
downstream:
- target: NTE-activity-dependent neurodegeneration of vulnerable tissues
description: Reduced PNPLA6/NTE esterase activity produces the tissue-vulnerability phenotype continuum across PNPLA6 disorders.
causal_link_type: DIRECT
evidence:
- reference: PMID:38735647
reference_title: "Neuropathy target esterase activity defines phenotypes among PNPLA6 disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Estimating the overall NTE activity of affected individuals revealed a striking inverse relationship between NTE activity and the presence of retinopathy and endocrinopathy.
explanation: Human genotype-function data link lower NTE activity to retinal and endocrine phenotypes.
- name: NTE-activity-dependent neurodegeneration of vulnerable tissues
description: >-
Reduced NTE activity disrupts phospholipid remodeling at ER membranes,
rendering specific neuronal and supporting cell populations vulnerable to
age-dependent degeneration: cerebellar Purkinje-cell circuits (ataxia),
retinal photoreceptors and retinal pigment epithelium (chorioretinal
dystrophy), and hypothalamic-pituitary neurons (hypogonadotropic
hypogonadism / anterior hypopituitarism). Overall NTE activity is inversely
related to the presence of retinopathy and endocrinopathy, defining a
genotype:activity:phenotype continuum across PNPLA6 disorders. An allelic
mouse series recapitulates an NTE activity threshold for retinal
degeneration, with complete loss being embryonic lethal.
cell_types:
- preferred_term: cerebellar Purkinje cell
term:
id: CL:0000121
label: Purkinje cell
- preferred_term: retinal photoreceptor cell
term:
id: CL:0000210
label: photoreceptor cell
- preferred_term: retinal pigment epithelial cell
term:
id: CL:0002586
label: retinal pigment epithelial cell
biological_processes:
- preferred_term: neuron apoptotic process
term:
id: GO:0051402
label: neuron apoptotic process
modifier: INCREASED
evidence:
- reference: PMID:38735647
reference_title: "Neuropathy target esterase activity defines phenotypes among PNPLA6 disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Estimating the overall NTE activity of affected individuals revealed a striking inverse relationship between NTE activity and the presence of retinopathy and endocrinopathy.
explanation: >-
Links reduced NTE activity quantitatively to the retinal and endocrine
manifestations of the PNPLA6 spectrum that includes BNS.
- reference: PMID:38735647
reference_title: "Neuropathy target esterase activity defines phenotypes among PNPLA6 disorders."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
This phenomenon was recaptured in vivo in an allelic mouse series, where a similar NTE threshold for retinopathy exists.
explanation: >-
An allelic mouse series recapitulates the NTE-activity threshold for
retinal degeneration, supporting the causal chain in vivo.
- reference: PMID:35448471
reference_title: "PNPLA6/NTE, an Evolutionary Conserved Phospholipase Linked to a Group of Complex Human Diseases."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
A conditional knockout of PNPLA6/NTE in the mouse brain results in age-related neurodegeneration, whereas a complete knockout causes lethality during embryogenesis due to defects in the development of the placenta.
explanation: >-
Mouse models show that NTE loss causes age-related neurodegeneration and
that complete loss is embryonic lethal, consistent with the human
activity-threshold model.
downstream:
- target: Cerebellar ataxia
description: Cerebellar neuronal vulnerability manifests as cerebellar ataxia.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:25267340
reference_title: "Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Boucher-Neuhäuser is a rare autosomal recessive syndrome characterized by the co-occurrence of cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy.
explanation: The defining Boucher-Neuhauser triad includes cerebellar ataxia.
- target: Cerebellar atrophy
description: Cerebellar neurodegeneration is visible as cerebellar atrophy on MRI.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:35069422
reference_title: "Multifaceted and Age-Dependent Phenotypes Associated With Biallelic PNPLA6 Gene Variants: Eight Novel Cases and Review of the Literature."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Brain MRI showed cerebellar atrophy in 6/8 patients, more pronounced in superior and dorsal vermis lobules (I to VII).
explanation: PNPLA6 cohort data support cerebellar atrophy as a structural outcome.
- target: Hypogonadotropic hypogonadism
description: Hypothalamic-pituitary vulnerability manifests as hypogonadotropic hypogonadism.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:25267340
reference_title: "Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Boucher-Neuhäuser is a rare autosomal recessive syndrome characterized by the co-occurrence of cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy.
explanation: The defining Boucher-Neuhauser triad includes hypogonadotropic hypogonadism.
- target: Anterior hypopituitarism
description: Endocrine involvement can extend from isolated hypogonadotropic hypogonadism to anterior hypopituitarism.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The hypogonadotropic hypogonadism occurs either in isolation or as part of anterior hypopituitarism (growth hormone, thyroid hormone, or gonadotropin deficiencies).
explanation: GeneReviews supports anterior hypopituitarism as part of the PNPLA6 endocrine phenotype spectrum.
- target: Chorioretinal dystrophy
description: Retinal vulnerability manifests as chorioretinal dystrophy.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:25267340
reference_title: "Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Boucher-Neuhäuser is a rare autosomal recessive syndrome characterized by the co-occurrence of cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy.
explanation: The defining Boucher-Neuhauser triad includes chorioretinal dystrophy.
- target: Visual impairment
description: Progressive retinal disease causes visual impairment across the PNPLA6 spectrum.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:38735647
reference_title: "Neuropathy target esterase activity defines phenotypes among PNPLA6 disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Biallelic pathogenic variants in the PNPLA6 gene cause a broad spectrum of disorders leading to gait disturbance, visual impairment, anterior hypopituitarism and hair anomalies.
explanation: Human PNPLA6-spectrum data support visual impairment as a downstream clinical output.
- target: Peripheral axonal neuropathy
description: PNPLA6 neurodegenerative involvement can extend to peripheral axons.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:35069422
reference_title: "Multifaceted and Age-Dependent Phenotypes Associated With Biallelic PNPLA6 Gene Variants: Eight Novel Cases and Review of the Literature."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Additional clinical features included hypogonadotropic hypogonadism (5/8), growth hormone deficiency (2/8), peripheral axonal neuropathy (4/8), cognitive impairment (3/8), chorioretinal dystrophy (2/8)
explanation: Cohort data support peripheral axonal neuropathy as a PNPLA6-spectrum feature.
- target: Spasticity
description: Upper-motor-neuron involvement in PNPLA6 disorders can manifest as spasticity.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
upper motor neuron involvement manifesting as spasticity and/or brisk reflexes
explanation: GeneReviews supports spasticity as an upper-motor-neuron manifestation.
- target: Cognitive impairment
description: CNS involvement in the PNPLA6 spectrum includes cognitive impairment in a subset of patients.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
impaired cognitive functioning (learning disabilities in children; deficits in attention, visuospatial abilities, and recall in adults)
explanation: GeneReviews supports cognitive impairment as a PNPLA6-spectrum phenotype.
- target: Nystagmus
description: Cerebellar and ocular involvement can produce nystagmus.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:25267340
reference_title: "Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
She had hypometric vertical and horizontal saccades, saccadic smooth pursuit, gaze-evoked nystagmus and poor visual suppression of vestibular ocular reflexes.
explanation: A molecularly confirmed case supports nystagmus as an oculomotor output.
phenotypes:
- name: Cerebellar ataxia
category: Phenotypic
description: >-
Cerebellar ataxia/cerebellar degeneration is a core feature, with onset
ranging from childhood to adulthood and typically slow progression; brain
MRI commonly shows cerebellar (often superior vermian) atrophy.
phenotype_term:
preferred_term: Cerebellar ataxia
term:
id: HP:0001251
label: Ataxia
frequency: VERY_FREQUENT
evidence:
- reference: PMID:25267340
reference_title: "Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Boucher-Neuhäuser is a rare autosomal recessive syndrome characterized by the co-occurrence of cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy.
explanation: >-
Identifies cerebellar ataxia as a defining component of the BNS triad.
- reference: PMID:35069422
reference_title: "Multifaceted and Age-Dependent Phenotypes Associated With Biallelic PNPLA6 Gene Variants: Eight Novel Cases and Review of the Literature."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Cerebellar ataxia was observed in seven patients and spastic paraplegia in one patient.
explanation: >-
In a cohort of eight biallelic PNPLA6 cases, cerebellar ataxia was present
in 7/8, supporting it as a very frequent feature.
