Achondrogenesis Type II
Achondrogenesis type II (ACG2), also known as Langer-Saldino type, is the most severe form of type 2 collagenopathy caused by dominant mutations in COL2A1. It is a lethal skeletal dysplasia characterized by severe micromelia, deficient ossification of the vertebral bodies and sacrum, a small thorax, and a large head. Death typically occurs in utero or shortly after birth. ACG2 represents the severe end of the COL2A1 mutation spectrum, which includes hypochondrogenesis, spondyloepiphyseal dysplasia congenita, and Stickler syndrome.
Ask OpenScientist
Ask a research question about Achondrogenesis Type II. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (1 reference)
Pathophysiology
3Show evidence (2 references)
Show evidence (3 references)
Show evidence (1 reference)
Histopathology
1Show evidence (1 reference)
Pathograph
Phenotypes
38Cardiovascular 1
Show evidence (1 reference)
Ear 1
Show evidence (1 reference)
Eye 4
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Head and Neck 6
Show evidence (3 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Limbs 2
Show evidence (3 references)
Show evidence (1 reference)
Metabolism 2
Show evidence (3 references)
Show evidence (1 reference)
Musculoskeletal 4
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Prenatal and Birth 1
Show evidence (2 references)
Respiratory 2
Show evidence (1 reference)
Show evidence (1 reference)
Growth 1
Show evidence (1 reference)
Other 14
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
1Show evidence (8 references)
Medical Actions
1Source YAML
click to showname: Achondrogenesis Type II
creation_date: '2026-02-06T03:25:37Z'
updated_date: '2026-04-28T00:00:00Z'
category: Mendelian
description: >
Achondrogenesis type II (ACG2), also known as Langer-Saldino type, is the most severe
form of type 2 collagenopathy caused by dominant mutations in COL2A1. It is a lethal
skeletal dysplasia characterized by severe micromelia, deficient ossification of
the vertebral bodies and sacrum, a small thorax, and a large head. Death typically
occurs in utero or shortly after birth.
ACG2 represents the severe end of the COL2A1 mutation spectrum, which includes
hypochondrogenesis, spondyloepiphyseal dysplasia congenita, and Stickler syndrome.
disease_term:
preferred_term: achondrogenesis type II
term:
id: MONDO:0008702
label: achondrogenesis type II
parents:
- Type 2 Collagenopathy
- Lethal Skeletal Dysplasia
inheritance:
- name: Autosomal Dominant (de novo)
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
de_novo_rate: Most cases (not quantified)
description: >
Almost all cases arise from de novo heterozygous mutations in COL2A1, as affected
individuals do not survive to reproduce. Germline mosaicism can lead to recurrence
in siblings.
evidence:
- reference: PMID:15054848
reference_title: "Recurrence of achondrogenesis type II within the same family: evidence for germline mosaicism."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "This mutation was not found in the parents. Although, we could not evaluate the presence of this mutation in the first fetus, we strongly believe that our data are in favor of germline mosaicism as the most likely explanation for the recurrence of type II achondrogenesis in both sibs."
explanation: Demonstrates that germline mosaicism can cause recurrence of ACG2 within families despite apparent de novo mutations.
prevalence:
- population: Global live births and published ACG2 case literature
measure_type: BIRTH_PREVALENCE
prevalence_class: BAND_1_9_PER_100000
rate_low: 1.666667
rate_high: 2.5
percentage: Unknown (achondrogenesis overall estimated at 1 in 40,000-60,000)
notes: >-
Exact subtype-specific prevalence for achondrogenesis type II is not well
established in population studies. The commonly cited estimate of 1 in
40,000-60,000 applies to achondrogenesis overall rather than ACG2
specifically. Available epidemiology is usually reported for
achondrogenesis as a group, while subtype-specific literature remains
limited to case reports and small reviews.
evidence:
- reference: PMID:31523626
reference_title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Achondrogenesis type 2 is the most severe chondrodysplasia caused by mutations in the COL2A1 gene. It is a rare lethal skeletal dysplasia."
explanation: Confirms that ACG2 is a rare lethal skeletal dysplasia, supporting its classification as an ultra-rare disorder, but does not provide frequency data.
- reference: PMID:7036745
reference_title: 'Achondrogenesis: a review with special consideration of achondrogenesis type II (Langer-Saldino).'
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We describe two dwarfed infants with large head, short neck and chest, prominent abdomen, and short limbs. Both died neonatally."
explanation: Type II-specific review literature remains based on isolated neonatal cases, supporting the inference that ACG2 itself is ultra-rare and not separately quantified in population datasets.
- reference: ORPHA:932
reference_title: Achondrogenesis (Orphanet structured-database record)
supports: SUPPORT
snippet: "1-9 / 100 000 | France | Prevalence at birth | PMID:2785882"
explanation: Orphanet cites a French population-based prevalence at birth of 1-9 per 100,000 for achondrogenesis as a group (not subtype-specific).
- reference: PMID:2785882
reference_title: Birth prevalence rates of skeletal dysplasias.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "thanatophoric dysplasia and achondrogenesis (0.28 0/000)"
explanation: French population-based study reporting combined birth prevalence of thanatophoric dysplasia and achondrogenesis at 0.28 per thousand; the achondrogenesis-specific rate is not separated.
pathophysiology:
- name: Type II Collagen Structural Defect
description: >
Mutations in COL2A1 disrupt the triple helix structure of type II collagen, the
major structural protein of cartilage. Glycine substitutions in the Gly-X-Y repeat
domain prevent proper helix formation, leading to intracellular retention and
degradation of abnormal procollagen chains.
genes:
- preferred_term: COL2A1
term:
id: hgnc:2200
label: COL2A1
molecular_functions:
- preferred_term: extracellular matrix structural constituent
term:
id: GO:0005201
label: extracellular matrix structural constituent
cell_types:
- preferred_term: Chondrocyte
term:
id: CL:0000138
label: chondrocyte
- preferred_term: Growth Plate Chondrocyte
term:
id: CL:1000217
label: growth plate cartilage chondrocyte
biological_processes:
- preferred_term: Collagen Biosynthesis
term:
id: GO:0032964
label: collagen biosynthetic process
- preferred_term: Cartilage Development
term:
id: GO:0051216
label: cartilage development
- preferred_term: Endochondral Bone Development
term:
id: GO:0060351
label: cartilage development involved in endochondral bone morphogenesis
evidence:
- reference: PMID:7829510
reference_title: "A COL2A1 mutation in achondrogenesis type II results in the replacement of type II collagen by type I and III collagens in cartilage."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A transition of G2853 to A in exon 41 produced a substitution of Gly769 by Ser within the triple helical domain of the alpha 1(II) chain of type II collagen, interrupting the mandatory Gly-X-Y triplet sequence required for the normal formation of stable triple helical type II collagen molecules"
explanation: Demonstrates that glycine substitutions in the Gly-X-Y repeat domain interrupt the formation of stable triple helical collagen molecules.
- reference: PMID:10745044
reference_title: "Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Five patients were heterozygous for a nucleotide change that predicted a glycine substitution in the triple helical domain (G313S, G517V, G571A, G910C, G943S)"
explanation: Documents multiple glycine substitution mutations in the COL2A1 triple helical domain causing lethal type II collagen disorders.
downstream:
- target: Extracellular Matrix Deficiency
causal_link_type: DIRECT
description: >
Disrupted type II collagen triple-helix formation leads to abnormal
cartilage extracellular matrix composition and loss of normal type II
collagen-rich cartilage architecture.
evidence:
- reference: PMID:7829510
reference_title: "A COL2A1 mutation in achondrogenesis type II results in the replacement of type II collagen by type I and III collagens in cartilage."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "resulting in the complete absence of type II collagen in the cartilage, which had a gelatinous composition"
explanation: Supports the link from COL2A1 structural disruption to loss of normal type II collagen in cartilage matrix.
- name: Extracellular Matrix Deficiency
description: >
Defective type II collagen secretion results in severely abnormal cartilage
extracellular matrix. The cartilage lacks normal fibrillar architecture and
fails to provide the template for endochondral ossification.
biological_processes:
- preferred_term: ECM Organization
term:
id: GO:0030198
label: extracellular matrix organization
evidence:
- reference: PMID:7829510
reference_title: "A COL2A1 mutation in achondrogenesis type II results in the replacement of type II collagen by type I and III collagens in cartilage."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "resulting in the complete absence of type II collagen in the cartilage, which had a gelatinous composition"
explanation: Demonstrates that the COL2A1 mutation results in complete loss of type II collagen from cartilage, leading to abnormal gelatinous matrix composition.
- reference: PMID:7829510
reference_title: "A COL2A1 mutation in achondrogenesis type II results in the replacement of type II collagen by type I and III collagens in cartilage."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Type I and III collagens were the major species found in cartilage tissue and synthesized by cultured chondrocytes along with cartilage type XI collagen"
explanation: Shows that mutant type II collagen is replaced by type I and III collagens, which are unable to support normal hyaline cartilage structure.
- reference: PMID:10745044
reference_title: "Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Overmodified type II collagen and the presence of type I collagen was found in the cartilage matrix of all seven cases"
explanation: Confirms that abnormal type II collagen processing and replacement by type I collagen is a consistent finding in lethal type II collagenopathies.
downstream:
- target: Type 2 Collagenopathy Spectrum
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
description: >
The degree of type I collagen replacement in cartilage is associated with
the severity continuum across lethal type II collagen disorders.
evidence:
- reference: PMID:10745044
reference_title: "Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Study of the clinical, radiographic, and morphological features of the seven cases supports evidence for a phenotypic continuum between achondrogenesis II-hypochondrogenesis and lethal SEDC and suggests a relationship between the amount of type I collagen in the cartilage and the severity of the phenotype."
explanation: Supports linking cartilage matrix collagen replacement to the severe type II collagenopathy phenotype spectrum.
- name: Type 2 Collagenopathy Spectrum
description: >
In ACG2, the near-complete loss of functional type II collagen leads to maximal
replacement by type I collagen in the cartilage matrix. This distinguishes ACG2
from milder COL2A1-related disorders (hypochondrogenesis, SEDC) where residual
type II collagen partially preserves cartilage architecture. The severity of
type I collagen replacement correlates directly with the degree of skeletal
underdevelopment and lethality in ACG2.
evidence:
- reference: PMID:10745044
reference_title: "Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Study of the clinical, radiographic, and morphological features of the seven cases supports evidence for a phenotypic continuum between achondrogenesis II-hypochondrogenesis and lethal SEDC and suggests a relationship between the amount of type I collagen in the cartilage and the severity of the phenotype."
explanation: Documents the phenotypic continuum from ACG2 through hypochondrogenesis to lethal SEDC and establishes the correlation between type I collagen content and disease severity.
downstream:
- target: Severe Micromelia
causal_link_type: DIRECT
- target: Delayed Vertebral Ossification
causal_link_type: DIRECT
- target: Small Thorax
causal_link_type: DIRECT
- target: Bell-Shaped Thorax
causal_link_type: DIRECT
- target: Macrocephaly
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Frontal Bossing
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Short Neck
causal_link_type: DIRECT
- target: Protuberant Abdomen
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Hydrops Fetalis
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Fetal Cystic Hygroma
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Micrognathia
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Polyhydramnios
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Short Long Bones
causal_link_type: DIRECT
- target: Abnormal Bone Ossification
causal_link_type: DIRECT
- target: Pierre-Robin Sequence
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Pulmonary Hypoplasia
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Short Ribs
causal_link_type: DIRECT
- target: Hypoplastic Ilia
causal_link_type: DIRECT
- target: Absent Vertebral Body Mineralization
causal_link_type: DIRECT
- target: Unossified Sacrum
causal_link_type: DIRECT
- target: Delayed Pubic Bone Ossification
causal_link_type: DIRECT
- target: Delayed Proximal Femoral Epiphyseal Ossification
causal_link_type: DIRECT
- target: Midface Retrusion
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Depressed Nasal Bridge
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Edema
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Hearing Impairment
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Myopia
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Skeletal Dysplasia
causal_link_type: DIRECT
- target: Short Stature
causal_link_type: DIRECT
- target: Abnormality of the Eye
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Cataract
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Retinal Detachment
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Lens Subluxation
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Abnormal Vitreous Humor Morphology
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Cardiorespiratory Arrest
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Metaphyseal Widening
causal_link_type: DIRECT
- target: Neonatal Respiratory Distress
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
- target: Pulmonary Hypertension
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
histopathology:
- name: Disorganized Growth Plate Cartilage
description: >
Growth cartilage shows disorganized epiphyseal and growth plate
architecture, with heterogeneous hypervascular and fibrous areas containing
enlarged hypertrophic-appearing chondrocytes.
diagnostic: true
evidence:
- reference: PMID:3309860
reference_title: "Achondrogenesis type II, abnormalities of extracellular matrix."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The normal architecture of the epiphyseal and growth plate cartilage was replaced by a morphologically heterogeneous tissue. Some areas were comprised of vascular canals surrounded by extensive fibrous tissue and enlarged cells that had the appearance and histochemical characteristics of hypertrophic chondrocytes."
explanation: Describes the hallmark disorganization of ACG2 epiphyseal and growth plate cartilage.
genetic:
- name: COL2A1 Mutations
gene_term:
preferred_term: COL2A1
term:
id: hgnc:2200
label: COL2A1
association: Causative
notes: >
Heterozygous mutations in COL2A1 encoding type II collagen alpha-1 chain.
Most are glycine substitutions in the triple helical domain that severely
disrupt collagen assembly, but splice-site and small in-frame deletion
variants have also been reported in the ACG2/hypochondrogenesis spectrum.
Common mutations include Gly to Ser, Arg, or Asp substitutions throughout
the triple helical domain.
variants:
- name: Triple-helical glycine substitutions
description: >
Predominant pathogenic variant class in molecularly characterized
achondrogenesis type II/hypochondrogenesis, replacing obligatory glycine
residues in the COL2A1 triple-helical Gly-X-Y repeat.
evidence:
- reference: PMID:10797431
reference_title: "Widely distributed mutations in the COL2A1 gene produce achondrogenesis type II/hypochondrogenesis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Ten of the mutations were single base substitutions that converted a codon for an obligate glycine to a codon for an amino acid with a bulkier side chain."
explanation: >
Aggregate molecular analysis of 12 ACG2/hypochondrogenesis patients
found glycine substitutions to be the dominant variant class.