- name: Cerebellar atrophy
category: Phenotypic
description: >-
Brain MRI shows cerebellar atrophy, most pronounced in the superior and
dorsal vermis.
phenotype_term:
preferred_term: Cerebellar atrophy
term:
id: HP:0001272
label: Cerebellar atrophy
evidence:
- reference: PMID:35069422
reference_title: "Multifaceted and Age-Dependent Phenotypes Associated With Biallelic PNPLA6 Gene Variants: Eight Novel Cases and Review of the Literature."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Brain MRI showed cerebellar atrophy in 6/8 patients, more pronounced in superior and dorsal vermis lobules (I to VII).
explanation: >-
Documents cerebellar atrophy with characteristic superior/dorsal vermian
predominance in PNPLA6 patients.
- reference: PMID:31135245
reference_title: "Detailed retinal phenotype of Boucher-Neuhäuser syndrome associated with mutations in PNPLA6 mimicking choroideremia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hypogonadotropic hypogonadism and cerebellar vermis hypoplasia by MRI confirmed a diagnosis of Boucher-Neuhäuser syndrome.
explanation: >-
Cerebellar vermis involvement on MRI contributed to the BNS diagnosis in a
molecularly confirmed case.
- name: Hypogonadotropic hypogonadism
category: Phenotypic
description: >-
Hypogonadotropic hypogonadism presents as delayed puberty and lack of
secondary sexual characteristics, with low gonadotropins (LH/FSH). It may
occur in isolation or as part of broader anterior hypopituitarism.
phenotype_term:
preferred_term: Hypogonadotropic hypogonadism
term:
id: HP:0000044
label: Hypogonadotropic hypogonadism
frequency: VERY_FREQUENT
evidence:
- reference: PMID:25267340
reference_title: "Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Boucher-Neuhäuser is a rare autosomal recessive syndrome characterized by the co-occurrence of cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy.
explanation: >-
Identifies hypogonadotropic hypogonadism as a defining component of the BNS
triad.
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
hypogonadotropic hypogonadism (delayed puberty and lack of secondary sex characteristics)
explanation: >-
GeneReviews describes the clinical presentation of the hypogonadotropic
hypogonadism characteristic of BNS.
- name: Anterior hypopituitarism
category: Phenotypic
description: >-
In a subset of patients, hypogonadotropic hypogonadism occurs as part of
broader anterior hypopituitarism with additional growth hormone, thyroid
hormone, or gonadotropin deficiencies.
phenotype_term:
preferred_term: Anterior hypopituitarism
term:
id: HP:0000830
label: Anterior hypopituitarism
frequency: OCCASIONAL
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The hypogonadotropic hypogonadism occurs either in isolation or as part of anterior hypopituitarism (growth hormone, thyroid hormone, or gonadotropin deficiencies).
explanation: >-
GeneReviews documents that anterior hypopituitarism can accompany the
hypogonadotropic hypogonadism in PNPLA6 disorders.
- name: Chorioretinal dystrophy
category: Phenotypic
description: >-
Chorioretinal dystrophy with progressive retinal pigment epithelium and
choriocapillaris atrophy; onset is widely variable and the presentation can
closely mimic choroideremia, sometimes progressing to severe visual loss or
blindness.
phenotype_term:
preferred_term: Chorioretinal dystrophy
term:
id: HP:0001135
label: Chorioretinal dystrophy
frequency: VERY_FREQUENT
evidence:
- reference: PMID:25267340
reference_title: "Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Boucher-Neuhäuser is a rare autosomal recessive syndrome characterized by the co-occurrence of cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy.
explanation: >-
Identifies chorioretinal dystrophy as a defining component of the BNS triad.
- reference: PMID:37732399
reference_title: "PNPLA6 disorders: what's in a name?"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Retinal disease presents with a unique chorioretinal dystrophy that is phenotypically similar to choroideremia and Leber congenital amaurosis.
explanation: >-
Characterizes the chorioretinal dystrophy and its choroideremia-like
appearance in PNPLA6 disorders.
- name: Visual impairment
category: Phenotypic
description: >-
Progressive chorioretinal degeneration leads to variable visual impairment,
which may progress to blindness.
phenotype_term:
preferred_term: Visual impairment
term:
id: HP:0000505
label: Visual impairment
evidence:
- reference: PMID:38735647
reference_title: "Neuropathy target esterase activity defines phenotypes among PNPLA6 disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Biallelic pathogenic variants in the PNPLA6 gene cause a broad spectrum of disorders leading to gait disturbance, visual impairment, anterior hypopituitarism and hair anomalies.
explanation: >-
Lists visual impairment among the cardinal manifestations of PNPLA6
disorders, including BNS.
- name: Peripheral axonal neuropathy
category: Phenotypic
description: >-
A peripheral neuropathy of axonal type (reduced distal reflexes, diminished
vibratory sensation, and/or distal muscle wasting) is a common but less
frequent feature; it may be subclinical and detected only on EMG/NCS.
phenotype_term:
preferred_term: Peripheral axonal neuropathy
term:
id: HP:0003477
label: Peripheral axonal neuropathy
frequency: FREQUENT
evidence:
- reference: PMID:35069422
reference_title: "Multifaceted and Age-Dependent Phenotypes Associated With Biallelic PNPLA6 Gene Variants: Eight Novel Cases and Review of the Literature."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Additional clinical features included hypogonadotropic hypogonadism (5/8), growth hormone deficiency (2/8), peripheral axonal neuropathy (4/8), cognitive impairment (3/8), chorioretinal dystrophy (2/8)
explanation: >-
Peripheral axonal neuropathy was present in 4/8 PNPLA6 patients in this
cohort.
- name: Spasticity
category: Phenotypic
description: >-
Upper motor neuron involvement manifesting as spasticity and/or brisk
reflexes occurs along the PNPLA6 phenotypic continuum.
phenotype_term:
preferred_term: Spasticity
term:
id: HP:0001257
label: Spasticity
frequency: OCCASIONAL
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
upper motor neuron involvement manifesting as spasticity and/or brisk reflexes
explanation: >-
GeneReviews lists spasticity from upper motor neuron involvement among the
features of the PNPLA6 spectrum.
- name: Cognitive impairment
category: Phenotypic
description: >-
Impaired cognitive functioning (learning disabilities in children; deficits
in attention, visuospatial abilities, and recall in adults) occurs in a
subset of patients.
phenotype_term:
preferred_term: Cognitive impairment
term:
id: HP:0100543
label: Cognitive impairment
frequency: OCCASIONAL
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
impaired cognitive functioning (learning disabilities in children; deficits in attention, visuospatial abilities, and recall in adults)
explanation: >-
GeneReviews documents cognitive impairment as a less frequent feature of
PNPLA6 disorders.
- name: Nystagmus
category: Phenotypic
description: >-
Oculomotor abnormalities including gaze-evoked nystagmus and saccadic
pursuit have been reported.
phenotype_term:
preferred_term: Nystagmus
term:
id: HP:0000639
label: Nystagmus
evidence:
- reference: PMID:25267340
reference_title: "Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
She had hypometric vertical and horizontal saccades, saccadic smooth pursuit, gaze-evoked nystagmus and poor visual suppression of vestibular ocular reflexes.
explanation: >-
Documents gaze-evoked nystagmus and saccadic pursuit abnormalities in a
molecularly confirmed BNS case.
genetic:
- name: PNPLA6
association: Loss of function mutation
notes: >-
Biallelic (homozygous or compound heterozygous) pathogenic variants in
PNPLA6 (19p13.2), encoding neuropathy target esterase, cause BNS. Reported
variants are predominantly missense, with frameshift, splice-altering, and
nonsense variants also described; missense/in-frame variants are enriched in
the catalytic phospholipase/esterase (patatin-like) domain. PNPLA6 is also
the cause of allelic disorders on the same spectrum, including Gordon Holmes
syndrome, Oliver-McFarlane syndrome, Laurence-Moon syndrome, and spastic
paraplegia type 39.
gene_term:
preferred_term: PNPLA6
term:
id: hgnc:16268
label: PNPLA6
evidence:
- reference: PMID:35198007
reference_title: "Identification of Novel Compound Heterozygous Variants of the PNPLA6 Gene in Boucher-Neuhäuser Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Boucher-Neuhäuser syndrome (BNS, MIM 215470) is a rare autosomal recessive syndrome caused by mutations in the PNPLA6 gene.
explanation: >-
Directly identifies PNPLA6 as the causal gene for BNS.
- reference: PMID:25267340
reference_title: "Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
both mutations (one novel and one known) fell in the phospholipase esterase domain, where most pathogenic mutations seem to cluster
explanation: >-
Documents that pathogenic PNPLA6 variants in BNS cluster in the
phospholipase esterase (catalytic) domain.
- reference: PMID:35069422
reference_title: "Multifaceted and Age-Dependent Phenotypes Associated With Biallelic PNPLA6 Gene Variants: Eight Novel Cases and Review of the Literature."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
A wide spectrum of neurodegenerative diseases has been associated with pathogenic variants in the PNPLA6 (patatin-like phospholipase domain-containing protein 6) gene, including spastic paraplegia type 39, Gordon-Holmes, Boucher-Neuhauser, Oliver-Mc Farlane, and Laurence-Moon syndromes.
explanation: >-
Establishes BNS as part of the broader allelic PNPLA6 disorder spectrum.
diagnosis:
- name: Molecular genetic testing
description: >-
Diagnosis is established in a proband with suggestive findings (cerebellar
ataxia, chorioretinal dystrophy, hypogonadotropic hypogonadism) by
identifying biallelic PNPLA6 pathogenic variants, typically via whole-exome
sequencing with Sanger segregation. Functional NTE esterase-activity assays
are an emerging adjunct for variant classification and phenotype prediction.
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The diagnosis of a PNPLA6 disorder is established in a proband with suggestive findings and biallelic PNPLA6 pathogenic variants identified on homologous alleles by molecular genetic testing.
explanation: >-
GeneReviews describes the molecular diagnostic approach for PNPLA6
disorders, including BNS.
- reference: PMID:35198007
reference_title: "Identification of Novel Compound Heterozygous Variants of the PNPLA6 Gene in Boucher-Neuhäuser Syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Gene sequencing is currently the primary diagnostic method.
explanation: >-
Confirms gene sequencing as the primary diagnostic modality for BNS.
treatments:
- name: Hormone replacement therapy
description: >-
Management is symptomatic. Hormone replacement therapy is given for the
endocrine deficiencies: at the expected time of puberty for hypogonadotropic
hypogonadism, during childhood/adolescence for growth hormone deficiency, and
as soon as identified for hypothyroidism.
treatment_term:
preferred_term: Hormone replacement therapy
term:
id: NCIT:C15599
label: Hormone Replacement Therapy
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hypogonadotropic hypogonadism. Hormone replacement therapy at the expected time of puberty.
explanation: >-
GeneReviews recommends hormone replacement therapy for the hypogonadotropic
hypogonadism of PNPLA6 disorders.
- name: Supportive and rehabilitative care
description: >-
Symptomatic, individually tailored multidisciplinary care: continuous
training of speech/swallowing, fine-motor skills, gait, and balance for
ataxia; physical therapy, assistive walking devices, and antispasticity
measures for spasticity; and low-vision aids and visual rehabilitation for
chorioretinal dystrophy. Patients should avoid alcohol, obesity, a sedentary
lifestyle, and medications/chemicals that exacerbate neuropathy.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Management is symptomatic and individually tailored. Ataxia. Continuous training of speech and swallowing, fine-motor skills, gait, and balance.
explanation: >-
GeneReviews describes the symptomatic, multidisciplinary supportive
management of PNPLA6 disorders.
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Agents/circumstances to avoid: Alcohol; obesity; inactive, sedentary lifestyle; exposure to medications or chemicals that exacerbate neuropathy.
explanation: >-
GeneReviews lists agents/circumstances to avoid, captured here as part of
supportive management guidance.
- name: Genetic counseling
description: >-
Genetic counseling is recommended given autosomal recessive inheritance.
Once the familial PNPLA6 variants are known, carrier testing for at-risk
relatives, prenatal testing, and preimplantation genetic testing are
possible.
treatment_term:
preferred_term: Genetic Counseling
term:
id: NCIT:C15240
label: Genetic Counseling
evidence:
- reference: PMID:25299038
reference_title: "PNPLA6 Disorders."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Once the PNPLA6 pathogenic variants in the family have been identified, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
explanation: >-
GeneReviews describes the genetic-counseling options available for PNPLA6
disorders.
datasets: []
References & Deep Research
References
1Deep Research
11. Disease Information
1.1 What is the disease?
BNS is a multisystem neurodegenerative syndrome that presents with the triad: - Cerebellar ataxia / cerebellar degeneration - Hypogonadotropic hypogonadism (HH) (often described clinically within anterior hypopituitarism) - Chorioretinal dystrophy / retinal degeneration
Primary literature defines BNS as “the triad of early-onset autosomal recessive cerebellar ataxia (ARCA), hypogonadotropic hypogonadism, and chorioretinal dystrophy.” (deik2014compoundheterozygouspnpla6 pages 1-2)
1.2 Key identifiers
- OMIM/MIM (disease): MIM #215470 (Boucher–Neuhäuser syndrome) (deik2014compoundheterozygouspnpla6 pages 1-2, he2022identificationofnovel pages 1-2)
- OMIM/MIM (gene): PNPLA6 MIM *603197 (deik2014compoundheterozygouspnpla6 pages 1-2)
- Other identifiers requested (Orphanet/ICD-10/ICD-11/MeSH/MONDO): Not found in the retrieved full-text excerpts; therefore cannot be asserted from this evidence set. (deik2014compoundheterozygouspnpla6 pages 1-2, he2022identificationofnovel pages 1-2)
1.3 Synonyms / alternative names
- BNS (common abbreviation) (he2022identificationofnovel pages 1-2, liampas2024twocasereports pages 1-5)
- Orthographic variants: Boucher–Neuhäuser, Boucher–Neuha¨user (deik2014compoundheterozygouspnpla6 pages 1-2)
- Historical phenotype description: “familial ataxia, hypogonadism and retinal degeneration” (reported as a historical descriptor in reviews of PNPLA6 phenotypes) (nanetti2022multifacetedandagedependent pages 9-10)
- Related/overlapping entities within PNPLA6 disorders: Gordon Holmes syndrome and others (deik2014compoundheterozygouspnpla6 pages 1-2, he2022identificationofnovel pages 1-2, liu2023pnpla6disorderswhat’s pages 3-4)
1.4 Evidence type (individual vs aggregated)
Evidence base is largely case reports/series aggregated in systematic reviews and cohort meta-analyses, including a large integrated dataset of published individuals with biallelic PNPLA6 variants (95 as of May 2023) and an expanded cohort of 118 individuals in later functional-genotype studies. (liu2023pnpla6disorderswhat’s pages 3-4, liu2024neuropathytargetesterase pages 4-6)
2. Etiology
2.1 Disease causal factors
Primary cause: biallelic pathogenic variants in PNPLA6, encoding neuropathy target esterase (NTE). BNS is repeatedly described as “a rare autosomal recessive syndrome caused by mutations in the PNPLA6 gene.” (he2022identificationofnovel pages 1-2)
Mechanistic framing: PNPLA6/NTE is an ER-associated patatin-like phospholipase/esterase involved in phospholipid homeostasis and trafficking; loss-of-function is supported by animal/cellular models. (liu2023pnpla6disorderswhat’s pages 1-3)
2.2 Risk factors
- Genetic risk: having biallelic pathogenic PNPLA6 variants (autosomal recessive). (he2022identificationofnovel pages 1-2, deik2014compoundheterozygouspnpla6 pages 2-5)
- Consanguinity can increase risk of homozygous variants (illustrated by affected siblings from a consanguineous family). (liampas2024twocasereports pages 1-5)
No environmental, infectious, or lifestyle risk factors specific to BNS were identified in the retrieved evidence.
2.3 Protective factors
No genetic or environmental protective factors were identified in the retrieved evidence.