- name: Splice-site and in-frame deletion variants
description: >
Less common COL2A1 variant classes reported in achondrogenesis type
II/hypochondrogenesis alongside the predominant glycine substitutions.
evidence:
- reference: PMID:10797431
reference_title: "Widely distributed mutations in the COL2A1 gene produce achondrogenesis type II/hypochondrogenesis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "One of the mutations was a change in a consensus RNA splice site. Another was an 18-base pair deletion of coding sequences."
explanation: >
The same 12-patient series documents non-glycine-substitution COL2A1
variant mechanisms in the severe ACG2/hypochondrogenesis spectrum.
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "COL2A1 | collagen type II alpha 1 chain | hgnc:2200 | Disease-causing germline mutation(s) in"
explanation: Orphanet confirms COL2A1 as the causative gene for achondrogenesis type 2 via disease-causing germline mutations.
- reference: PMID:15054848
reference_title: "Recurrence of achondrogenesis type II within the same family: evidence for germline mosaicism."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Achondrogenesis type II is a lethal skeletal dysplasia caused by new dominant mutations within the type II collagen gene (COL2A1)"
explanation: Confirms that ACG2 is caused by dominant mutations in the COL2A1 gene.
- reference: PMID:15054848
reference_title: "Recurrence of achondrogenesis type II within the same family: evidence for germline mosaicism."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Molecular analysis of genomic DNA extracted from amniotic cells of the second fetus revealed heterozygosity for a 1340G > A missense mutation (G316D) in the COL2A1 gene"
explanation: Demonstrates identification of a glycine substitution (G316D) in the COL2A1 gene causing ACG2.
- reference: PMID:10745044
reference_title: "Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Achondrogenesis II-hypochondrogenesis and severe spondyloepiphyseal dysplasia congenita (SEDC) are lethal forms of dwarfism caused by dominant mutations in the type II collagen gene (COL2A1)"
explanation: Confirms that dominant COL2A1 mutations cause achondrogenesis II-hypochondrogenesis.
- reference: PMID:10797431
reference_title: "Widely distributed mutations in the COL2A1 gene produce achondrogenesis type II/hypochondrogenesis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Mutations in the COL2A1 gene were found in all 12 patients."
explanation: >
Aggregate sequencing of 12 ACG2/hypochondrogenesis patients supports
COL2A1 as the central molecular cause across the severe end of the type
II collagenopathy spectrum.
- reference: CGGV:assertion_14640c06-c66e-4dc7-86ea-9b0850e51474-2020-10-05T040000.000Z
reference_title: "COL2A1 / achondrogenesis type II (Definitive)"
supports: SUPPORT
evidence_source: OTHER
snippet: "COL2A1 | HGNC:2200 | achondrogenesis type II | MONDO:0008702 | AD | Definitive"
explanation: ClinGen classifies the COL2A1-achondrogenesis type II gene-disease relationship as definitive with autosomal dominant inheritance.
- reference: PMID:31523626
reference_title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Next generation sequencing (NGS) showed a heterozygous missense variation, c.2546G>A, p.Gly849Asp on the COL2A1 gene that was confirmed by Sanger sequencing"
explanation: Documents a novel de novo heterozygous glycine-substitution variant (p.Gly849Asp) in the COL2A1 triple-helical domain causing ACG2.
- reference: PMID:36376277
reference_title: Novel missense COL2A1 variant in a fetus with achondrogenesis type II.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "This change results in a predicted substitution of glycine 996 with asparagine acid"
explanation: Documents an additional de novo COL2A1 glycine-substitution variant (glycine 996, p.Gly996Asp) causing ACG2, reinforcing the dominant-negative glycine-substitution mechanism.
phenotypes:
- name: Severe Micromelia
description: >
Severe prenatal shortening of the limbs involving all segments.
frequency: FREQUENT
phenotype_term:
preferred_term: Severe Micromelia
term:
id: HP:0002983
label: Micromelia
modifier: ABNORMAL
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0002983 | Micromelia | Frequent (79-30%)"
explanation: Orphanet lists micromelia as a frequent feature of achondrogenesis type 2.
- reference: PMID:15054848
reference_title: "Recurrence of achondrogenesis type II within the same family: evidence for germline mosaicism."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "severe micromelia and generalized edema were noted on ultrasound at 21 weeks' gestation"
explanation: Documents severe micromelia as a key ultrasound finding in ACG2.
- reference: PMID:36376277
reference_title: "Novel missense COL2A1 variant in a fetus with achondrogenesis type II."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We present a fetus with cystic hygroma and severe shortening of the limbs at 14 weeks of gestation."
explanation: Supports that marked limb shortening can already be present in the early second trimester.
- name: Delayed Vertebral Ossification
description: >
Prenatal imaging may show absent or markedly delayed ossification of the
vertebral bodies.
frequency: FREQUENT
phenotype_term:
preferred_term: Delayed vertebral ossification
term:
id: HP:0031096
label: Delayed vertebral ossification
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0031096 | Delayed vertebral ossification | Frequent (79-30%)"
explanation: Orphanet lists delayed vertebral ossification as a frequent feature of achondrogenesis type 2.
- reference: PMID:12124695
reference_title: "Achondrogenesis type II with normally developed extremities: a case report."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Intrauterine sonographic examination of the vertebrae is very important and the absence of vertebral body ossification may be the unique finding of achondrogenesis type II."
explanation: Directly supports absent or delayed vertebral body ossification as a diagnostically important prenatal phenotype.
- name: Small Thorax
description: >
Prenatal imaging can show a small thorax accompanying severe limb
shortening.
frequency: FREQUENT
phenotype_term:
preferred_term: Small thorax
term:
id: HP:0000774
label: Narrow chest
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000774 | Narrow chest | Frequent (79-30%)"
explanation: Orphanet lists narrow chest as a frequent feature of achondrogenesis type 2.
- reference: PMID:20387359
reference_title: "Antenatal diagnosis of achondrogenesis type II."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Achondrogenesis is a lethal congenital chondrodystrophy characterized by extreme micromelia, small thorax and polyhydramnios."
explanation: Supports a small thorax as part of the prenatal phenotype described for achondrogenesis type II.
- name: Bell-Shaped Thorax
description: >
Radiographs may show a narrow bell-shaped chest, a specific thoracic
configuration contributing to severe respiratory compromise.
phenotype_term:
preferred_term: Bell-shaped thorax
term:
id: HP:0001591
label: Bell-shaped thorax
evidence:
- reference: PMID:31523626
reference_title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Radiographical findings: short tubular bone with widened metaphyses and non ossified cervical vertebrae, short unfractured ribs, narrow bell-shaped chest, lack of ossification in pelvis and normal ossification of the skull."
explanation: >
Full-text radiographic description in a molecularly confirmed ACG2 neonate
supports a narrow bell-shaped thorax as a specific thoracic phenotype.
- name: Macrocephaly
description: >
A large head has been described in both prenatal and neonatal reports of
achondrogenesis type II.
frequency: VERY_FREQUENT
phenotype_term:
preferred_term: Large head
term:
id: HP:0000256
label: Macrocephaly
evidence:
- reference: ORPHA:932
reference_title: Achondrogenesis (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000256 | Macrocephaly | Very frequent (99-80%)"
explanation: Orphanet lists macrocephaly as a very frequent feature of achondrogenesis (parent group).
- reference: PMID:7036745
reference_title: "Achondrogenesis: a review with special consideration of achondrogenesis type II (Langer-Saldino)."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We describe two dwarfed infants with large head, short neck and chest, prominent abdomen, and short limbs. Both died neonatally."
explanation: Supports a large head as part of the neonatal phenotype in type II achondrogenesis.
- reference: PMID:20387359
reference_title: "Antenatal diagnosis of achondrogenesis type II."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Prenatal ultrasonography at 22-weeks gestation revealed a fetus with large head, short neck and chest, prominent abdomen and short limbs."
explanation: Supports large head on prenatal ultrasound in achondrogenesis type II.
- name: Frontal Bossing
description: >
Prominent forehead is part of the characteristic craniofacial appearance.
phenotype_term:
preferred_term: Frontal bossing
term:
id: HP:0002007
label: Frontal bossing
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "distinctive facial features such as a prominent forehead, a small chin, a cleft palate (in some)"
explanation: >
Orphanet's ACG2 definition lists prominent forehead among the distinctive
facial features.
- reference: PMID:31523626
reference_title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A physical examination revealed extremely short extremities, abdominal distention, a small chest, a prominent forehead and a flat nasal bridge"
explanation: >
Molecularly confirmed ACG2 neonate directly supports prominent forehead,
mapped to frontal bossing.
- name: Short Neck
description: >
A short neck has been described on prenatal ultrasound.
frequency: VERY_FREQUENT
phenotype_term:
preferred_term: Short neck
term:
id: HP:0000470
label: Short neck
evidence:
- reference: ORPHA:932
reference_title: Achondrogenesis (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000470 | Short neck | Very frequent (99-80%)"
explanation: Orphanet lists short neck as a very frequent feature of achondrogenesis (parent group).
- reference: PMID:20387359
reference_title: "Antenatal diagnosis of achondrogenesis type II."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Prenatal ultrasonography at 22-weeks gestation revealed a fetus with large head, short neck and chest, prominent abdomen and short limbs."
explanation: Directly supports short neck as part of the prenatal gestalt in ACG2.
- name: Protuberant Abdomen
description: >
Prenatal imaging and neonatal examination may show a prominent or
distended abdomen.
phenotype_term:
preferred_term: Protuberant abdomen
term:
id: HP:0001538
label: Protuberant abdomen
evidence:
- reference: PMID:20387359
reference_title: "Antenatal diagnosis of achondrogenesis type II."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Prenatal ultrasonography at 22-weeks gestation revealed a fetus with large head, short neck and chest, prominent abdomen and short limbs."
explanation: Supports a prominent abdomen as part of the prenatal phenotype in ACG2.
- name: Hydrops Fetalis
description: >
Prenatal hydrops has been reported in affected fetuses, including
presentations with septated cystic hygroma.
frequency: VERY_FREQUENT
phenotype_term:
preferred_term: Hydrops fetalis
term:
id: HP:0001789
label: Hydrops fetalis
evidence:
- reference: ORPHA:932
reference_title: Achondrogenesis (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0001789 | Hydrops fetalis | Very frequent (99-80%)"
explanation: Orphanet lists hydrops fetalis as a very frequent feature of achondrogenesis (parent group).
- reference: PMID:41373627
reference_title: "Prenatal Imaging of Micrognathia, Micromelia, and Fetal Hydrops Leading to the Diagnosis of Achondrogenesis Type II with a COL2A1 Missense Mutation."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "This case report describes a fetus with achondrogenesis type II, a severe and lethal type II collagen disorder, presenting with micrognathia and hydrops."
explanation: Directly supports fetal hydrops as a prenatal presentation of ACG2.
- reference: PMID:17994563
reference_title: "A familial case of achondrogenesis type II caused by a dominant COL2A1 mutation and \"patchy\" expression in the mosaic father."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The couple had a fourth pregnancy, and at 11 weeks fetal hydrops with a septated cystic hygroma were obvious."
explanation: Supports that hydrops can be present very early in gestation and may occur with septated cystic hygroma.
- name: Fetal Cystic Hygroma
description: >
Prenatal sonography may identify fetal hygroma or septated cystic
hygroma early in gestation.
phenotype_term:
preferred_term: fetal cystic hygroma
term:
id: HP:0010878
label: Fetal cystic hygroma
evidence:
- reference: PMID:17994563
reference_title: "A familial case of achondrogenesis type II caused by a dominant COL2A1 mutation and \"patchy\" expression in the mosaic father."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The couple had a fourth pregnancy, and at 11 weeks fetal hydrops with a septated cystic hygroma were obvious."
explanation: Directly supports fetal cystic hygroma as an early prenatal manifestation of ACG2.
- reference: PMID:36376277
reference_title: "Novel missense COL2A1 variant in a fetus with achondrogenesis type II."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We present a fetus with cystic hygroma and severe shortening of the limbs at 14 weeks of gestation."
explanation: Independently supports cystic hygroma during the fetal presentation of ACG2.
- name: Micrognathia
description: >
Micrognathia has been documented as part of the craniofacial phenotype.
frequency: VERY_FREQUENT
phenotype_term:
preferred_term: Micrognathia
term:
id: HP:0000347
label: Micrognathia
evidence:
- reference: ORPHA:932
reference_title: Achondrogenesis (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000347 | Micrognathia | Very frequent (99-80%)"
explanation: Orphanet lists micrognathia as a very frequent feature of achondrogenesis (parent group).
- reference: PMID:41373627
reference_title: "Prenatal Imaging of Micrognathia, Micromelia, and Fetal Hydrops Leading to the Diagnosis of Achondrogenesis Type II with a COL2A1 Missense Mutation."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "This case report describes a fetus with achondrogenesis type II, a severe and lethal type II collagen disorder, presenting with micrognathia and hydrops."
explanation: Directly supports micrognathia as part of the prenatal craniofacial phenotype.
- name: Polyhydramnios
description: >
Polyhydramnios has been described as part of the prenatal presentation.
phenotype_term:
preferred_term: Polyhydramnios
term:
id: HP:0001561
label: Polyhydramnios
evidence:
- reference: ORPHA:932
reference_title: Achondrogenesis (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0001561 | Polyhydramnios | Frequent (79-30%)"
explanation: Orphanet lists polyhydramnios as a frequent feature of achondrogenesis (parent group).
- reference: PMID:20387359
reference_title: "Antenatal diagnosis of achondrogenesis type II."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Achondrogenesis is a lethal congenital chondrodystrophy characterized by extreme micromelia, small thorax and polyhydramnios."
explanation: Supports polyhydramnios as a reported prenatal manifestation in ACG2.
- name: Short Long Bones
description: >
Marked shortening of the long bones is a hallmark radiographic feature.
frequency: VERY_FREQUENT
phenotype_term:
preferred_term: Short long bone
term:
id: HP:0003026
label: Short long bone
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0003026 | Short long bone | Very frequent (99-80%)"
explanation: Orphanet lists short long bone as a very frequent feature of achondrogenesis type 2.