2.4 Gene–environment interactions
Direct gene–environment interaction evidence for BNS is not present in the retrieved texts. Mechanistically, PNPLA6/NTE is historically linked to organophosphate-induced delayed neuropathy (OPIDN), which establishes environmental inhibition of NTE as neurotoxic, but this is not evidence of a specific GxE interaction for Mendelian BNS. (liu2023pnpla6disorderswhat’s pages 8-9)
3. Phenotypes
3.1 Core phenotype triad (with suggested HPO terms)
1) Cerebellar ataxia / cerebellar degeneration - Characteristics: variable onset (childhood to adulthood), often slowly progressive; MRI may show superior vermian/cerebellar atrophy. (deik2014compoundheterozygouspnpla6 pages 2-5, nanetti2022multifacetedandagedependent pages 3-5) - Suggested HPO: HP:0001251 (Ataxia); HP:0001272 (Cerebellar atrophy)
2) Hypogonadotropic hypogonadism / anterior hypopituitarism - Characteristics: may be recognized in the first two decades; hormone testing often shows low gonadotropins (LH/FSH) and broader pituitary hormone issues in the PNPLA6 spectrum. (deik2014compoundheterozygouspnpla6 pages 2-5, liampas2024twocasereports pages 1-5, liu2023pnpla6disorderswhat’s pages 3-4) - Suggested HPO: HP:0000044 (Hypogonadotropic hypogonadism); HP:0000871 (Hypopituitarism)
3) Chorioretinal dystrophy / retinal degeneration - Characteristics: onset widely variable (reported 1–64 years across PNPLA6 disorders); may mimic choroideremia; progression can lead to severe vision loss/blindness. (o’neil2019detailedretinalphenotype pages 6-7, liampas2024twocasereports pages 1-5, liu2023pnpla6disorderswhat’s pages 3-4) - Suggested HPO: HP:0000510 (Chorioretinal dystrophy); HP:0000546 (Retinal dystrophy); HP:0000505 (Visual impairment)
3.2 Additional, variably present features
- Peripheral axonal neuropathy (less common in some BNS descriptions; present in cohorts of PNPLA6 disorders). (liampas2024twocasereports pages 1-5, nanetti2022multifacetedandagedependent pages 3-5)
- HPO: HP:0003477 (Axonal neuropathy)
- Oculomotor abnormalities (e.g., gaze-evoked nystagmus, saccadic pursuit) reported in case descriptions. (deik2014compoundheterozygouspnpla6 pages 2-5)
- HPO: HP:0000639 (Nystagmus)
- Cognitive impairment observed in a subset of PNPLA6 cohort cases. (nanetti2022multifacetedandagedependent pages 3-5)
- HPO: HP:0100543 (Cognitive impairment)
- Hair anomalies occur in a minority of PNPLA6 disorders (more characteristic of Oliver–McFarlane end of spectrum). (liu2023pnpla6disorderswhat’s pages 3-4)
- HPO examples: HP:0002213 (Trichomegaly), HP:0001596 (Alopecia)
3.3 Frequency / statistics (best available from retrieved sources)
- Review-level counts: 95 published individuals with biallelic PNPLA6 variants as of May 2023 (PNPLA6 disorders overall). (liu2023pnpla6disorderswhat’s pages 3-4)
- Cerebellar atrophy/ataxia in up to ~90% of reported PNPLA6 cases. (liu2023pnpla6disorderswhat’s pages 3-4)
- Case-series (8 novel PNPLA6 cases): 7/8 progressive cerebellar syndrome; 5/8 HH; 2/8 chorioretinal dystrophy; 4/8 peripheral axonal neuropathy and/or spasticity; 3/8 cognitive impairment. (nanetti2022multifacetedandagedependent pages 3-5)
3.4 Quality of life impact
Direct QoL instrument results (EQ-5D/SF-36/PROMIS) were not found in the retrieved texts. Based on clinical manifestations, major impacts include mobility limitations from ataxia and disability from progressive visual loss. (liampas2024twocasereports pages 1-5, liu2023pnpla6disorderswhat’s pages 3-4)
4. Genetic / Molecular Information
4.1 Causal gene(s)
- PNPLA6 (patatin-like phospholipase domain-containing protein 6), encoding NTE. (he2022identificationofnovel pages 1-2, liu2023pnpla6disorderswhat’s pages 1-3)
4.2 Pathogenic variants (examples from primary literature)
BNS is caused by biallelic PNPLA6 variants (homozygous or compound heterozygous). Examples in retrieved sources include: - Compound heterozygous PNPLA6 mutations (e.g., p.Ser1045Leu and p.Ser1173Arg) in a genetically confirmed BNS case with late-onset ataxia. (deik2014compoundheterozygouspnpla6 pages 2-5) - Compound heterozygous variants including a frameshift and missense in a BNS case with predominantly retinal presentation (p.Arg1031GlnfsTer38 / p.Arg1183Gln). (o’neil2019detailedretinalphenotype pages 6-7) - Novel compound heterozygous variants identified by WES (c.2241del/p.Met748TrpfsTer65 and c.2986A>G/p.Thr996Ala) with RNA-level validation showing decreased PNPLA6 mRNA. (he2022identificationofnovel pages 1-2) - Homozygous missense c.3323G>A (p.Arg1108Gln) in two siblings. (liampas2024twocasereports pages 1-5)
Variant types reported across PNPLA6 disorders include missense, frameshift, splice-altering, and nonsense variants. In the Brain 2024 systematic cohort, 106 unique variants were summarized (70 missense; 35 predicted loss-of-function; 1 in-frame deletion), with enrichment of missense/in-frame variants in the catalytic domain. (liu2024neuropathytargetesterase pages 4-6)
4.3 Functional consequences
Multiple sources support loss-of-function as a principal mechanism, operationalized as reduced NTE hydrolase/esterase activity for many disease-associated variants. (liu2023pnpla6disorderswhat’s pages 1-3, liu2023neuropathytargetesterase pages 1-6)
4.4 Modifier genes / epigenetics / chromosomal abnormalities
No modifier genes, epigenetic signatures, or chromosomal abnormalities specific to BNS were identified in the retrieved evidence.
5. Environmental Information
No validated non-genetic environmental causes of Mendelian BNS were identified in the retrieved sources. However, PNPLA6/NTE is historically implicated in organophosphate neurotoxicity (OPIDN), indicating that environmental inhibition of NTE is neurotoxic in other contexts and motivates biomarker measurement in exposure studies. (liu2023pnpla6disorderswhat’s pages 8-9, NCT00671866 chunk 1)
6. Mechanism / Pathophysiology
6.1 Molecular function and pathways
PNPLA6 encodes NTE, described as a patatin-like serine hydrolase on the cytoplasmic face of the ER with: - Phospholipase B activity (deacylation of glycerophospholipids) - Strong lysophospholipase activity - Roles in phosphatidylcholine (PC) homeostasis (CDP-choline/Kennedy pathway), membrane trafficking, and axonal integrity. (liu2023pnpla6disorderswhat’s pages 1-3)
Suggested GO terms (biological process / molecular function; provisional) - GO:0006644 (phospholipid metabolic process) - GO:0004620 (phospholipase activity) - GO:0047499 (phospholipase B activity) - GO:0052689 (carboxylic ester hydrolase activity) - GO:0005789 (endoplasmic reticulum membrane; cellular component)
6.2 Causal chain (conceptual)
1) Biallelic PNPLA6 variants → 2) Reduced NTE enzymatic activity and disrupted phospholipid remodeling/homeostasis at ER membranes → 3) tissue vulnerability in retina (photoreceptors/RPE), cerebellum (Purkinje cell systems), and pituitary–gonadal axis → 4) clinical triad of retinopathy, ataxia, hypogonadotropic hypogonadism.
A key 2024 advance is that residual NTE activity can be used as a quantitative intermediate phenotype that predicts retinopathy and endocrinopathy risk. (liu2024neuropathytargetesterase pages 1-2, liu2024neuropathytargetesterase pages 7-9)
6.3 Recent (2023–2024) genotype:activity:phenotype relationships
In Brain (May 2024), Liu et al. report: - Cohort: 23 new + 95 reported individuals. (liu2024neuropathytargetesterase pages 1-2) - Variant functional assay: measured esterase activity for 46 disease-associated and 20 common variants; reclassified 36 as pathogenic and 10 as likely pathogenic. (liu2024neuropathytargetesterase pages 1-2) - Clinical subtype activity differences: SPG39 mean residual activity ~51% (n=12) vs BNHS ~28% (n=19) and OMCS/LNMS ~28% (n=26). (liu2024neuropathytargetesterase pages 4-6) - Mouse allelic series: <40% NTE activity was embryonic lethal; retinal degeneration onset ~40–50% residual activity. (liu2024neuropathytargetesterase pages 7-9)
These results support expert interpretation that PNPLA6 syndromic labels represent a continuum and that functional enzyme activity can serve both diagnostic and prognostic roles, potentially enabling trial stratification. (liu2024neuropathytargetesterase pages 1-2)
6.4 Model organism evidence (authoritative sources)
- Chickens (OPIDN): OPIDN requires ~70% inhibition of NTE activity via “aging.” (liu2023pnpla6disorderswhat’s pages 8-9)
- Drosophila (swiss-cheese; sws): sws loss causes rapid age-dependent neurodegeneration with ~20% more PC, ER stress signatures, and partial rescue by TUDCA (ER stress inhibitor). (liu2023pnpla6disorderswhat’s pages 8-9)
- Mouse: global Pnpla6 knockout embryonic lethal; neuronal conditional KO shows hippocampal vacuolization, neuronal loss (including Purkinje cells) and axonal lesions. (liu2023pnpla6disorderswhat’s pages 8-9)
Suggested CL terms (cell types; provisional) - Purkinje cell: CL:0000121 - Retinal photoreceptor cell: CL:0000210 - Retinal pigment epithelial cell: CL:0000088
7. Anatomical Structures Affected
Organ/system level (with UBERON suggestions; provisional)
- Cerebellum (UBERON:0002037) (deik2014compoundheterozygouspnpla6 pages 2-5, liu2023pnpla6disorderswhat’s pages 3-4)
- Retina / choroid / RPE (UBERON:0000966 for retina; RPE is part of eye tissues) (o’neil2019detailedretinalphenotype pages 6-7, liu2023pnpla6disorderswhat’s pages 3-4)
- Pituitary gland / hypothalamic–pituitary axis (UBERON:0000007 for pituitary) (liu2023pnpla6disorderswhat’s pages 3-4)
Subcellular localization
- Endoplasmic reticulum membrane (GO:0005789) for PNPLA6/NTE. (liu2023pnpla6disorderswhat’s pages 1-3)
8. Temporal Development
Onset
Across PNPLA6 disorders (including BNS): - Gait disturbance: 1–55 years - Visual impairment: 1–64 years - Anterior hypopituitarism: birth–25 years - Hair anomalies: birth–18 years
These ranges reflect substantial heterogeneity and age-dependent penetrance of sub-phenotypes. (liu2023pnpla6disorderswhat’s pages 3-4)
Progression
Progression can be slow: - In an 8-patient PNPLA6 series, cerebellar symptoms had mean onset 31 years (range 9–55) and were “very slow” in progression; most had progressive cerebellar syndrome. (nanetti2022multifacetedandagedependent pages 3-5)
9. Inheritance and Population
Inheritance
- Autosomal recessive inheritance due to biallelic PNPLA6 variants is consistently reported. (he2022identificationofnovel pages 1-2, liampas2024twocasereports pages 1-5)
Epidemiology
No prevalence/incidence per population was identified in the retrieved full texts. Best available “epidemiology-like” statistic is literature case count: - 95 published individuals with biallelic PNPLA6 variants (as of May 2023). (liu2023pnpla6disorderswhat’s pages 3-4)
Population genetics
No carrier frequency estimates or founder variants were identified in the retrieved evidence.
10. Diagnostics
10.1 Clinical tests / evaluations used in practice
Across case reports and reviews, diagnosis is typically assembled from multisystem evaluation: - Brain MRI: cerebellar atrophy, often vermian/superior cerebellar involvement. (deik2014compoundheterozygouspnpla6 pages 2-5, nanetti2022multifacetedandagedependent pages 3-5) - Ophthalmology: fundus exam, OCT, ERG, visual fields/perimetry; can reveal outer retinal loss and chorioretinal atrophy; presentations may mimic choroideremia, necessitating careful differential diagnosis. (deik2014compoundheterozygouspnpla6 pages 2-5, o’neil2019detailedretinalphenotype pages 6-7) - Endocrine testing: gonadotropins (LH/FSH), sex steroids; broader pituitary hormone panels depending on presentation. (deik2014compoundheterozygouspnpla6 pages 2-5, he2022identificationofnovel pages 1-2) - Neurophysiology: EMG/NCS for mild/subclinical axonal neuropathy. (deik2014compoundheterozygouspnpla6 pages 2-5)
10.2 Genetic testing
- Whole-exome sequencing (WES) with segregation testing is commonly used; authors explicitly state “Gene sequencing is currently the primary diagnostic method.” (he2022identificationofnovel pages 1-2)
- Emerging 2024 evidence supports adding functional NTE activity assays for variant classification and phenotype prediction. (liu2024neuropathytargetesterase pages 1-2)
10.3 Differential diagnosis
Not comprehensively enumerated in retrieved texts. One practical point is that PNPLA6-associated chorioretinal dystrophy can mimic choroideremia-like presentations, and thus PNPLA6 should be considered in diffuse chorioretinal atrophies with subtle systemic signs. (o’neil2019detailedretinalphenotype pages 6-7)
11. Outcome / Prognosis
Quantitative survival/life expectancy estimates were not found in the retrieved evidence.
Best available natural-history/prognosis statements: - Vision: chorioretinal dystrophy “leads to variable decreased visual acuity, even blindness.” (liampas2024twocasereports pages 1-5) - Some PNPLA6 disorders (milder spectrum) retain ambulation into adulthood with minimal/moderate aid. (liu2023pnpla6disorderswhat’s pages 3-4) - Slow progression of cerebellar syndrome has been reported with long disease duration in case series. (nanetti2022multifacetedandagedependent pages 3-5)
12. Treatment
No disease-modifying therapy is established in the retrieved evidence. Management is supportive and multidisciplinary.
12.1 Pharmacotherapy / endocrine replacement
- Case report supportive regimen included hormone replacement therapy and vitamin supplementation (B12, C, E). (liampas2024twocasereports pages 1-5)
Suggested MAXO terms (provisional) - MAXO:0000258 (Hormone replacement therapy) - MAXO:0000747 (Vitamin supplementation)
12.2 Rehabilitation / supportive care
Direct rehabilitation trial data were not present in the retrieved texts. Based on multi-system involvement, supportive care typically includes neurology/ataxia management, vision support, and endocrine replacement/surveillance. (liu2023pnpla6disorderswhat’s pages 3-4, liampas2024twocasereports pages 1-5)
12.3 Experimental/clinical trials
No BNS-specific interventional trials were identified in the retrieved clinical-trials search. - An observational trial on organophosphate exposure measures NTE biomarkers but is not a therapeutic trial for BNS and lists “Peripheral Neuropathy” as condition (NCT00671866). (NCT00671866 chunk 1)
13. Prevention
No primary prevention exists for Mendelian BNS beyond reproductive/genetic counseling.
Secondary prevention conceptually includes: - Early recognition of retinal degeneration and pituitary hormone deficiencies for timely supportive interventions. (o’neil2019detailedretinalphenotype pages 6-7, liu2023pnpla6disorderswhat’s pages 3-4)
14. Other Species / Natural Disease
Naturally occurring BNS as a veterinary entity was not identified in the retrieved evidence.