- name: Abnormal Bone Ossification
description: >
Generalized abnormality of bone ossification reflecting the failure of
endochondral ossification due to defective type II collagen.
frequency: VERY_FREQUENT
phenotype_term:
preferred_term: Abnormal bone ossification
term:
id: HP:0011849
label: Abnormal bone ossification
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0011849 | Abnormal bone ossification | Very frequent (99-80%)"
explanation: Orphanet lists abnormal bone ossification as a very frequent feature of achondrogenesis type 2.
- name: Pierre-Robin Sequence
description: >
Craniofacial involvement including micrognathia can manifest as
Pierre-Robin sequence in some cases.
frequency: FREQUENT
phenotype_term:
preferred_term: Pierre-Robin sequence
term:
id: HP:0000201
label: Pierre-Robin sequence
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000201 | Pierre-Robin sequence | Frequent (79-30%)"
explanation: Orphanet lists Pierre-Robin sequence as a frequent feature of achondrogenesis type 2.
- name: Pulmonary Hypoplasia
description: >
Underdeveloped lungs secondary to the small thorax, contributing to
perinatal lethality.
frequency: FREQUENT
phenotype_term:
preferred_term: Pulmonary hypoplasia
term:
id: HP:0002089
label: Pulmonary hypoplasia
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0002089 | Pulmonary hypoplasia | Frequent (79-30%)"
explanation: Orphanet lists pulmonary hypoplasia as a frequent feature of achondrogenesis type 2.
- name: Short Ribs
description: >
Shortened ribs contribute to the narrow thorax and pulmonary hypoplasia.
frequency: FREQUENT
phenotype_term:
preferred_term: Short ribs
term:
id: HP:0000773
label: Short ribs
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000773 | Short ribs | Frequent (79-30%)"
explanation: Orphanet lists short ribs as a frequent feature of achondrogenesis type 2.
- name: Hypoplastic Ilia
description: >
Hypoplasia of the iliac bones is a characteristic radiographic finding.
frequency: FREQUENT
phenotype_term:
preferred_term: Hypoplastic ilia
term:
id: HP:0000946
label: Hypoplastic ilia
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000946 | Hypoplastic ilia | Frequent (79-30%)"
explanation: Orphanet lists hypoplastic ilia as a frequent feature of achondrogenesis type 2.
- name: Absent Vertebral Body Mineralization
description: >
Absence of mineralization of the vertebral bodies on radiography.
frequency: FREQUENT
phenotype_term:
preferred_term: Absent vertebral body mineralization
term:
id: HP:0004605
label: Absent vertebral body mineralization
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0004605 | Absent vertebral body mineralization | Frequent (79-30%)"
explanation: Orphanet lists absent vertebral body mineralization as a frequent feature of achondrogenesis type 2.
- name: Unossified Sacrum
description: >
Absence of sacral ossification on radiography.
frequency: FREQUENT
phenotype_term:
preferred_term: Unossified sacrum
term:
id: HP:0030290
label: Unossified sacrum
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0030290 | Unossified sacrum | Frequent (79-30%)"
explanation: Orphanet lists unossified sacrum as a frequent feature of achondrogenesis type 2.
- name: Delayed Pubic Bone Ossification
description: >
Delayed ossification of the pubic bones observed on radiography.
frequency: FREQUENT
phenotype_term:
preferred_term: Delayed pubic bone ossification
term:
id: HP:0008788
label: Delayed pubic bone ossification
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0008788 | Delayed pubic bone ossification | Frequent (79-30%)"
explanation: Orphanet lists delayed pubic bone ossification as a frequent feature of achondrogenesis type 2.
- name: Delayed Proximal Femoral Epiphyseal Ossification
description: >
Delayed ossification of the proximal femoral epiphysis.
frequency: FREQUENT
phenotype_term:
preferred_term: Delayed proximal femoral epiphyseal ossification
term:
id: HP:0008828
label: Delayed proximal femoral epiphyseal ossification
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0008828 | Delayed proximal femoral epiphyseal ossification | Frequent (79-30%)"
explanation: Orphanet lists delayed proximal femoral epiphyseal ossification as a frequent feature of achondrogenesis type 2.
- name: Midface Retrusion
description: >
Midface hypoplasia contributing to the flat facial profile.
frequency: FREQUENT
phenotype_term:
preferred_term: Midface retrusion
term:
id: HP:0011800
label: Midface retrusion
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0011800 | Midface retrusion | Frequent (79-30%)"
explanation: Orphanet lists midface retrusion as a frequent feature of achondrogenesis type 2.
- name: Depressed Nasal Bridge
description: >
Flat or depressed nasal bridge has been reported as part of the neonatal
craniofacial presentation.
phenotype_term:
preferred_term: Depressed nasal bridge
term:
id: HP:0005280
label: Depressed nasal bridge
evidence:
- reference: PMID:31523626
reference_title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "A physical examination revealed extremely short extremities, abdominal distention, a small chest, a prominent forehead and a flat nasal bridge"
explanation: >
The full-text neonatal examination reports flat nasal bridge in a
molecularly confirmed ACG2 case; HPO treats flat nasal bridge as a synonym
of depressed nasal bridge.
- name: Edema
description: >
Generalized soft tissue edema (anasarca) is characteristic.
frequency: FREQUENT
phenotype_term:
preferred_term: Edema
term:
id: HP:0000969
label: Edema
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000969 | Edema | Frequent (79-30%)"
explanation: Orphanet lists edema as a frequent feature of achondrogenesis type 2.
- name: Hearing Impairment
description: >
Part of the type 2 collagenopathy spectrum; type II collagen is present
in the inner ear. Direct clinical observation in ACG2 is confounded by
perinatal lethality.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Hearing impairment
term:
id: HP:0000365
label: Hearing impairment
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000365 | Hearing impairment | Occasional (29-5%)"
explanation: Orphanet lists hearing impairment as an occasional feature of achondrogenesis type 2.
- name: Myopia
description: >
Ocular involvement as part of the type 2 collagenopathy spectrum;
type II collagen is a major component of the vitreous. Direct clinical
observation in ACG2 is confounded by perinatal lethality.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Myopia
term:
id: HP:0000545
label: Myopia
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000545 | Myopia | Occasional (29-5%)"
explanation: Orphanet lists myopia as an occasional feature of achondrogenesis type 2.
- name: Skeletal Dysplasia
description: >
Generalized skeletal dysplasia reflecting the widespread role of
type II collagen in endochondral bone development.
frequency: FREQUENT
phenotype_term:
preferred_term: Skeletal dysplasia
term:
id: HP:0002652
label: Skeletal dysplasia
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0002652 | Skeletal dysplasia | Frequent (79-30%)"
explanation: Orphanet lists skeletal dysplasia as a frequent feature of achondrogenesis type 2.
- name: Short Stature
description: >
Severe short stature reflecting generalized skeletal underdevelopment.
frequency: FREQUENT
phenotype_term:
preferred_term: Short stature
term:
id: HP:0004322
label: Short stature
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0004322 | Short stature | Frequent (79-30%)"
explanation: Orphanet lists short stature as a frequent feature of achondrogenesis type 2.
- name: Abnormality of the Eye
description: >
Ocular abnormalities as part of the type 2 collagenopathy spectrum;
type II collagen is a major structural component of the vitreous humor.
Direct clinical observation in ACG2 is confounded by perinatal lethality.
frequency: FREQUENT
phenotype_term:
preferred_term: Abnormality of the eye
term:
id: HP:0000478
label: Abnormality of the eye
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000478 | Abnormality of the eye | Frequent (79-30%)"
explanation: Orphanet lists abnormality of the eye as a frequent feature of achondrogenesis type 2.
- name: Cataract
description: >
Cataract as an occasional ocular finding in the type 2 collagenopathy
spectrum. Direct clinical observation in ACG2 is confounded by perinatal
lethality.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Cataract
term:
id: HP:0000518
label: Cataract
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000518 | Cataract | Occasional (29-5%)"
explanation: Orphanet lists cataract as an occasional feature of achondrogenesis type 2.
- name: Retinal Detachment
description: >
Retinal detachment related to vitreous abnormalities from defective
type II collagen. Direct clinical observation in ACG2 is confounded by
perinatal lethality.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Retinal detachment
term:
id: HP:0000541
label: Retinal detachment
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0000541 | Retinal detachment | Occasional (29-5%)"
explanation: Orphanet lists retinal detachment as an occasional feature of achondrogenesis type 2.
- name: Lens Subluxation
description: >
Occasional lens subluxation related to defective type II collagen in
ocular connective tissue. Direct clinical observation in ACG2 is
confounded by perinatal lethality.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Lens subluxation
term:
id: HP:0001132
label: Lens subluxation
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0001132 | Lens subluxation | Occasional (29-5%)"
explanation: Orphanet lists lens subluxation as an occasional feature of achondrogenesis type 2.
- name: Abnormal Vitreous Humor Morphology
description: >
Vitreous abnormalities due to type II collagen being the major
structural protein of the vitreous humor. Direct clinical observation
in ACG2 is confounded by perinatal lethality.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Abnormal vitreous humor morphology
term:
id: HP:0004327
label: Abnormal vitreous humor morphology
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0004327 | Abnormal vitreous humor morphology | Occasional (29-5%)"
explanation: Orphanet lists abnormal vitreous humor morphology as an occasional feature of achondrogenesis type 2.
- name: Cardiorespiratory Arrest
description: >
Cardiorespiratory arrest contributing to perinatal lethality,
secondary to pulmonary hypoplasia and thoracic restriction.
frequency: OCCASIONAL
phenotype_term:
preferred_term: Cardiorespiratory arrest
term:
id: HP:0006543
label: Cardiorespiratory arrest
evidence:
- reference: ORPHA:93296
reference_title: Achondrogenesis type 2 (Orphanet structured-database record)
supports: SUPPORT
snippet: "HP:0006543 | Cardiorespiratory arrest | Occasional (29-5%)"
explanation: Orphanet lists cardiorespiratory arrest as an occasional feature of achondrogenesis type 2.
- name: Metaphyseal Widening
description: >
Radiographs show short tubular bones with widened, flared metaphyses, a
characteristic radiographic feature in achondrogenesis type II.
phenotype_term:
preferred_term: Metaphyseal widening
term:
id: HP:0003016
label: Metaphyseal widening
evidence:
- reference: PMID:31523626
reference_title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "an X-ray examination revealed a short tubular bone structure, metaphyseal widening, short ribs, a small chest and a lack of ossification in the pelvis"
explanation: Radiographic examination in a molecularly confirmed ACG2 neonate documented metaphyseal widening of the short tubular bones.
- name: Neonatal Respiratory Distress
description: >
Liveborn affected neonates develop severe respiratory distress immediately
after birth secondary to pulmonary hypoplasia and thoracic restriction, a
major contributor to perinatal death.
phenotype_term:
preferred_term: Neonatal respiratory distress
term:
id: HP:0002643
label: Neonatal respiratory distress
evidence:
- reference: PMID:31523626
reference_title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "respiratory distress can occur immediately after birth with severe forms of the disorder"
explanation: Full-text case report documents severe neonatal respiratory distress as an immediate postnatal manifestation contributing to lethality.
- name: Pulmonary Hypertension
description: >
Persistent pulmonary hypertension of the newborn, secondary to pulmonary
hypoplasia, has been documented postnatally and required inhaled nitric
oxide therapy.
phenotype_term:
preferred_term: Pulmonary hypertension
term:
id: HP:0002092
label: Pulmonary arterial hypertension
evidence:
- reference: PMID:31523626
reference_title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "An echocardiography performed on postnatal day 4 revealed pulmonary hypertension"
explanation: Postnatal echocardiography in a molecularly confirmed ACG2 neonate revealed pulmonary hypertension, managed with inhaled nitric oxide.
treatments:
- name: Supportive Care
description: >
No effective treatment exists. Supportive care and palliative measures
for affected neonates. Genetic counseling for families.
diagnosis:
- name: Prenatal Ultrasound
description: >
Severe skeletal abnormalities including micromelia, narrow thorax,
and deficient ossification detectable on prenatal ultrasound,
often in the second trimester.
evidence:
- reference: PMID:15054848
reference_title: "Recurrence of achondrogenesis type II within the same family: evidence for germline mosaicism."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "severe micromelia and generalized edema were noted on ultrasound at 21 weeks' gestation"
explanation: Demonstrates that ACG2 can be detected on prenatal ultrasound by 21 weeks gestation.
- name: Molecular Genetic Testing
description: >
Confirmation by COL2A1 sequencing identifying pathogenic variants.
Essential for accurate diagnosis and recurrence risk counseling.
evidence:
- reference: PMID:15054848
reference_title: "Recurrence of achondrogenesis type II within the same family: evidence for germline mosaicism."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Molecular analysis of genomic DNA extracted from amniotic cells of the second fetus revealed heterozygosity for a 1340G > A missense mutation (G316D) in the COL2A1 gene"
explanation: Demonstrates molecular genetic testing via amniocentesis can confirm the diagnosis prenatally.
notes: >
ACG2 exists on a phenotypic continuum with hypochondrogenesis and severe spondyloepiphyseal
dysplasia congenita. The amount of type I collagen replacement in cartilage correlates
with phenotypic severity.
datasets:
references:
- reference: DOI:10.1002/ar.24086
title: Genetic Disorders of the Extracellular Matrix
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: DOI:10.1002/dvdy.24131
title: Mechanisms and models of endoplasmic reticulum stress in chondrodysplasia
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: DOI:10.1002/jemt.1070280505
title: 'Cartilage proteoglycans: Structure and potential functions'
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: DOI:10.1073/pnas.070050097
title: Destabilization of osteogenesis imperfecta collagen-like model peptides correlates with the identity of the residue replacing glycine
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: DOI:10.1111/dgd.12203
title: 'Fate of growth plate hypertrophic chondrocytes: Death or lineage extension?'
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: DOI:10.3389/fcell.2021.664168
title: Molecular Mechanisms of Chondrocyte Proliferation and Differentiation
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: DOI:10.3389/fmolb.2021.593310
title: The Smad Dependent TGF-β and BMP Signaling Pathway in Bone Remodeling and Therapies
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:11698112
title: The interactions of cartilage proteoglycans with collagens are determined by their structures.
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:1374906
title: Characterization of a type II collagen gene (COL2A1) mutation identified in cultured chondrocytes from human hypochondrogenesis.
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:1515761
title: 'Collagen type II in Langer-Saldino achondrogenesis: absence of major abnormalities in a less severe case.'
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:15365990
title: Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders.
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:15895462
title: The phenotypic spectrum of COL2A1 mutations.
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:17437277
title: Missense and silent mutations in COL2A1 result in Stickler syndrome but via different molecular mechanisms.
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:17638425
title: 'Achondrogenesis Type IA (Houston-Harris): a still-unresolved molecular phenotype.'
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:18773462
title: 'Collagen fibrillogenesis in tendon development: current models and regulation of fibril assembly.'
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:30854241
title: Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin β1-SMAD1 interaction.
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:12124695
title: "Achondrogenesis type II with normally developed extremities: a case report."
findings: []
- reference: PMID:17994563
title: "A familial case of achondrogenesis type II caused by a dominant COL2A1 mutation and \"patchy\" expression in the mosaic father."
findings: []
- reference: PMID:20387359
title: Antenatal diagnosis of achondrogenesis type II.
findings: []
- reference: PMID:31523626
title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene.
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:3309860
title: Achondrogenesis type II, abnormalities of extracellular matrix.
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:36376277
title: Novel missense COL2A1 variant in a fetus with achondrogenesis type II.
findings: []
- reference: PMID:41373627
title: Prenatal Imaging of Micrognathia, Micromelia, and Fetal Hydrops Leading to the Diagnosis of Achondrogenesis Type II with a COL2A1 Missense Mutation.
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:15054848
title: "Recurrence of achondrogenesis type II within the same family: evidence for germline mosaicism."
found_in:
- Achondrogenesis_Type_II-deep-research-cyberian-codex.md
- Achondrogenesis_Type_II-deep-research-perplexity.md
findings: []
- reference: PMID:10797431
title: Widely distributed mutations in the COL2A1 gene produce achondrogenesis type II/hypochondrogenesis.
found_in:
- Achondrogenesis_Type_II-deep-research-falcon.md
findings: []
- reference: DOI:10.1002/ajmg.a.20597
title: "Recurrence of achondrogenesis type II within the same family: Evidence for germline mosaicism"
found_in:
- Achondrogenesis_Type_II-deep-research-falcon.md
findings: []
- reference: DOI:10.1002/humu.22915
title: Mutation Update for COL2A1 Gene Variants Associated with Type II Collagenopathies
found_in:
- Achondrogenesis_Type_II-deep-research-falcon.md
findings: []
- reference: DOI:10.1038/s41439-022-00218-5
title: Novel missense COL2A1 variant in a fetus with achondrogenesis type II
found_in:
- Achondrogenesis_Type_II-deep-research-falcon.md
findings: []
- reference: DOI:10.1111/cge.13680
title: Integrated analysis of COL2A1 variant data and classification of type II collagenopathies
found_in:
- Achondrogenesis_Type_II-deep-research-falcon.md
findings: []
- reference: DOI:10.1186/s43055-021-00479-0
title: A case report of achondrogenesis type II (Langer-Saldino achondrogenesis)
found_in:
- Achondrogenesis_Type_II-deep-research-falcon.md
findings: []
- reference: DOI:10.1201/9781003166948
title: Fetal and Perinatal Skeletal Dysplasias
found_in:
- Achondrogenesis_Type_II-deep-research-falcon.md
findings: []
- reference: DOI:10.2478/bjmg-2019-0001
title: Achondrogenesis Type 2 in a Newborn with a Novel Mutation on the COL2A1 Gene
found_in:
- Achondrogenesis_Type_II-deep-research-falcon.md
findings: []
References & Deep Research
References
32Deep Research
3Disorder
- Name: Achondrogenesis Type II
- Category: Mendelian
- Existing deep-research providers: cyberian-codex, perplexity
- Existing evidence reference count in YAML: 33
Key Pathophysiology Nodes
- Type II Collagen Structural Defect
- Extracellular Matrix Deficiency
- Type 2 Collagenopathy Spectrum
- Deep research literature mapping
Citation Inventory (for evidence mapping)
- DOI:10.1002/ar.24086
- DOI:10.1002/dvdy.24131
- DOI:10.1002/jemt.1070280505
- DOI:10.1073/pnas.070050097
- DOI:10.1111/dgd.12203
- DOI:10.3389/fcell.2021.664168
- DOI:10.3389/fmolb.2021.593310
- PMID:11698112
- PMID:1374906
- PMID:15054848
- PMID:1515761
- PMID:15365990
- PMID:15895462
- PMID:17437277
- PMID:17638425
- PMID:18773462
- PMID:30854241
- PMID:31523626
- PMID:3309860
- PMID:41373627
Achondrogenesis Type II (ACG2): Disease Characteristics Research Report
1. Disease Information
Overview / definition
Achondrogenesis type II (ACG2), also known as Langer–Saldino achondrogenesis, is a lethal congenital skeletal dysplasia on the severe end of the type II collagenopathy spectrum, typically presenting prenatally with extreme short limbs and a small thorax and resulting in stillbirth or death in the early neonatal period due to pulmonary hypoplasia/respiratory failure. (maheshwari2021acasereport pages 1-3, dogan2019achondrogenesistype2 pages 3-4, kobayashi2022novelmissensecol2a1 pages 1-2)
Direct abstract-supported definition example: “Achondrogenesis Type 2 is a lethal skeletal dysplasia …” with typical findings including “short arms and legs, a small chest with short ribs, lung hypoplasia …” (dogan2019achondrogenesistype2 pages 3-4)
Key identifiers
- OMIM: 200610 (Achondrogenesis type II) (kobayashi2022novelmissensecol2a1 pages 1-2, barathouari2016mutationupdatefor pages 1-2)
- Gene (OMIM): COL2A1 MIM 108300 (barathouari2016mutationupdatefor pages 1-2)
- MeSH / Orphanet / ICD-10 / ICD-11 / MONDO: Not found in retrieved sources for this run (see artifact table). (kobayashi2022novelmissensecol2a1 pages 1-2)
Synonyms / alternative names
- Achondrogenesis type II (ACG2) (kobayashi2022novelmissensecol2a1 pages 1-2)
- Langer–Saldino achondrogenesis (maheshwari2021acasereport pages 1-3)
- Achondrogenesis type II / hypochondrogenesis spectrum (korkko2000widelydistributedmutations pages 1-2)
Evidence sources used
The retrieved evidence includes both aggregated resources (variant/disease spectrum reviews and integrated variant analyses) and individual case reports/series (prenatal imaging and newborn presentations). (maheshwari2021acasereport pages 1-3, korkko2000widelydistributedmutations pages 1-2, barathouari2016mutationupdatefor pages 1-2, zhang2020integratedanalysisof pages 1-6)
2. Etiology
Disease causal factors
Primary cause: Germline pathogenic variants in COL2A1 (type II procollagen) causing type II collagenopathy with severe disruption of cartilage matrix and endochondral ossification. (barathouari2016mutationupdatefor pages 1-2, zhang2020integratedanalysisof pages 1-6)
Risk factors
- Genetic: Presence of a heterozygous pathogenic COL2A1 variant is causal. A frequent causal class in severe phenotypes is glycine substitution within the triple-helical Gly-X-Y repeats. (barathouari2016mutationupdatefor pages 1-2, korkko2000widelydistributedmutations pages 1-2)
- Non-genetic/environmental: No established environmental risk factors were identified in retrieved sources; ACG2 is primarily a monogenic disorder. (barathouari2016mutationupdatefor pages 1-2)
Protective factors / gene–environment interactions
No protective factors or gene–environment interactions were identified in the retrieved sources for ACG2. (barathouari2016mutationupdatefor pages 1-2)
3. Phenotypes (clinical + radiographic)
Typical timing and severity
ACG2 is typically prenatal-onset with severe skeletal abnormalities detectable on ultrasound and/or other fetal imaging, and the phenotype is severe and usually lethal. (maheshwari2021acasereport pages 1-3, kobayashi2022novelmissensecol2a1 pages 1-2)
Key prenatal phenotypes
Prenatal findings may include: * Severe limb shortening / micromelia (e.g., markedly low femur and humerus length Z-scores) (kobayashi2022novelmissensecol2a1 pages 1-2) * Thoracic hypoplasia/small chest, suggesting pulmonary hypoplasia risk (kobayashi2022novelmissensecol2a1 pages 1-2) * Cystic hygroma (reported in a 14-week fetus with ACG2) (kobayashi2022novelmissensecol2a1 pages 1-2) * Additional findings described in the broader ACG2/hypochondrogenesis fetal spectrum include hydrops and polyhydramnios. (korkko2000widelydistributedmutations pages 1-2)
Neonatal phenotypes
In liveborn infants, typical features include: * Short trunk, small/narrow chest, prominent/distended abdomen, and micromelia (dogan2019achondrogenesistype2 pages 3-4) * Severe respiratory distress due to pulmonary hypoplasia (dogan2019achondrogenesistype2 pages 3-4)
Hallmark radiographic phenotype (skeletal survey)
Classic radiographic findings reported across ACG2 cases include: * Very short long bones with widened/flared metaphyses (dogan2019achondrogenesistype2 pages 3-4) * Non-ossification or markedly decreased ossification of vertebral bodies (including cervical; and in some reports near-complete vertebral non-ossification) (maheshwari2021acasereport pages 1-3, dogan2019achondrogenesistype2 pages 3-4) * Short ribs (often without fractures) and narrow bell-shaped thorax (dogan2019achondrogenesistype2 pages 3-4) * Poor pelvic ossification with relatively preserved skull ossification (helpful discriminator vs some type I achondrogenesis forms) (dogan2019achondrogenesistype2 pages 3-4, dogan2019achondrogenesistype2 pages 4-5)
A representative post-termination radiograph showing these features (including vertebral non-ossification, flared metaphyses, cupped ribs, and iliac “paraglider” appearance) is available in the retrieved image evidence. (maheshwari2021acasereport media 016b7757)
Suggested HPO terms (examples)
Based on retrieved clinical/radiographic descriptions: * Micromelia, Short limb, Short long bones (maheshwari2021acasereport pages 1-3, kobayashi2022novelmissensecol2a1 pages 1-2) * Narrow thorax / thoracic hypoplasia, Short ribs, Bell-shaped thorax (dogan2019achondrogenesistype2 pages 3-4) * Pulmonary hypoplasia, Respiratory distress (dogan2019achondrogenesistype2 pages 3-4) * Abnormal vertebral ossification / absent vertebral ossification, Platyspondyly (conceptually consistent with near-complete absent vertebral ossification described) (maheshwari2021acasereport pages 1-3, maheshwari2021acasereport media 016b7757) * Cystic hygroma, Hydrops fetalis, Polyhydramnios (korkko2000widelydistributedmutations pages 1-2, kobayashi2022novelmissensecol2a1 pages 1-2)
Quality-of-life impact
Because ACG2 is typically perinatally lethal, longer-term quality-of-life outcomes are generally not applicable; for rare short-term survivors, quality of life is dominated by respiratory failure and intensive care needs. (dogan2019achondrogenesistype2 pages 3-4)
4. Genetic / Molecular Information
Causal gene(s)
- COL2A1 (type II procollagen alpha-1 chain) is the principal causal gene for ACG2. (barathouari2016mutationupdatefor pages 1-2, zhang2020integratedanalysisof pages 1-6)
Inheritance pattern
ACG2 is described as autosomal dominant; many cases are de novo, but recurrence can occur due to parental germline mosaicism. (maheshwari2021acasereport pages 1-3, faivre2004recurrenceofachondrogenesis pages 4-5, kobayashi2022novelmissensecol2a1 pages 1-2)
Variant spectrum and classes (ACG2-relevant)
Aggregated evidence indicates that severe COL2A1 phenotypes (including ACG2/hypochondrogenesis) are enriched for dominant-negative glycine substitutions in the triple helix: * “One-third of the mutations are dominant-negative mutations that affect the glycine residue in the G-X-Y repeats … [and] are common in achondrogenesis type II and hypochondrogenesis.” (barathouari2016mutationupdatefor pages 1-2)
A fetal case series found ACG2/HCG mutations distributed across the gene and reported classes including: * Glycine substitutions (10/12 in that cohort) * Splice-site mutation * In-frame deletion (18 bp) (korkko2000widelydistributedmutations pages 1-2)
Example pathogenic/likely pathogenic variants (HGVS)
Examples from retrieved primary reports: * NM_001844.5:c.2987G>A (p.Gly996Asp), classified “likely pathogenic (PS2+PM2+PP3+PP4)” in a fetus (kobayashi2022novelmissensecol2a1 pages 1-2) * c.2546G>A (p.Gly849Asp) (novel heterozygous missense) in a newborn (dogan2019achondrogenesistype2 pages 3-4)
Functional consequences and mechanistic interpretation
The dominant-negative triple-helix glycine substitutions are described as disrupting collagen structure: * “These mutations disrupt the collagen triple helix …” (barathouari2016mutationupdatefor pages 1-2)
Mechanistically, this is consistent with defective cartilage extracellular matrix formation and impaired endochondral ossification, producing the profound under-ossification and growth failure characteristic of ACG2. (barathouari2016mutationupdatefor pages 1-2, dogan2019achondrogenesistype2 pages 3-4)
Modifier genes / epigenetics / chromosomal abnormalities
No modifier genes, epigenetic drivers, or recurrent chromosomal abnormalities specific to ACG2 were identified in retrieved sources. (barathouari2016mutationupdatefor pages 1-2)
5. Environmental Information
No established environmental or infectious contributors are described in the retrieved sources; ACG2 is primarily a monogenic collagen disorder due to COL2A1 variants. (barathouari2016mutationupdatefor pages 1-2)
6. Mechanism / Pathophysiology
Causal chain (high-level)
- COL2A1 variant (often glycine substitution in triple helix; dominant-negative) (barathouari2016mutationupdatefor pages 1-2)
- Disrupted type II collagen triple-helix formation/structure and cartilage matrix integrity (barathouari2016mutationupdatefor pages 1-2)
- Abnormal cartilage growth plate biology (histology can show hypercellularity/enlarged chondrocytes and disrupted growth plate zonation) (faivre2004recurrenceofachondrogenesis pages 4-5)
- Failure of normal endochondral ossification → poor vertebral/pelvic ossification, markedly short long bones, short ribs, small thorax (maheshwari2021acasereport media 016b7757, dogan2019achondrogenesistype2 pages 3-4)
- Pulmonary hypoplasia from severely restricted thoracic development → perinatal respiratory failure and lethality (dogan2019achondrogenesistype2 pages 3-4)
Pathway/process ontology suggestions
- GO biological processes (suggestions): extracellular matrix organization; collagen fibril organization; endochondral ossification; cartilage development (supported conceptually by COL2A1 cartilage role and growth plate pathology descriptions) (faivre2004recurrenceofachondrogenesis pages 4-5, zhang2020integratedanalysisof pages 1-6, barathouari2016mutationupdatefor pages 1-2)