However, PNPLA6 is evolutionarily conserved with an orthologue in Drosophila (swiss-cheese; sws) used to model neurodegeneration and lipid dysregulation. (liu2023pnpla6disorderswhat’s pages 8-9)
15. Model Organisms
Key models and how they recapitulate disease biology
- Drosophila sws mutants: age-dependent neurodegeneration, altered phospholipids, ER stress; partial pharmacologic rescue (TUDCA). (liu2023pnpla6disorderswhat’s pages 8-9)
- Mouse Pnpla6 allelic series (2024): demonstrates activity thresholds for viability and retinal degeneration and provides translational evidence for NTE activity as a biomarker. (liu2024neuropathytargetesterase pages 7-9)
Recent developments and expert analysis (2023–2024)
Conceptual shift: discrete syndromes → continuous PNPLA6 spectrum
A 2023 ophthalmic-genetics review frames PNPLA6-related conditions (including BNS) as “PNPLA6-opathies” across a spectrum and highlights a choroideremia-like retinal phenotype and multisystem involvement requiring integrated diagnosis. (liu2023pnpla6disorderswhat’s pages 3-4)
Functional stratification: NTE activity as diagnostic/prognostic biomarker (2024)
The 2024 Brain study provides a quantitative model linking biallelic genotype to residual NTE activity and to the probability of retinopathy/endocrinopathy, and proposes NTE as a biomarker “paving the way for therapeutic trials.” (liu2024neuropathytargetesterase pages 1-2)
Figure evidence supporting these claims is available from cropped panels showing (i) variant domain associations with retinopathy/endocrinopathy and (ii) activity-phenotype correlations and (iii) activity thresholds for retinopathy in vivo. (liu2024neuropathytargetesterase media cbbab372, liu2024neuropathytargetesterase media f1acab95, liu2024neuropathytargetesterase media d77bd8b8)
Key sources table (prioritizing 2023–2024)
| Year | Citation (first author, journal) | Study type (review/case series/mechanistic) | Key contributions for BNS (definition/triad; cohort size; key stats like NTE activity means/thresholds; diagnostic insights) | URL/DOI | Notes (PMID if known—leave blank if not present in text) |
|---|---|---|---|---|---|
| 2024 | Liu, Brain | Mechanistic + cohort + systematic review | PNPLA6 disorders placed on a continuous spectrum including Boucher–Neuhäuser syndrome (BNHS). Cohort: 23 new patients + 95 previously reported individuals. Functional assay measured 46 disease-associated and 20 common variants; 36 variants reclassified as pathogenic and 10 as likely pathogenic. Synthetic residual NTE activity differed by phenotype: SPG39 mean ~51% (n=12) vs BNHS ~28% (n=19) and OMCS/LNMS ~28% (n=26); lower activity associated with retinopathy/endocrinopathy, with reported threshold-type observations for endocrine/ophthalmic disease below ~32% and retinal degeneration around 40–50% residual activity; mouse data supported retinopathy threshold and embryonic lethality below 40% activity. Supports NTE activity as a biomarker for therapeutic trials. (liu2024neuropathytargetesterase pages 1-2, liu2024neuropathytargetesterase pages 4-6, liu2023neuropathytargetesterase pages 10-14, liu2024neuropathytargetesterase pages 7-9) | https://doi.org/10.1093/brain/awae055 | |
| 2023 | Liu, Ophthalmic Genetics | Review | Defines five PNPLA6 disorders, including Boucher–Neuhäuser syndrome. States PNPLA6/NTE is involved in phospholipid homeostasis and trafficking; animal and cellular models support loss-of-function. As of May 2023, 95 published individuals with biallelic PNPLA6 variants. Cerebellar atrophy/ataxia reported in up to ~90% of cases. BNHS distinguished from Gordon–Holmes by added chorioretinal dystrophy. Diagnostic insights: neurological imaging for cerebellar atrophy, ERG/retinal imaging/visual fields for chorioretinal dystrophy, hormonal testing for anterior hypopituitarism, and exam for hair anomalies. (liu2023pnpla6disorderswhat’s pages 3-4, liu2023pnpla6disorderswhat’s pages 4-6, liu2023pnpla6disorderswhat’s pages 1-3) | https://doi.org/10.1080/13816810.2023.2254830 | |
| 2024 | Liampas, Molecular Biology Reports | Case reports | Two siblings with a novel homozygous PNPLA6 missense variant. Reiterates classical BNS triad: hypogonadotropic hypogonadism, spinocerebellar ataxia, and chorioretinal dystrophy; notes peripheral axonal neuropathy can occur. Provides temporal guidance: HH often in first two decades; ataxia usually before early adulthood but can be late; chorioretinal dystrophy usually before age 50 and may progress to severe visual loss/blindness. Diagnostic workup included MRI, ophthalmologic assessment, endocrine evaluation, electrophysiology, WES and Sanger segregation. (liampas2024twocasereports pages 1-5) | https://doi.org/10.1007/s11033-024-09515-4 | |
| 2022 | He, Frontiers in Genetics | Case report + variant analysis/systematic review | Defines BNS as a rare autosomal recessive PNPLA6 disorder with the triad of cerebellar ataxia, chorioretinal dystrophy, and hypogonadotropic hypogonadism; gives MIM 215470. Reports a 17-year-old with progressive night blindness from age 4, primary amenorrhea, absent secondary sexual development, retinal pigmentary degeneration, and CHH without current ataxia. Identified compound heterozygous PNPLA6 variants by WES; RT-PCR showed reduced PNPLA6 mRNA. Diagnostic insight: detailed ophthalmic exam, endocrine panels, pituitary/pelvic imaging, bone age X-ray, WES/Sanger; authors note gene sequencing is currently the primary diagnostic method. (he2022identificationofnovel pages 1-2) | https://doi.org/10.3389/fgene.2022.810537 | |
| 2022 | Nanetti, Frontiers in Neurology | Case series + literature review | Reviews age-dependent PNPLA6 phenotypes and includes BN presentations. Across eight new PNPLA6 cases: 7/8 had cerebellar ataxia, 5/8 hypogonadotropic hypogonadism, 2/8 chorioretinal dystrophy; cerebellar symptoms mean onset 31 years (range 9–55) with very slow progression. Notes early-onset presentations may start with chorioretinal dystrophy, juvenile cases with HH, adult cases with ataxia. MRI may show cerebellar atrophy (often superior/dorsal vermis) and severe cerebellar atrophy in BN cases. Recommends multidisciplinary assessment and PNPLA6 screening in late-onset/cANVAS-like ataxia when RFC1 expansions are absent. (nanetti2022multifacetedandagedependent pages 9-10, nanetti2022multifacetedandagedependent pages 3-5) | https://doi.org/10.3389/fneur.2021.793547 | |
| 2019 | O’Neil, Ophthalmic Genetics | Deep phenotyping case report | Shows that PNPLA6-associated BNS may present with predominantly retinal findings and subtle systemic abnormalities, mimicking choroideremia. Ophthalmic workup included SD-OCT, ERG, kinetic fields, autofluorescence imaging; systemic confirmation came from hypogonadotropic hypogonadism and cerebellar vermis hypoplasia on MRI. Highlights the need to consider PNPLA6/BNS in diffuse chorioretinal atrophies and to combine ophthalmic phenotyping with systemic review and genetic testing. (o’neil2019detailedretinalphenotype pages 6-7) | https://doi.org/10.1080/13816810.2019.1605392 | |
| 2014 | Deik, Journal of Neurology | Case report + genetics | Landmark report confirming compound heterozygous PNPLA6 mutations as a cause of BNS with late-onset ataxia. Uses MIM #215470 for BNS and emphasizes the triad of autosomal recessive cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy. Diagnostic workup included fundoscopy/OCT, ERG, visual fields, endocrine testing (low LH/FSH), brain MRI showing superior cerebellar/vermian atrophy, EMG/NCS showing mild distal axonal neuropathy, and PNPLA6 sequencing. Also notes possible cognitive involvement and subclinical polyneuropathy. (deik2014compoundheterozygouspnpla6 pages 11-12, deik2014compoundheterozygouspnpla6 pages 2-5, deik2014compoundheterozygouspnpla6 pages 1-2) | https://doi.org/10.1007/s00415-014-7516-3 | |
| 2022 | Kretzschmar, Metabolites | Mechanistic review | Reviews PNPLA6/NTE as an evolutionarily conserved phospholipase first linked to organophosphate-induced delayed neuropathy and later to inherited disorders including BNS. Summarizes normal role in lipid homeostasis and model-system evidence: mouse brain conditional knockout causes age-related neurodegeneration, complete knockout is embryonic lethal, and Drosophila swiss-cheese loss causes progressive locomotor defects and neurodegeneration. Useful for mechanistic context underlying BNS/PNPLA6 loss-of-function. (liu2023pnpla6disorderswhat’s pages 8-9) | https://doi.org/10.3390/metabo12040284 |
Table: This table summarizes the most useful recent and foundational sources for Boucher–Neuhäuser syndrome within the broader PNPLA6 disorder spectrum. It highlights how each paper contributes evidence on definition, genotype–phenotype correlations, diagnostics, and mechanistic understanding.
Direct abstract quotes (as available in retrieved context)
- He et al. 2022 (Frontiers in Genetics) abstract includes: “Boucher–Neuhäuser syndrome (BNS, MIM 215470) is a rare autosomal recessive syndrome caused by mutations in the PNPLA6 gene.” (he2022identificationofnovel pages 1-2)
- O’Neil et al. 2019 abstract includes: “PNPLA6-associated retinal degenerations can present with predominantly retinal findings and subtle systemic abnormalities and should be considered in the differential diagnosis of diffuse chorioretinal atrophies.” (o’neil2019detailedretinalphenotype pages 6-7)
(Additional verbatim abstract text for Liu et al. 2024 and Liu & Hufnagel 2023 was not captured in the provided excerpts; therefore, only non-verbatim extraction is reported for those papers.) (liu2024neuropathytargetesterase pages 1-2, liu2023pnpla6disorderswhat’s pages 3-4)
Evidence gaps relative to the requested template
- MONDO / Orphanet / ICD-10 / ICD-11 / MeSH identifiers: not present in retrieved full texts; would require direct querying of those databases.
- Prevalence/incidence estimates: not provided in retrieved literature excerpts; current best quantitative evidence is published case counts.
- Controlled treatment studies: no interventional trials specific to BNS identified in retrieved evidence.
References
-
(deik2014compoundheterozygouspnpla6 pages 1-2): A. Deik, Brooke Johannes, J. Rucker, E. Sánchez, S. Brodie, E. Deegan, K. Landy, Y. Kajiwara, S. Scelsa, R. Saunders-Pullman, R. Saunders-Pullman, and C. Paisán-Ruiz. Compound heterozygous pnpla6 mutations cause boucher–neuhäuser syndrome with late-onset ataxia. Journal of Neurology, 261:2411-2423, Sep 2014. URL: https://doi.org/10.1007/s00415-014-7516-3, doi:10.1007/s00415-014-7516-3. This article has 44 citations and is from a domain leading peer-reviewed journal.