- Cell types (CL suggestions): chondrocyte (growth plate / cartilage) (faivre2004recurrenceofachondrogenesis pages 4-5)
Molecular profiling / single-cell / spatial / functional genomics
No ACG2-specific omics profiling (transcriptomic/proteomic/metabolomic) or single-cell/spatial studies were identified in retrieved sources for this run. (barathouari2016mutationupdatefor pages 1-2)
7. Anatomical Structures Affected
Organ/system level
Primary involvement is the skeletal system (long bones, ribs, spine, pelvis) with secondary lethal involvement of the respiratory system due to pulmonary hypoplasia. (dogan2019achondrogenesistype2 pages 3-4)
Tissue/cell level
Primary tissue pathology localizes to cartilage and the growth plate (disordered chondrocyte morphology/zoning). (faivre2004recurrenceofachondrogenesis pages 4-5)
UBERON suggestions
- vertebral column/vertebral bodies; rib; pelvis/ilium; limb long bones; cartilage; lung (maheshwari2021acasereport media 016b7757, dogan2019achondrogenesistype2 pages 3-4)
8. Temporal Development
- Onset: Congenital/prenatal; may be detectable in the first or second trimester depending on severity and imaging (e.g., 14-week fetus with severe limb shortening and cystic hygroma). (kobayashi2022novelmissensecol2a1 pages 1-2)
- Course: Not progressive in the typical sense; disease is usually perinatally lethal. (dogan2019achondrogenesistype2 pages 3-4)
9. Inheritance and Population
Epidemiology
A radiology case report review states an estimated frequency of approximately 0.2 per 100,000 births for achondrogenesis type II. (maheshwari2021acasereport pages 1-3)
De novo occurrence and recurrence risk
Although many cases occur de novo, familial recurrence has been documented and attributed to germline mosaicism, which is important for counseling. (faivre2004recurrenceofachondrogenesis pages 4-5)
Population demographics (sex ratio, geographic distribution) were not available in the retrieved sources for this run. (barathouari2016mutationupdatefor pages 1-2)
10. Diagnostics
Prenatal diagnosis
Imaging: Obstetric ultrasound is the primary screening and diagnostic modality. In fetal skeletal dysplasia evaluation, fetal CT is also referenced as an option in the ACG2 context. (kobayashi2022novelmissensecol2a1 pages 1-2)
A quantitative example from a prenatal imaging case report: an extremely low femur length/abdominal circumference ratio (FL/AC) of 0.08 (vs typical 0.20–0.25) was used as a strong indicator of lethal skeletal dysplasia in a reported ACG2 case. (maheshwari2021acasereport pages 1-3)
Postnatal/postmortem confirmation
Radiographic skeletal survey demonstrates the diagnostic pattern of under-ossification and limb/rib/pelvic abnormalities. (maheshwari2021acasereport media 016b7757, dogan2019achondrogenesistype2 pages 3-4)
Genetic testing
ACG2 can be confirmed by NGS-based molecular diagnosis (COL2A1 sequencing within targeted panels or exome approaches) with confirmatory Sanger sequencing in reported cases. (dogan2019achondrogenesistype2 pages 3-4)
Differential diagnosis
Important differentials discussed in retrieved sources include: * thanatophoric dysplasia * osteogenesis imperfecta type II * hypophosphatasia congenita and distinction from achondrogenesis type I subtypes can be supported by radiographic patterns (e.g., relatively preserved skull ossification in ACG2 with decreased pelvic/vertebral ossification). (maheshwari2021acasereport pages 1-3, dogan2019achondrogenesistype2 pages 4-5)
11. Outcome / Prognosis
ACG2 is consistently described as lethal, with death occurring in utero or in the early neonatal period, largely due to pulmonary hypoplasia and respiratory failure. (dogan2019achondrogenesistype2 pages 3-4, barathouari2016mutationupdatefor pages 1-2)
A documented liveborn course illustrates the severity: a newborn with ACG2 required high-frequency ventilation and died in the neonatal period. (dogan2019achondrogenesistype2 pages 3-4)
12. Treatment
Disease-modifying therapy
No disease-modifying pharmacologic or molecular therapy for ACG2 was identified in retrieved sources for this run. (barathouari2016mutationupdatefor pages 1-2)
Supportive/palliative care
Management for liveborn infants is generally supportive (e.g., respiratory support in intensive care), but overall survival is poor. (dogan2019achondrogenesistype2 pages 3-4)
MAXO term suggestions (examples)
- respiratory support
- palliative care
- neonatal intensive care
- genetic counseling
- prenatal molecular diagnosis
13. Prevention
Because ACG2 is genetic, “prevention” primarily consists of reproductive risk reduction and early detection: * Genetic counseling is emphasized in mutation-spectrum reviews as a critical component of care (“an early diagnosis is critical … and genetic counseling to affected families”). (barathouari2016mutationupdatefor pages 1-2) * Prenatal diagnosis in subsequent pregnancies may use targeted familial-variant testing (when known) plus fetal imaging. * Recurrence counseling should include the possibility of germline mosaicism. (faivre2004recurrenceofachondrogenesis pages 4-5)
Pregnancy termination after definitive diagnosis is discussed in lethal skeletal dysplasia case management contexts. (maheshwari2021acasereport pages 1-3)
14. Other Species / Natural Disease
No naturally occurring ACG2-equivalent disease in other species was identified in the retrieved sources for this run. (barathouari2016mutationupdatefor pages 1-2)
15. Model Organisms
No model organism studies specific to achondrogenesis type II were identified in the retrieved sources for this run (the retrieved literature focused on human case reports/series and human variant-spectrum analyses). (barathouari2016mutationupdatefor pages 1-2)
Summary tables (knowledge-base ready)
| Section | Item | Details | Ontology suggestions | Evidence |
|---|---|---|---|---|
| Disease identifiers & synonyms | Primary disease name | Achondrogenesis type II; lethal congenital/perinatal skeletal dysplasia within the type II collagenopathy spectrum | MONDO: Not found in retrieved sources | (kobayashi2022novelmissensecol2a1 pages 1-2, maheshwari2021acasereport pages 1-3, barathouari2016mutationupdatefor pages 1-2) |
| Disease identifiers & synonyms | Named subtype / synonyms | Langer-Saldino achondrogenesis; ACG2; achondrogenesis type II/hypochondrogenesis spectrum | MONDO: Not found in retrieved sources | (maheshwari2021acasereport pages 1-3, korkko2000widelydistributedmutations pages 1-2) |
| Disease identifiers & synonyms | Database identifiers available in retrieved evidence | OMIM #200610 for achondrogenesis type II; COL2A1 MIM #108300 | MeSH: Not found in retrieved sources; Orphanet: Not found in retrieved sources; ICD-10: Not found in retrieved sources; ICD-11: Not found in retrieved sources | (kobayashi2022novelmissensecol2a1 pages 1-2, barathouari2016mutationupdatefor pages 1-2) |
| Disease identifiers & synonyms | Evidence provenance | Retrieved information is aggregated from disease-level and mutation review resources plus individual prenatal/newborn case reports, radiology reports, and molecular case series; not from EHR datasets | — | (kobayashi2022novelmissensecol2a1 pages 1-2, maheshwari2021acasereport pages 1-3, korkko2000widelydistributedmutations pages 1-2, barathouari2016mutationupdatefor pages 1-2) |
| Genetic / molecular | Causal gene | COL2A1 (collagen type II alpha 1 chain), encoding the alpha-1 chain of type II procollagen, the major collagen of cartilage | HGNC gene symbol: COL2A1 | (kobayashi2022novelmissensecol2a1 pages 1-2, barathouari2016mutationupdatefor pages 1-2, zhang2020integratedanalysisof pages 1-6) |
| Genetic / molecular | Inheritance | Typically autosomal dominant; many cases are de novo; recurrence can occur due to parental germline/somatic mosaicism | — | (maheshwari2021acasereport pages 1-3, faivre2004recurrenceofachondrogenesis pages 4-5, kobayashi2022novelmissensecol2a1 pages 1-2) |
| Genetic / molecular | Typical pathogenic variant classes | Predominantly heterozygous glycine substitutions in the triple-helical Gly-X-Y repeat; also splice-site variants and in-frame deletions reported | Sequence ontology suggestions: missense_variant, splice_acceptor_variant, inframe_deletion | (korkko2000widelydistributedmutations pages 1-2, barathouari2016mutationupdatefor pages 1-2) |
| Genetic / molecular | Example variants (HGVS) | c.2987G>A (p.Gly996Asp), likely pathogenic; c.2546G>A (p.Gly849Asp), novel; c.1267-2_1269del causing exon 21 skipping/in-frame deletion; familial recurrence reported with G316D | — | (kobayashi2022novelmissensecol2a1 pages 1-2, dogan2019achondrogenesistype2 pages 3-4, faivre2004recurrenceofachondrogenesis pages 4-5) |
| Genetic / molecular | Mechanistic notes | Glycine substitutions disrupt collagen triple-helix folding/assembly (dominant-negative effect), impair cartilage matrix formation, and underlie severe/lethal phenotypes; ACG2/hypochondrogenesis are among the most severe COL2A1 phenotypes, associated with neonatal death | GO: collagen trimer assembly; extracellular matrix organization; CL: chondrocyte | (barathouari2016mutationupdatefor pages 1-2, zhang2020integratedanalysisof pages 1-6) |
| Phenotype / anatomy | Core prenatal findings | Severe micromelia/short limbs, short humerus/femur, thoracic hypoplasia or small/narrow chest, cystic hygroma, hydrops/polyhydramnios, increased nuchal fold; may be detectable from first/second trimester | HPO: Short limb, Micromelia, Narrow thorax, Cystic hygroma, Hydrops fetalis, Polyhydramnios; UBERON: limb, thoracic cage | (kobayashi2022novelmissensecol2a1 pages 1-2, korkko2000widelydistributedmutations pages 1-2) |
| Phenotype / anatomy | Core neonatal/physical findings | Short trunk, prominent/distended abdomen, relatively large head/prominent forehead, small chin/midface hypoplasia, severe respiratory distress due to pulmonary hypoplasia | HPO: Short trunk, Abnormal abdomen morphology, Frontal bossing, Micrognathia, Midface retrusion, Respiratory distress, Pulmonary hypoplasia; UBERON: abdomen, lung, skull | (maheshwari2021acasereport pages 1-3, dogan2019achondrogenesistype2 pages 3-4) |
| Phenotype / anatomy | Hallmark radiographic findings | Very short long bones with widened/flared metaphyses; non-ossification or markedly reduced ossification of vertebral bodies (including cervical/sacral), lack of pelvic ossification, short unfractured ribs, narrow bell-shaped thorax, cupped ribs, iliac “paraglider” appearance; skull ossification usually relatively preserved compared with type I achondrogenesis | HPO: Platyspondyly/abnormal vertebral ossification, Short rib, Bell-shaped thorax, Short long bone, Flared metaphysis, Abnormal iliac bone morphology; UBERON: vertebral body, rib, ilium, pelvis | (maheshwari2021acasereport pages 1-3, dogan2019achondrogenesistype2 pages 3-4, maheshwari2021acasereport media 016b7757) |
| Phenotype / anatomy | Tissue/cell focus | Primary pathology localizes to cartilage/growth plate; histology shows hypercellular, hypervascular cartilage with enlarged chondrocytes and disorganized growth plate maturation | CL: chondrocyte; UBERON: cartilage tissue, growth plate; GO: endochondral ossification | (faivre2004recurrenceofachondrogenesis pages 4-5, barathouari2016mutationupdatefor pages 1-2) |
| Diagnostics | Prenatal imaging | Detailed obstetric ultrasound is first-line; fetal CT and sometimes MRI may assist characterization of skeletal dysplasia and thoracic/rib/ossification abnormalities | MAXO: prenatal imaging evaluation; fetal ultrasonography; computed tomography | (kobayashi2022novelmissensecol2a1 pages 1-2, hall2024fetalandperinatal pages 61-63) |
| Diagnostics | Postnatal / postmortem confirmation | Skeletal survey/radiography confirms classic ossification and limb/rib/pelvic abnormalities; pathology/histology may support classification | MAXO: radiographic skeletal survey; pathologic examination | (maheshwari2021acasereport pages 1-3, faivre2004recurrenceofachondrogenesis pages 4-5, maheshwari2021acasereport media 016b7757) |
| Diagnostics | Molecular testing strategy | Molecular confirmation by NGS-based testing (single-gene COL2A1, targeted skeletal dysplasia panels, clinical exome/WES); variants commonly confirmed by Sanger sequencing; exome sequencing increases diagnostic yield in fetuses with short long bones after negative karyotype/CMA | MAXO: sequence analysis of COL2A1; exome sequencing; confirmatory Sanger sequencing | (kobayashi2022novelmissensecol2a1 pages 1-2, dogan2019achondrogenesistype2 pages 3-4) |
| Diagnostics | Differential diagnosis clues | Distinguish from achondrogenesis type I, thanatophoric dysplasia, osteogenesis imperfecta type II, and hypophosphatasia congenita; preserved skull ossification with poor vertebral/pelvic ossification favors ACG2 over some type I forms | MAXO: differential diagnostic assessment | (maheshwari2021acasereport pages 1-3, dogan2019achondrogenesistype2 pages 4-5, dogan2019achondrogenesistype2 pages 3-4) |
| Management / prognosis / prevention | Prognosis | Usually lethal: many affected fetuses are stillborn or die in utero, immediately after birth, or in the early neonatal period; death is driven largely by pulmonary hypoplasia/respiratory failure | HPO: Perinatal lethality; Pulmonary hypoplasia | (maheshwari2021acasereport pages 1-3, dogan2019achondrogenesistype2 pages 3-4, barathouari2016mutationupdatefor pages 1-2, kobayashi2022novelmissensecol2a1 pages 1-2) |
| Management / prognosis / prevention | Supportive care | No disease-modifying therapy identified in retrieved evidence; neonatal management is supportive/palliative, including respiratory support when liveborn, but survival is generally poor | MAXO: respiratory support; palliative care; neonatal intensive care | (dogan2019achondrogenesistype2 pages 3-4, kobayashi2022novelmissensecol2a1 pages 1-2) |
| Management / prognosis / prevention | Prevention / reproductive options | Genetic counseling is recommended; prenatal diagnosis in future pregnancies via targeted molecular testing and fetal imaging; recurrence counseling should include possibility of germline mosaicism; pregnancy termination may be considered where legally/ethically appropriate after definitive diagnosis | MAXO: genetic counseling; prenatal molecular diagnosis; prenatal ultrasound monitoring; reproductive counseling | (maheshwari2021acasereport pages 1-3, faivre2004recurrenceofachondrogenesis pages 4-5, barathouari2016mutationupdatefor pages 1-2) |
| Research / real-world implementation | Current implementations / studies | A natural history study in children with type II collagen disorders and short stature is recruiting (NCT05408715), but retrieved evidence does not indicate an interventional trial specific to lethal ACG2 | MAXO: natural history study enrollment | (hall2024fetalandperinatal pages 61-63) |
Table: This table condenses the retrieved evidence on Achondrogenesis type II into knowledge-base-ready fields covering identifiers, genetics, phenotype, diagnostics, and management. It is useful as a structured source map with ontology suggestions and per-row citation IDs.
Key real-world implementations and recent developments (emphasis on 2023–2024 where available)
- A 2024 comprehensive resource on fetal and perinatal skeletal dysplasias highlights the role of multimodal fetal imaging and multidisciplinary evaluation for prenatal-onset skeletal dysplasia conditions and explicitly lists ACG2 within the COL2A1-related spectrum. (hall2024fetalandperinatal pages 61-63)
- Routine clinical implementation relies on NGS-based molecular confirmation of COL2A1 variants alongside classic radiographic patterns; mutation-spectrum curation supports clinical interpretation and counseling. (barathouari2016mutationupdatefor pages 1-2, dogan2019achondrogenesistype2 pages 3-4)
URLs and publication dates (retrieved sources cited)
- Barat-Houari et al. Human Mutation (Jan 2016). https://doi.org/10.1002/humu.22915 (barathouari2016mutationupdatefor pages 1-2)
- Doğan et al. Balkan J Med Genet (Jun 2019). https://doi.org/10.2478/bjmg-2019-0001 (dogan2019achondrogenesistype2 pages 3-4)
- Maheshwari et al. Egyptian J Radiol Nucl Med (Apr 2021). https://doi.org/10.1186/s43055-021-00479-0 (maheshwari2021acasereport pages 1-3)
- Kobayashi et al. Human Genome Variation (Nov 2022). https://doi.org/10.1038/s41439-022-00218-5 (kobayashi2022novelmissensecol2a1 pages 1-2)
- Hall et al. Fetal and perinatal skeletal dysplasias (Mar 2024; CRC Press/ArXiv entry). https://doi.org/10.1201/9781003166948 (hall2024fetalandperinatal pages 61-63)
Limitations of this report (evidence availability in this run)
- Standard identifiers beyond OMIM (Orphanet, ICD-10/11, MeSH, MONDO) were not present in the retrieved full texts; they should be added via dedicated ontology/database lookup workflows. (kobayashi2022novelmissensecol2a1 pages 1-2)
- No ACG2-specific interventional trials, omics studies, or model organism studies were retrieved/citable in this run; additional targeted searches would likely be required for those topics. (barathouari2016mutationupdatefor pages 1-2)
References
-
(maheshwari2021acasereport pages 1-3): Saurabh Maheshwari, Dilip Ingole, Samar Chatterjee, Uddandam Rajesh, and Varun Anand. A case report of achondrogenesis type ii (langer-saldino achondrogenesis). Egyptian Journal of Radiology and Nuclear Medicine, Apr 2021. URL: https://doi.org/10.1186/s43055-021-00479-0, doi:10.1186/s43055-021-00479-0. This article has 2 citations.
-
(dogan2019achondrogenesistype2 pages 3-4): P. Doğan, IG Varal, O. Gorukmez, M. Akkurt, and A. Akdağ. Achondrogenesis type 2 in a newborn with a novel mutation on the col2a1 gene. Balkan Journal of Medical Genetics, 22:89-94, Jun 2019. URL: https://doi.org/10.2478/bjmg-2019-0001, doi:10.2478/bjmg-2019-0001. This article has 7 citations.
-
(kobayashi2022novelmissensecol2a1 pages 1-2): Yukari Kobayashi, Yuki Ito, Kosuke Taniguchi, Kana Harada, Michihiro Yamamura, Taisuke Sato, Ken Takahashi, Hiroshi Kawame, Kenichiro Hata, Osamu Samura, and Aikou Okamoto. Novel missense col2a1 variant in a fetus with achondrogenesis type ii. Human Genome Variation, Nov 2022. URL: https://doi.org/10.1038/s41439-022-00218-5, doi:10.1038/s41439-022-00218-5. This article has 4 citations.
-
(barathouari2016mutationupdatefor pages 1-2): Mouna Barat-Houari, Guillaume Sarrabay, Vincent Gatinois, Aurélie Fabre, Bruno Dumont, David Genevieve, and Isabelle Touitou. Mutation update for col2a1 gene variants associated with type ii collagenopathies. Human Mutation, 37:7-15, Jan 2016. URL: https://doi.org/10.1002/humu.22915, doi:10.1002/humu.22915. This article has 173 citations and is from a domain leading peer-reviewed journal.
-
(korkko2000widelydistributedmutations pages 1-2): J. Körkkö, J. Körkkö, D. Cohn, L. Ala‐kokko, L. Ala‐kokko, D. Krakow, D. Krakow, and D. Prockop. Widely distributed mutations in the col2a1 gene produce achondrogenesis type ii/hypochondrogenesis. American journal of medical genetics, 92 2:95-100, May 2000. URL: https://doi.org/10.1002/(sici)1096-8628(20000515)92:2<95::aid-ajmg3>3.0.co;2-9, doi:10.1002/(sici)1096-8628(20000515)92:2<95::aid-ajmg3>3.0.co;2-9. This article has 76 citations.
-
(zhang2020integratedanalysisof pages 1-6): Boyan Zhang, Yue Zhang, Naichao Wu, Jianing Li, He Liu, and Jincheng Wang. Integrated analysis of col2a1 variant data and classification of type ii collagenopathies. Dec 2020. URL: https://doi.org/10.1111/cge.13680, doi:10.1111/cge.13680. This article has 61 citations and is from a peer-reviewed journal.
-
(dogan2019achondrogenesistype2 pages 4-5): P. Doğan, IG Varal, O. Gorukmez, M. Akkurt, and A. Akdağ. Achondrogenesis type 2 in a newborn with a novel mutation on the col2a1 gene. Balkan Journal of Medical Genetics, 22:89-94, Jun 2019. URL: https://doi.org/10.2478/bjmg-2019-0001, doi:10.2478/bjmg-2019-0001. This article has 7 citations.
-
(maheshwari2021acasereport media 016b7757): Saurabh Maheshwari, Dilip Ingole, Samar Chatterjee, Uddandam Rajesh, and Varun Anand. A case report of achondrogenesis type ii (langer-saldino achondrogenesis). Egyptian Journal of Radiology and Nuclear Medicine, Apr 2021. URL: https://doi.org/10.1186/s43055-021-00479-0, doi:10.1186/s43055-021-00479-0. This article has 2 citations.
-
(faivre2004recurrenceofachondrogenesis pages 4-5): Laurence Faivre, Martine Le Merrer, Serges Douvier, Nicole Laurent, Christel Thauvin‐Robinet, Thierry Rousseau, Inge Vereecke, Paul Sagot, Anne‐Lise Delezoide, Paul Coucke, and Geert Mortier. Recurrence of achondrogenesis type ii within the same family: evidence for germline mosaicism. American Journal of Medical Genetics Part A, 126A:308-312, Apr 2004. URL: https://doi.org/10.1002/ajmg.a.20597, doi:10.1002/ajmg.a.20597. This article has 27 citations.
-
(hall2024fetalandperinatal pages 61-63): Christine M Hall, Amaka C Offiah, Francesca Forzano, Mario Lituania, Gen Nishimura, and Valerie Cormier-Daire. Fetal and perinatal skeletal dysplasias. ArXiv, Mar 2024. URL: https://doi.org/10.1201/9781003166948, doi:10.1201/9781003166948. This article has 26 citations.
Artifacts
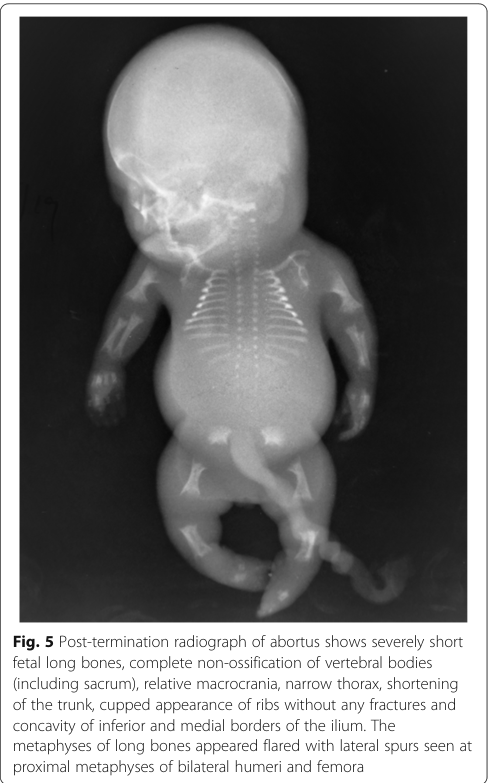
Comprehensive Pathophysiology of Achondrogenesis Type II: Molecular Mechanisms and Disease Progression
Achondrogenesis Type II (ACG2), also known as Langer-Saldino achondrogenesis, represents the most severe form of skeletal dysplasia caused by heterozygous mutations in the COL2A1 gene, which encodes type II collagen.[1][3][4] This lethal condition manifests as severe micromelic dwarfism, incomplete ossification of the vertebral bodies and pelvis, and profound underdevelopment of the lungs, with approximately 85% of affected infants dying before birth or within the first few days of life due to respiratory failure.[28] The fundamental pathophysiology of ACG2 involves the production of structurally abnormal type II collagen molecules that fail to form proper fibrillar networks within the extracellular matrix of cartilage and other connective tissues, triggering cellular stress responses, impaired chondrocyte differentiation, and disrupted endochondral ossification. This report provides an in-depth examination of the molecular mechanisms, cellular consequences, and disease progression mechanisms that characterize achondrogenesis type II, integrating findings from recent molecular genetics, developmental biology, and clinical studies to establish a comprehensive understanding of this devastating genetic disorder.
Core Pathophysiological Mechanisms of Achondrogenesis Type II
The Genetic Foundation: COL2A1 Gene Mutations
Achondrogenesis Type II is caused exclusively by heterozygous mutations in the COL2A1 gene, which is located on chromosome 12 (12q13.11-q13.2) and consists of 54 exons spanning over 31.5 kilobases.[2][5][41] The COL2A1 gene provides comprehensive instructions for synthesizing the alpha-1(II) chain, the basic structural component of type II collagen, a homotrimeric protein composed of three identical alpha-1(II) polypeptide chains, each containing 1060 amino acid residues.[2][41] Unlike the autosomal recessive inheritance patterns observed in achondrogenesis types 1A and 1B, ACG2 exhibits autosomal dominant inheritance, meaning that a single mutated copy of the COL2A1 gene in each cell is sufficient to cause the severe phenotype.[1][4][16] Importantly, most cases of ACG2 (approximately 80% of achondrogenesis cases) arise from de novo mutations that occur spontaneously during the formation of reproductive cells in an affected individual's parent or during early embryonic development, meaning that affected individuals typically have no family history of the condition.[4][13][28] However, germline and somatic mosaicism have been documented in rare familial cases, creating complex recurrence risks for parents of affected individuals.[37][40]
The molecular spectrum of COL2A1 mutations in ACG2 encompasses a diverse array of genetic alterations, including point mutations (missense, nonsense, deletion, insertion, and frameshift mutations) and complex rearrangements, with more than 400 mutations currently described in public databases and scientific literature.[2][12] Among these mutations, missense mutations constitute the most common type, accounting for over 70% of all pathogenic variants identified in COL2A1-related disorders.[2][12] These missense mutations frequently target the highly conserved glycine residues within the Gly-X-Y repeat motif that characterizes the triple-helical domain of type II collagen, where the X and Y positions are typically occupied by proline and hydroxyproline residues respectively.[2][41] Glycine substitutions in the Gly-X-Y repeat are particularly disruptive because glycine, being the smallest amino acid with only a hydrogen atom as its side chain, is uniquely suited to fit within the tightly packed interior of the collagen triple helix.[2][31][34] When glycine residues are replaced by larger amino acids such as serine, alanine, arginine, aspartate, cysteine, glutamate, or valine, the resulting structural distortion severely destabilizes the triple helix.[31][34] Studies examining host-guest triple-helical peptides have demonstrated that any substitution for glycine results in dramatic destabilization of the triple helix, with melting temperature (Tm) decreasing from approximately 45°C for normal collagen to approximately 10°C for alanine and serine substitutions, and to below 0°C for arginine, valine, glutamate, and aspartate substitutions.[31]
A particularly illustrative example comes from recent case reports documenting novel mutations in ACG2. One documented case revealed a heterozygous missense variation c.2546G>A, p.Gly849Asp in COL2A1, which had never been previously described in scientific literature before its identification through next-generation sequencing.[3][43] This glycine-to-aspartate substitution at position 849 in the triple-helical domain exemplifies the dominant-negative mechanism characteristic of ACG2, where the altered collagen chain integrates into the trimeric collagen molecule and destabilizes the entire structure through its compromised geometry. In another familial case, a father with proven somatic mosaicism harbored a c.1037G>T (p.Gly346Val) mutation that resulted in glycine substitution with valine, a bulkier hydrophobic residue, and this mutation was transmitted to multiple affected offspring, producing the characteristic lethal phenotype.[40] These documented mutations illustrate the fundamental principle that glycine-substituting mutations in the triple-helical domain produce the most severe phenotypes through dominant-negative mechanisms, whereas other types of mutations may lead to milder presentations through different pathogenic mechanisms.