-
(he2022identificationofnovel pages 1-2): Junyu He, Xin Liu, Liyi Liu, Shaohao Zeng, Shuanghong Shan, and Zhihong Liao. Identification of novel compound heterozygous variants of the pnpla6 gene in boucher–neuhäuser syndrome. Frontiers in Genetics, Feb 2022. URL: https://doi.org/10.3389/fgene.2022.810537, doi:10.3389/fgene.2022.810537. This article has 8 citations and is from a peer-reviewed journal.
-
(liu2023pnpla6disorderswhat’s pages 3-4): James Liu and Robert B. Hufnagel. Pnpla6 disorders: what’s in a name? Ophthalmic Genetics, 44:530-538, Sep 2023. URL: https://doi.org/10.1080/13816810.2023.2254830, doi:10.1080/13816810.2023.2254830. This article has 15 citations and is from a peer-reviewed journal.
-
(liu2024neuropathytargetesterase pages 1-2): James Liu, Yi He, Cara Lwin, Marina Han, Bin Guan, Amelia Naik, Chelsea Bender, Nia Moore, Laryssa A Huryn, Yuri V Sergeev, Haohua Qian, Yong Zeng, Lijin Dong, Pinghu Liu, Jingqi Lei, Carl J Haugen, Lev Prasov, Ruifang Shi, Hélène Dollfus, Petros Aristodemou, Yannik Laich, Andrea H Németh, John Taylor, Susan Downes, Maciej R Krawczynski, Isabelle Meunier, Melissa Strassberg, Jessica Tenney, Josephine Gao, Matthew A Shear, Anthony T Moore, Jacque L Duncan, Beatriz Menendez, Sarah Hull, Andrea L Vincent, Carly E Siskind, Elias I Traboulsi, Craig Blackstone, Robert A Sisk, Virginia Miraldi Utz, Andrew R Webster, Michel Michaelides, Gavin Arno, Matthis Synofzik, and Robert B Hufnagel. Neuropathy target esterase activity defines phenotypes among pnpla6 disorders. Brain : a journal of neurology, 147:2085-2097, May 2024. URL: https://doi.org/10.1093/brain/awae055, doi:10.1093/brain/awae055. This article has 10 citations.
-
(liu2024neuropathytargetesterase pages 4-6): James Liu, Yi He, Cara Lwin, Marina Han, Bin Guan, Amelia Naik, Chelsea Bender, Nia Moore, Laryssa A Huryn, Yuri V Sergeev, Haohua Qian, Yong Zeng, Lijin Dong, Pinghu Liu, Jingqi Lei, Carl J Haugen, Lev Prasov, Ruifang Shi, Hélène Dollfus, Petros Aristodemou, Yannik Laich, Andrea H Németh, John Taylor, Susan Downes, Maciej R Krawczynski, Isabelle Meunier, Melissa Strassberg, Jessica Tenney, Josephine Gao, Matthew A Shear, Anthony T Moore, Jacque L Duncan, Beatriz Menendez, Sarah Hull, Andrea L Vincent, Carly E Siskind, Elias I Traboulsi, Craig Blackstone, Robert A Sisk, Virginia Miraldi Utz, Andrew R Webster, Michel Michaelides, Gavin Arno, Matthis Synofzik, and Robert B Hufnagel. Neuropathy target esterase activity defines phenotypes among pnpla6 disorders. Brain : a journal of neurology, 147:2085-2097, May 2024. URL: https://doi.org/10.1093/brain/awae055, doi:10.1093/brain/awae055. This article has 10 citations.
-
(liampas2024twocasereports pages 1-5): Andreas Liampas, Paschalis Nicolaou, Christina Votsi, Anthi Georghiou, Kyproula Christodoulou, George A Tanteles, and Marios Pantzaris. Two case reports of a novel missense mutation in the pnpla6 gene in two siblings with chorioretinal dystrophy, hypogonadotropic hypogonadism, and cerebellar ataxia. Molecular biology reports, 51 1:590, Apr 2024. URL: https://doi.org/10.1007/s11033-024-09515-4, doi:10.1007/s11033-024-09515-4. This article has 0 citations and is from a peer-reviewed journal.
-
(nanetti2022multifacetedandagedependent pages 9-10): Lorenzo Nanetti, Daniela Di Bella, Stefania Magri, Mario Fichera, Elisa Sarto, Anna Castaldo, Alessia Mongelli, Silvia Baratta, Silvia Fenu, Marco Moscatelli, Maria Teresa Bonati, Andrea Martinuzzi, Caterina Mariotti, and Franco Taroni. Multifaceted and age-dependent phenotypes associated with biallelic pnpla6 gene variants: eight novel cases and review of the literature. Frontiers in Neurology, Jan 2022. URL: https://doi.org/10.3389/fneur.2021.793547, doi:10.3389/fneur.2021.793547. This article has 18 citations and is from a peer-reviewed journal.
-
(liu2023pnpla6disorderswhat’s pages 1-3): James Liu and Robert B. Hufnagel. Pnpla6 disorders: what’s in a name? Ophthalmic Genetics, 44:530-538, Sep 2023. URL: https://doi.org/10.1080/13816810.2023.2254830, doi:10.1080/13816810.2023.2254830. This article has 15 citations and is from a peer-reviewed journal.
-
(deik2014compoundheterozygouspnpla6 pages 2-5): A. Deik, Brooke Johannes, J. Rucker, E. Sánchez, S. Brodie, E. Deegan, K. Landy, Y. Kajiwara, S. Scelsa, R. Saunders-Pullman, R. Saunders-Pullman, and C. Paisán-Ruiz. Compound heterozygous pnpla6 mutations cause boucher–neuhäuser syndrome with late-onset ataxia. Journal of Neurology, 261:2411-2423, Sep 2014. URL: https://doi.org/10.1007/s00415-014-7516-3, doi:10.1007/s00415-014-7516-3. This article has 44 citations and is from a domain leading peer-reviewed journal.
-
(liu2023pnpla6disorderswhat’s pages 8-9): James Liu and Robert B. Hufnagel. Pnpla6 disorders: what’s in a name? Ophthalmic Genetics, 44:530-538, Sep 2023. URL: https://doi.org/10.1080/13816810.2023.2254830, doi:10.1080/13816810.2023.2254830. This article has 15 citations and is from a peer-reviewed journal.
-
(nanetti2022multifacetedandagedependent pages 3-5): Lorenzo Nanetti, Daniela Di Bella, Stefania Magri, Mario Fichera, Elisa Sarto, Anna Castaldo, Alessia Mongelli, Silvia Baratta, Silvia Fenu, Marco Moscatelli, Maria Teresa Bonati, Andrea Martinuzzi, Caterina Mariotti, and Franco Taroni. Multifaceted and age-dependent phenotypes associated with biallelic pnpla6 gene variants: eight novel cases and review of the literature. Frontiers in Neurology, Jan 2022. URL: https://doi.org/10.3389/fneur.2021.793547, doi:10.3389/fneur.2021.793547. This article has 18 citations and is from a peer-reviewed journal.
-
(o’neil2019detailedretinalphenotype pages 6-7): Erin O’Neil, Leona Serrano, Drew Scoles, Kayla E Cunningham, Grace Han, John Chiang, Jean Bennett, and Tomas S. Aleman. Detailed retinal phenotype of boucher-neuhäuser syndrome associated with mutations in pnpla6 mimicking choroideremia. Ophthalmic Genetics, 40:267-275, May 2019. URL: https://doi.org/10.1080/13816810.2019.1605392, doi:10.1080/13816810.2019.1605392. This article has 17 citations and is from a peer-reviewed journal.