Triple-Helical Structure and Its Disruption in ACG2
The normal type II collagen molecule represents a remarkable structural feat of biomolecular engineering, consisting of three alpha-1(II) chains that wind together to form a stable, tightly packed triple helix with a distinctive left-handed twist at the individual chain level and a right-handed superhelical arrangement of the three chains.[2][41] The triple-helical conformation is stabilized by extensive hydrogen bonding between the polypeptide backbones of adjacent chains and by the geometric constraints imposed by the Gly-X-Y repeat pattern, where the small glycine residue can fit into the crowded interior of the helix while larger amino acids cannot.[31][34] The triple-helical region spans approximately 1000 amino acids, interrupted only at the termini by non-helical telopeptide regions: a 19-residue N-telopeptide and a 27-residue C-telopeptide that are crucial for initiating triple-helix formation and for subsequent cross-linking of mature collagen fibers.[2][41] This architectural precision is essential for type II collagen's biological functions, and disruption of the Gly-X-Y pattern through point mutations directly compromises the structural integrity of the triple helix.
In achondrogenesis type II, glycine-to-amino acid substitutions produce several interconnected consequences for collagen structure. Mutant type II collagen molecules exhibit altered electrophoretic mobility, indicating changes in their charge and hydrodynamic properties, and demonstrate relatively low thermostability compared with normal collagen, suggesting that the destabilized triple helix is more prone to unfolding and denaturation.[2][12][41] Additionally, mutant collagen molecules show markedly slow rates of secretion into the extracellular space, as the poorly formed triple helix is recognized by cellular quality control mechanisms and retained within the endoplasmic reticulum.[2][12] When mutant type II collagen chains do manage to reach the extracellular space, they participate in abnormal fibril assembly, forming malformed fibrils that cannot properly interact with other elements of the extracellular matrix such as proteoglycans, other collagens, and matricellular proteins.[2][12][41] The consequence is that the normally robust extracellular matrix scaffold, which is supposed to comprise approximately 95% collagen and constitute approximately 60% of the dry weight of mature cartilage, is severely compromised in its structural integrity and biomechanical competence.[2]
Molecular and Genetic Basis: Type II Collagen and its Pathophysiological Roles
Biosynthesis and Normal Function of Type II Collagen
Type II collagen, encoded by the COL2A1 gene, serves as the predominant structural component of hyaline cartilage extracellular matrix, where it provides tensile strength and shape stability to this specialized tissue.[2][5][41] Beyond cartilage, type II collagen is also the major protein component of the nucleus pulposus of intervertebral discs, the vitreous humor of the eye (approximately 70% of vitreous total protein content), and the structural elements of the inner ear, tissues that together underscore the widespread importance of type II collagen for normal development and function of multiple organ systems.[2][5][27][41] During normal skeletal development, type II collagen is synthesized by proliferating chondrocytes within the growth plates until these cells differentiate into hypertrophic chondrocytes, at which point the synthesis of type II collagen is downregulated in favor of type X collagen synthesis.[2][41]
The normal biosynthetic pathway for type II collagen begins with the transcription of the COL2A1 gene and translation of the primary transcript into pro-alpha-1(II) chains, which undergo extensive post-translational modifications including hydroxylation of proline and lysine residues, glycosylation of certain hydroxylysine residues, and removal of signal peptides.[5][41] Three pro-alpha-1(II) chains then associate in the endoplasmic reticulum through interactions involving their C-propeptide domains and triple-helix formation, which proceeds directionally from the C-terminal toward the N-terminal region, creating stable procollagen molecules.[2][5][41][58] These procollagen molecules are transported through the secretory pathway, modified further by enzymes in the Golgi apparatus (a particularly critical step that is disrupted in achondrogenesis type 1A due to TRIP11 mutations affecting Golgi function), and secreted into the extracellular space where they undergo processing to remove the terminal propeptides and create mature collagen molecules.[2] The mature collagen molecules then spontaneously self-assemble into fibrils, which further associate laterally to form larger fibrils and fibers that are cross-linked through lysine and hydroxylysine residue interactions to achieve maximum structural stability and biomechanical strength.[2][41]
Remarkably, type II collagen functions not merely as a passive structural scaffold but also as an active extracellular signaling molecule with profound regulatory effects on chondrocyte biology.[2][7][41][20][23] Specifically, type II collagen acts as an autocrine factor of proliferation and differentiation via multiple downstream effectors and a potent suppressor of chondrocyte hypertrophy and apoptosis through negative regulation of SMAD1 activity, a finding that has major implications for understanding how COL2A1 mutations disrupt normal endochondral ossification processes.[2][7][41] The primary cellular receptor for type II collagen is integrin β1 (ITGB1), which mediates chondrocyte-extracellular matrix interactions and initiates intracellular signaling cascades that influence chondrocyte differentiation, metabolism, and survival.[7][23] Recent studies have demonstrated that upon interaction between COL2A1 and ITGB1, the integrin receptor competes with bone morphogenetic protein (BMP) receptors for binding to SMAD1 and phosphorylates ERK1/2, both of which mechanisms suppress BMP-SMAD1-mediated chondrocyte hypertrophy.[7][23] This regulatory function becomes dramatically impaired in ACG2 when mutant type II collagen fails to assemble properly, depriving chondrocytes of the critical suppressive signals that normally maintain them in an appropriate state of differentiation and proliferation.
Mutation-Specific Pathogenic Mechanisms
The diversity of COL2A1 mutations in achondrogenesis type II results in disease through two primary molecular mechanisms: dominant-negative effects and haploinsufficiency, though dominant-negative effects account for the overwhelming majority of severe type II collagenopathies including ACG2.[2][12][33] The dominant-negative mechanism operates when mutant collagen chains incorporate into trimeric collagen molecules alongside normal chains, forming hybrid molecules in which the presence of even a single defective chain severely compromises the stability and function of the entire complex.[2][12][33][34] This explains why ACG2, caused by dominant-negative mutations, produces such severe phenotypes despite the presence of one normal COL2A1 allele producing normal collagen chains: the abnormal chains "poison" the collagen fibrils through their deleterious effects on triple-helix stability and proper fibril assembly.
In contrast, haploinsufficiency results from mutations that cause premature termination of translation through nonsense mutations, out-of-frame deletions, or splice-site mutations that lead to non-sense mediated decay of the mutant transcript and reduced synthesis of normal collagen.[2][12][33][36] These haploinsufficiency mutations lead to milder phenotypes because 50% reduction in normal collagen production is better tolerated than the presence of destabilizing mutant chains, which explains why conditions such as Stickler syndrome, associated with truncation mutations causing haploinsufficiency, present less severe skeletal involvement than ACG2.[2][12][33][36][44] Missense mutations that substitute amino acids other than glycine result in generally milder phenotypes compared with glycine substitutions, because these mutations typically cause localized protein instability and impaired proper function of type II collagen without completely disrupting triple-helix formation, resulting in production of some partially functional collagen molecules.[2][12][33]
Particularly severe phenotypes result from glycine-to-nonserine substitutions in the triple-helical domain, such as glycine-to-arginine, glycine-to-aspartate, or glycine-to-valine substitutions, which produce alternating zones of severe skeletal dysplasia.[2][12][26] Glycine-to-serine substitutions, though still producing severe phenotypes, appear to generate somewhat milder manifestations compared with other glycine replacements, suggesting that the specific chemical properties of the substituted amino acid determine the degree of triple-helix destabilization.[26][31] C-propeptide domain mutations represent an important subset of COL2A1 mutations that produce distinctive clinical phenotypes with prominent brachydactyly (short digits) and different patterns of skeletal involvement compared with triple-helical mutations, likely because the C-propeptide region plays a specialized role in assembly of stable trimeric collagen molecules and may possess signaling functions distinct from the triple-helical domain.[2][9][36]
Cellular and Molecular Mechanisms: Disrupted Chondrocyte Function and Cartilage Matrix Assembly
Endoplasmic Reticulum Stress and Chondrocyte Dysfunction
One of the most critical molecular consequences of COL2A1 mutations in achondrogenesis type II involves intracellular retention of misfolded procollagen and type II collagen in the endoplasmic reticulum (ER), leading to ER stress that severely compromises chondrocyte function and survival.[2][11][12][41] In normal chondrocytes, procollagen molecules fold properly in the ER and transit smoothly through the secretory pathway, but mutant type II collagen chains that fail to form stable triple helices are recognized by cellular quality control mechanisms (including unfolded protein response pathways) and retained within the ER.[2][11][12][41] This retention causes accumulation of misfolded protein aggregates within the ER lumen, triggering classical endoplasmic reticulum stress responses that include upregulation of heat-shock proteins, activation of the unfolded protein response (UPR) involving ATF4 and IRE1α signaling, and engagement of apoptotic pathways if the stress becomes severe enough.[2][11][12][41]
Transgenic mouse models bearing COL2A1 mutations demonstrate this mechanism directly: chondrocytes in affected animals show greatly extended cisternae of rough endoplasmic reticulum with obvious retention of procollagen and other secretory pathway proteins such as fibronectin, confirming that the mutation directly prevents proper trafficking of type II collagen.[2][12] This ER stress retention causes multiple downstream consequences for chondrocyte biology. First, endoplasmic reticulum stress sufficient to reduce proliferation rate at the growth plates, as documented in experimental systems and animal models, directly decreases the population expansion of chondrocytes necessary for normal bone elongation.[2][12][41] Second, ER stress triggers absence or marked reduction in the mRNA expression of critical chondrocyte marker genes, including Cdkn1a (cyclin-dependent kinase inhibitor involved in cell cycle regulation), Ihh (Indian hedgehog, crucial for growth plate morphogenesis), Fgfr3 (fibroblast growth factor receptor 3, which signals in response to FGF ligands), COL10A1 (type X collagen, a marker of hypertrophic differentiation), and Runx2 (runt-related transcription factor 2, essential for osteoblast differentiation).[2][12][41] This coordinated downregulation of multiple essential growth plate genes suggests that ER stress triggers a global suppression of the genetic programs required for normal chondrocyte development and endochondral ossification.
Abnormal Chondrocyte Differentiation and Growth Plate Disorganization
The consequence of ER stress and reduced expression of growth plate regulatory genes is profound disruption of the normal sequence and coordination of chondrocyte differentiation events within the growth plate, fundamentally altering the morphological organization and functional capacity of this critical developmental structure.[2][12][41] In normal growth plates, chondrocytes undergo a highly orchestrated differentiation program progressing from resting chondrocytes in the resting zone, through proliferating cells in the proliferative zone that arrange themselves into characteristic columns parallel to the axis of bone elongation, followed by pre-hypertrophic chondrocytes and finally hypertrophic chondrocytes in the hypertrophic zone that undergo terminal differentiation, calcify their surrounding cartilage matrix, and prepare the tissue for vascular invasion and osteoblast recruitment.[19][22][39][42] This columnar organization is essential for efficient endochondral ossification and normal bone elongation rates.
In ACG2, this normal growth plate architecture is severely disrupted. Pathological studies of growth plates from achondrogenesis type II fetuses reveal growth plates with severely reduced or completely absent columnar-zone formation, with chondrocytes showing marked disorganization and enlargement due to intracellular vacuolization.[2][14][41][45] Rather than forming orderly columns, chondrocytes are scattered haphazardly throughout aberrant cartilage tissue, and the characteristic progression of chondrocyte differentiation from resting to proliferative to hypertrophic states becomes indistinguishable.[2][14][41] Proliferative and hypertrophic zones of cartilage are either markedly shorter than normal or completely indistinguishable, indicating severe disruption of the temporal and spatial regulation of chondrocyte differentiation.[2][12][41] Furthermore, deposition of cartilage matrix is notably impaired, with collagen fibrils present in significantly reduced numbers and showing less elaborate architecture compared with normal cartilage, directly reflecting the failure of normal type II collagen to assemble into proper fibrillar networks.[2][12][41]
The abnormal chondrocyte differentiation that results from ACG2 mutations negatively affects linear bone growth through multiple mechanisms: the altered relationships between chondrocytes prevent them from providing proper mechanical and chemical signals to neighboring cells; the reduction in growth factors produced by abnormally differentiating chondrocytes impairs the endocrine-like actions that normally regulate growth plate function; and the disrupted extracellular matrix fails to bind and present growth factors like Indian hedgehog and BMP ligands in their normal spatiotemporal patterns.[2][12][41] The consequence is a growth plate that is fundamentally unable to direct normal bone elongation, resulting in the severe micromelic dwarfism that characterizes achondrogenesis type II.
Disrupted Extracellular Matrix Composition and Architecture
Beyond the intracellular retention of mutant collagen, the extracellular matrix itself becomes profoundly abnormal in achondrogenesis type II due to the inability of mutant type II collagen to form proper fibrillar networks and interact normally with other matrix components.[2][12][41][45] Histochemical and immunohistochemical analyses of cartilage from ACG2 cases reveal that while the tissue contains types I and II collagen, cartilage proteoglycans (primarily aggrecan), fibronectin, and various glycoconjugates, the normal spatial organization and fibrillar architecture are severely disrupted.[2][14][45] Normal hyaline cartilage derives 95% of its collagenous content from type II collagen, which self-assembles into fibrils that form the tensile load-bearing network of the tissue, but in ACG2, the mutant type II collagen molecules fail to form proper fibrils, and this failure leads to striking changes in matrix ultrastructure.[2][45]
At the ultrastructural level, electron microscopy studies of ACG2 cartilage demonstrate obvious reduction in collagen fibrils throughout the entire growth plate, with the fibrils that do form appearing structurally abnormal, having irregular cross-sections and reduced diameters compared with normal collagen fibrils.[2][41][45] This abnormal fibril morphology reflects the incorporation of defective collagen chains into fibrillar aggregates that lack the precise geometric organization of normal collagen fibrils, which are characterized by a distinctive quarter-stagger alignment pattern that contributes to their mechanical strength and water-binding capacity.[2] The consequence is that the extracellular matrix becomes gelatinous and soft, lacking the structural integrity normally provided by well-organized type II collagen fibrils.[2][3][15][45] This gelatinous matrix cannot properly support the weight of the developing fetus or infant, cannot transmit mechanical forces properly between cells and bones, and cannot bind and present growth factors and other regulatory molecules in their normal concentrations and spatial distributions.