-
(liu2023neuropathytargetesterase pages 1-6): James Liu, Yi He, Cara Lwin, Marina Han, Bin Guan, Amelia Naik, Chelsea Bender, Nia Moore, Laryssa A. Huryn, Yuri Sergeev, Haohua Qian, Yong Zeng, Lijin Dong, Pinghu Liu, Jingqi Lei, Carl J. Haugen, Lev Prasov, Ruifang Shi, Hélène Dollfus, Petros Aristodemou, Yannik Laich, Andrea H. Németh, John Taylor, Susan Downes, Maciej Krawczynski, Isabelle Meunier, Melissa Strassberg, Jessica Tenney, Josephine Gao, Matthew A. Shear, Anthony T. Moore, Jacque L. Duncan, Beatriz Menendez, Sarah Hull, Andrea Vincent, Carly E. Siskind, Elias I. Traboulsi, Craig Blackstone, Robert Sisk, Virginia Utz, Andrew R. Webster, Michel Michaelides, Gavin Arno, Matthis Synofzik, and Robert B Hufnagel. Neuropathy target esterase activity predicts retinopathy among pnpla6 disorders. bioRxiv, Jun 2023. URL: https://doi.org/10.1101/2023.06.09.544373, doi:10.1101/2023.06.09.544373. This article has 1 citations.
-
(NCT00671866 chunk 1): Neurotoxic Health Hazards of Long-Term Low-Level Exposure to Organophosphate (OP) Compounds in in Hula Valley. Shaare Zedek Medical Center. ClinicalTrials.gov Identifier: NCT00671866
-
(liu2024neuropathytargetesterase pages 7-9): James Liu, Yi He, Cara Lwin, Marina Han, Bin Guan, Amelia Naik, Chelsea Bender, Nia Moore, Laryssa A Huryn, Yuri V Sergeev, Haohua Qian, Yong Zeng, Lijin Dong, Pinghu Liu, Jingqi Lei, Carl J Haugen, Lev Prasov, Ruifang Shi, Hélène Dollfus, Petros Aristodemou, Yannik Laich, Andrea H Németh, John Taylor, Susan Downes, Maciej R Krawczynski, Isabelle Meunier, Melissa Strassberg, Jessica Tenney, Josephine Gao, Matthew A Shear, Anthony T Moore, Jacque L Duncan, Beatriz Menendez, Sarah Hull, Andrea L Vincent, Carly E Siskind, Elias I Traboulsi, Craig Blackstone, Robert A Sisk, Virginia Miraldi Utz, Andrew R Webster, Michel Michaelides, Gavin Arno, Matthis Synofzik, and Robert B Hufnagel. Neuropathy target esterase activity defines phenotypes among pnpla6 disorders. Brain : a journal of neurology, 147:2085-2097, May 2024. URL: https://doi.org/10.1093/brain/awae055, doi:10.1093/brain/awae055. This article has 10 citations.
-
(liu2024neuropathytargetesterase media cbbab372): James Liu, Yi He, Cara Lwin, Marina Han, Bin Guan, Amelia Naik, Chelsea Bender, Nia Moore, Laryssa A Huryn, Yuri V Sergeev, Haohua Qian, Yong Zeng, Lijin Dong, Pinghu Liu, Jingqi Lei, Carl J Haugen, Lev Prasov, Ruifang Shi, Hélène Dollfus, Petros Aristodemou, Yannik Laich, Andrea H Németh, John Taylor, Susan Downes, Maciej R Krawczynski, Isabelle Meunier, Melissa Strassberg, Jessica Tenney, Josephine Gao, Matthew A Shear, Anthony T Moore, Jacque L Duncan, Beatriz Menendez, Sarah Hull, Andrea L Vincent, Carly E Siskind, Elias I Traboulsi, Craig Blackstone, Robert A Sisk, Virginia Miraldi Utz, Andrew R Webster, Michel Michaelides, Gavin Arno, Matthis Synofzik, and Robert B Hufnagel. Neuropathy target esterase activity defines phenotypes among pnpla6 disorders. Brain : a journal of neurology, 147:2085-2097, May 2024. URL: https://doi.org/10.1093/brain/awae055, doi:10.1093/brain/awae055. This article has 10 citations.
-
(liu2024neuropathytargetesterase media f1acab95): James Liu, Yi He, Cara Lwin, Marina Han, Bin Guan, Amelia Naik, Chelsea Bender, Nia Moore, Laryssa A Huryn, Yuri V Sergeev, Haohua Qian, Yong Zeng, Lijin Dong, Pinghu Liu, Jingqi Lei, Carl J Haugen, Lev Prasov, Ruifang Shi, Hélène Dollfus, Petros Aristodemou, Yannik Laich, Andrea H Németh, John Taylor, Susan Downes, Maciej R Krawczynski, Isabelle Meunier, Melissa Strassberg, Jessica Tenney, Josephine Gao, Matthew A Shear, Anthony T Moore, Jacque L Duncan, Beatriz Menendez, Sarah Hull, Andrea L Vincent, Carly E Siskind, Elias I Traboulsi, Craig Blackstone, Robert A Sisk, Virginia Miraldi Utz, Andrew R Webster, Michel Michaelides, Gavin Arno, Matthis Synofzik, and Robert B Hufnagel. Neuropathy target esterase activity defines phenotypes among pnpla6 disorders. Brain : a journal of neurology, 147:2085-2097, May 2024. URL: https://doi.org/10.1093/brain/awae055, doi:10.1093/brain/awae055. This article has 10 citations.
-
(liu2024neuropathytargetesterase media d77bd8b8): James Liu, Yi He, Cara Lwin, Marina Han, Bin Guan, Amelia Naik, Chelsea Bender, Nia Moore, Laryssa A Huryn, Yuri V Sergeev, Haohua Qian, Yong Zeng, Lijin Dong, Pinghu Liu, Jingqi Lei, Carl J Haugen, Lev Prasov, Ruifang Shi, Hélène Dollfus, Petros Aristodemou, Yannik Laich, Andrea H Németh, John Taylor, Susan Downes, Maciej R Krawczynski, Isabelle Meunier, Melissa Strassberg, Jessica Tenney, Josephine Gao, Matthew A Shear, Anthony T Moore, Jacque L Duncan, Beatriz Menendez, Sarah Hull, Andrea L Vincent, Carly E Siskind, Elias I Traboulsi, Craig Blackstone, Robert A Sisk, Virginia Miraldi Utz, Andrew R Webster, Michel Michaelides, Gavin Arno, Matthis Synofzik, and Robert B Hufnagel. Neuropathy target esterase activity defines phenotypes among pnpla6 disorders. Brain : a journal of neurology, 147:2085-2097, May 2024. URL: https://doi.org/10.1093/brain/awae055, doi:10.1093/brain/awae055. This article has 10 citations.
-
(liu2023neuropathytargetesterase pages 10-14): James Liu, Yi He, Cara Lwin, Marina Han, Bin Guan, Amelia Naik, Chelsea Bender, Nia Moore, Laryssa A. Huryn, Yuri Sergeev, Haohua Qian, Yong Zeng, Lijin Dong, Pinghu Liu, Jingqi Lei, Carl J. Haugen, Lev Prasov, Ruifang Shi, Hélène Dollfus, Petros Aristodemou, Yannik Laich, Andrea H. Németh, John Taylor, Susan Downes, Maciej Krawczynski, Isabelle Meunier, Melissa Strassberg, Jessica Tenney, Josephine Gao, Matthew A. Shear, Anthony T. Moore, Jacque L. Duncan, Beatriz Menendez, Sarah Hull, Andrea Vincent, Carly E. Siskind, Elias I. Traboulsi, Craig Blackstone, Robert Sisk, Virginia Utz, Andrew R. Webster, Michel Michaelides, Gavin Arno, Matthis Synofzik, and Robert B Hufnagel. Neuropathy target esterase activity predicts retinopathy among pnpla6 disorders. bioRxiv, Jun 2023. URL: https://doi.org/10.1101/2023.06.09.544373, doi:10.1101/2023.06.09.544373. This article has 1 citations.
-
(liu2023pnpla6disorderswhat’s pages 4-6): James Liu and Robert B. Hufnagel. Pnpla6 disorders: what’s in a name? Ophthalmic Genetics, 44:530-538, Sep 2023. URL: https://doi.org/10.1080/13816810.2023.2254830, doi:10.1080/13816810.2023.2254830. This article has 15 citations and is from a peer-reviewed journal.
-
(deik2014compoundheterozygouspnpla6 pages 11-12): A. Deik, Brooke Johannes, J. Rucker, E. Sánchez, S. Brodie, E. Deegan, K. Landy, Y. Kajiwara, S. Scelsa, R. Saunders-Pullman, R. Saunders-Pullman, and C. Paisán-Ruiz. Compound heterozygous pnpla6 mutations cause boucher–neuhäuser syndrome with late-onset ataxia. Journal of Neurology, 261:2411-2423, Sep 2014. URL: https://doi.org/10.1007/s00415-014-7516-3, doi:10.1007/s00415-014-7516-3. This article has 44 citations and is from a domain leading peer-reviewed journal.