The disorganized extracellular matrix also fails to support proper cell-matrix interactions mediated through integrins and other adhesion molecules. Normal interactions between chondrocytes and type II collagen through integrin β1 trigger signaling cascades that normally suppress chondrocyte hypertrophy and apoptosis through mechanisms involving SMAD1 inhibition and ERK1/2 activation.[7][23] The failure of these interactions due to absent or improperly organized type II collagen deprives chondrocytes of essential suppressive signals, likely contributing to the abnormal chondrocyte differentiation and increased apoptosis observed in ACG2 cartilage. Furthermore, other matrix components like proteoglycans and fibronectin cannot interact properly with abnormally structured type II collagen fibrils, as these interactions are highly dependent on the correct three-dimensional architecture of the collagen scaffold.[56] The result is loss of the normal synergistic interactions between matrix components that confer mechanical resilience, osmotic stability, and biological activity to healthy cartilage tissue.
Key Affected Cell Types and Tissues
Chondrocytes as the Primary Target Cells
Chondrocytes, the specialized cells that produce cartilage matrix, are the cellular populations most critically affected in achondrogenesis type II, as these cells are responsible for producing type II collagen in massive quantities during development and maintaining this crucial protein throughout life.[2][12][41][45] All forms of achondrogenesis preferentially affect skeletal development because chondrocytes are the cells that synthesize the vast majority of the body's type II collagen, and any disruption of this synthesis rapidly cascades into impaired skeletal development.[2][12][41] In addition to the general effects on all chondrocytes, the differentiation stages of chondrocytes show differential vulnerability: proliferating chondrocytes in the growth plate appear particularly affected by ER stress and reduced growth factor signaling, leading to abnormal cell cycle progression and impaired proliferation; pre-hypertrophic and hypertrophic chondrocytes appear to undergo premature or abnormal differentiation, contributing to the disorganized growth plate architecture.[2][12][41]
The fate of hypertrophic chondrocytes in normal development represents a recent area of significant revision in our understanding of skeletal biology. Traditionally, hypertrophic chondrocytes were thought to undergo apoptosis as terminal differentiation, to be followed by vascular invasion and replacement by osteoblasts derived from perichondrial precursors.[22] However, modern lineage-tracing studies using genetic tags to permanently mark and track cells have demonstrated that some hypertrophic chondrocytes survive and directly differentiate into osteoblasts and osteocytes, contributing substantially to the trabecular and cortical bone formed by endochondral ossification.[22] This revised understanding suggests that ACG2 may disrupt not only normal chondrocyte development but also the transition of hypertrophic chondrocytes to osteogenic fates, potentially explaining why ossification is so severely deficient in these infants beyond merely the failure of cartilage matrix production.
Fibroblasts and Other Connective Tissue Cells
While chondrocytes are the primary target cells, type II collagen is also produced by fibroblasts in certain contexts, particularly during wound healing and tissue remodeling.[2][5] In achondrogenesis type II, fibroblasts and other connective tissue cells are affected as secondary targets, as type II collagen from mutant alleles would be incorporated into any tissue attempting to produce this collagen isoform. However, because type II collagen is primarily synthesized in cartilage under normal conditions, systemic effects on fibroblasts appear less prominent than effects specifically on cartilage development. Nonetheless, the vitreous humor of the eye contains substantial amounts of type II collagen, and ocular complications are well-documented in some type II collagenopathies, though the specific ocular involvement in ACG2 may vary due to the lethality of the condition preventing survival to ages where some complications might develop.[27]
Anatomical Locations and Tissue-Specific Manifestations
The Skeletal System: Primary Site of Pathology
The skeletal system represents the primary anatomical location affected by achondrogenesis type II, as cartilage serves as the template for endochondral bone formation throughout the skeleton.[1][4][16][17][25][28] All forms of achondrogenesis feature short arms and legs (micromelia), a narrow chest (thoracic constriction), and underdeveloped lungs (pulmonary hypoplasia), consequences that follow directly from the failure of cartilage to develop normally.[1][4][16][17] In achondrogenesis type II specifically, skeletal findings include markedly reduced or absent ossification of vertebral bodies, sacrum, and pubic bones, while the skull typically shows normal or only slightly reduced ossification, a distinctive pattern that helps differentiate ACG2 from other skeletal dysplasias.[1][3][4][13][15][25] The ribs are characteristically short without fractures (in contrast to achondrogenesis type 1A, where rib fractures are typical), and the costochondral junctions show severe disorganization, reflecting the failure of cartilage to develop properly at the junctures between ribs and costal cartilage.[1][3][4][13][25]
The growth plates of long bones show the pathological changes described previously, with severe disorganization, reduced matrix deposition, and impaired chondrocyte differentiation, resulting in severely shortened long bones with metaphyseal widening visible on radiographs.[1][3][4][15][25] The vertebral column shows characteristic incomplete ossification, with individual vertebral bodies failing to form bone normally and instead consisting primarily of unossified cartilage.[1][3][4][15][25] This incomplete ossification of the vertebral column contributes to spinal instability and potentially impacts the development of neural structures, though the severity of ACG2 typically prevents comprehensive evaluation of neurological consequences. The pelvis is severely hypoplastic, with markedly reduced ossification of the pubic and ischial bones, reflecting global failure of endochondral ossification throughout this region.[1][3][4][13][15][25]
The chest is characteristically small and narrow, with a bell-shaped or barrel appearance in some cases, creating a severely restrictive ribcage that cannot expand normally to accommodate lung development.[1][4][15][25][28] This chest wall deformity represents one of the most clinically significant consequences of ACG2, as it directly leads to the pulmonary hypoplasia that causes respiratory failure and death in most affected infants. The narrowed thorax mechanically restricts lung expansion, preventing the lungs from reaching their normal volume and preventing the proper development of alveolar structures necessary for gas exchange.[28][51][53] Even infants who survive the immediate perinatal period face severe limitations in their pulmonary function due to the restrictive ribcage phenotype.
The Respiratory System: Secondary but Critical Target
While the lungs themselves are not directly affected by type II collagen mutations, they become severely compromised as a consequence of chest wall abnormalities that physically restrict their development, creating a severe mechanical restriction that leads to pulmonary hypoplasia (underdeveloped lungs) that represents the primary cause of death in achondrogenesis type II.[28][51][53][54] The severely narrowed thorax cannot expand to the volume required for normal lung development, and the underdeveloped lungs cannot generate sufficient gas exchange to oxygenate the blood and eliminate carbon dioxide. Affected infants present immediately after birth with severe respiratory distress and require intensive respiratory support, often including high-frequency oscillation ventilation (HFOV) rather than conventional mechanical ventilation, because the abnormal chest mechanics prevent adequate tidal volume generation even with conventional ventilator settings.[3][25][28]
The pathophysiology of respiratory failure in ACG2 involves multiple interconnected factors beyond simple lung hypoplasia. First, the mechanical properties of the ribcage are fundamentally altered due to abnormal cartilage development, resulting in a severely overcompliant chest wall that offers little outward recoil to counterbalance the opposing elastic forces of the lungs, leading to decreased functional residual capacity and tendency toward atelectasis.[51] Second, laryngotracheobronchomalacia (softening of the larynx, trachea, and bronchi due to insufficient cartilaginous support) may develop as a consequence of type II collagen involvement in the structural cartilage of these airways, further compromising airway patency and increasing airway resistance.[51] Third, the severely restricted thoracic volume limits the lung volume that can be achieved even with aggressive mechanical ventilation, creating a fundamental ceiling to oxygenation and ventilation capacity that cannot be overcome through increased ventilator support.[28][51]
Facial and Craniofacial Features
Achondrogenesis Type II features characteristic facial abnormalities that, while not directly involving skeletal development in the same way as the limbs and chest, reflect broader disruptions in cranial and facial cartilage development and tissue differentiation processes. Distinctive facial features include a prominent forehead (frontal bossing), a small chin (micrognathia), and a flattened facial profile, with the small chin particularly noteworthy as it may contribute to airway obstruction and feeding difficulties.[1][4][13][16][25][49] Cleft palate occurs in some cases, reflecting the involvement of type II collagen in palatal cartilage development, and represents an additional anatomical factor that can compromise airway patency and feeding ability.[1][4][16][25][49] In some cases, cystic hygroma (accumulation of lymphatic fluid in the neck region) has been documented prenatally, and increased nuchal thickness and fetal hydrops (generalized body edema) may develop in utero, likely due to obstruction of lymphatic drainage by the severely abnormal skeletal and connective tissue development.[1][4][13][25][49] One case report emphasizes that micrognathia with a flattened facial profile and hydrops fetalis are consistently described as characteristic prenatal ultrasound features of achondrogenesis type II, even though quantitative epidemiological data on the incidence of these specific features remain limited.[49]
Disease Progression and Clinical Timeline
Prenatal Course and Prenatal Diagnosis
Achondrogenesis Type II typically becomes detectable during prenatal imaging as early as 14-17 weeks of gestation using ultrasound screening, when the characteristic skeletal abnormalities first become apparent on ultrasonography.[1][4][16] Early prenatal detection is often triggered by the observation of severe shortening of long bones (micromelia) and abnormal limb proportions, which prompt more detailed skeletal evaluation.[4][49][50][53] As pregnancy progresses, additional findings become apparent, including poor mineralization of the vertebral bodies and pelvis, short ribs, and a characteristically narrow chest with reduced thoracic volume.[49][50][53] In many cases, fetal hydrops develops during the second and third trimesters, manifesting as increased nuchal translucency, skin edema, ascites, and pleural or pericardial effusions, likely due to impaired lymphatic drainage and possibly cardiac compromise secondary to the severe skeletal deformities.[1][4][13][25][49]
Prenatal diagnosis can be confirmed through genetic testing using next-generation sequencing (NGS) of fetal DNA obtained via cordocentesis, amniocentesis, or chorionic villus sampling, which can identify heterozygous COL2A1 mutations.[3][25][49][50] The application of targeted exome sequencing focusing on known skeletal dysplasia genes, or whole exome sequencing, allows rapid molecular confirmation of the diagnosis and enables specific genetic counseling regarding recurrence risk.[3] In some cases, somatic or germline mosaicism has been documented in unaffected parents of affected infants, meaning that parents with a COL2A1 mosaicism may have a recurrence risk higher than the standard 1% de novo risk, potentially up to 5-25% depending on the degree of mosaicism in the parental germline cells.[37][40] Post-mortem imaging with radiography or CT scanning in terminated pregnancies can reveal the characteristic skeletal dysplasia findings and supports precise phenotypic characterization for genetic interpretation.[50]
Neonatal Course and Early Postnatal Period
Infants with achondrogenesis type II born alive or born alive following delayed intrauterine death present at birth with obvious clinical features of severe skeletal dysplasia, including extreme micromelia (very short, abnormally positioned limbs), markedly short stature, a disproportionately large head, prominent forehead, small chin, a small thorax, a prominent/distended abdomen, and severe respiratory distress.[3][25][28][49] The APGAR scores are characteristically low due to severe respiratory depression, with affected infants often unable to achieve spontaneous respiration and requiring immediate endotracheal intubation and mechanical ventilation.[3][25][49] Birth weight may be near normal in some cases (creating the distinctive appearance of a normal-sized head with severely shortened limbs), while length is severely reduced, often at or below the 3rd percentile for gestational age, and head circumference may be at the 90-97th percentile due to the relative macrocephaly characteristic of the condition.[3][25][49]
Immediately after birth, affected infants face a cascade of life-threatening complications directly related to the anatomical abnormalities produced by defective type II collagen.[28][51][53][54] Most critically, respiratory failure develops due to the combination of pulmonary hypoplasia and chest wall restriction, requiring aggressive mechanical ventilation, often including high-frequency oscillation ventilation (HFOV) or other advanced ventilator strategies to achieve adequate gas exchange.[3][25][28][51] Even with aggressive respiratory support, the mechanical limitations of the severely restricted thorax typically prevent adequate oxygenation and ventilation, leading to hypoxemia and hypercarbia (elevated blood carbon dioxide) that progressively worsen over hours to days.[3][25][28] Some infants develop pulmonary hypertension as a consequence of chronic hypoxemia, further compromising cardiac output and oxygen delivery.[3][25] Additional complications may include difficulty establishing peripheral vascular access due to severe generalized edema, necessitating central venous catheterization (umbilical catheter) for intravenous therapy and parenteral nutrition.[3][25][49]
Progression to Death and Characteristic Survival Times
Unfortunately, achondrogenesis type II uniformly results in perinatal lethality, with up to 85% of affected fetuses dying before birth or within the first few hours to days after birth, and virtually no surviving infants beyond the first few weeks of life.[28][53] The prognosis for achondrogenesis type II is universally poor, with death occurring either in utero (as stillbirth), immediately at or within minutes of birth due to apnea or inability to establish respiration, or within hours to days of birth despite intensive medical support.[28][53] The median survival time for liveborn infants with achondrogenesis type II is extremely short, typically in the range of less than 24 hours to several days**, with only rare cases of infants surviving beyond 2-3 weeks, and even then only with continuous intensive respiratory support in the setting of a neonatal intensive care unit.[3][28][49]
The progressive mechanism of death in surviving infants involves inexorable worsening of respiratory failure due to the physical limitations imposed by the severely restricted thorax, which prevents adequate lung expansion regardless of ventilator support, combined with metabolic acidosis developing from tissue hypoxia and anaerobic metabolism. Over a period of hours to days, arterial oxygen saturation progressively declines despite escalating ventilator support, arterial carbon dioxide rises above normal levels (respiratory acidosis), and mixed acidosis develops as anaerobic metabolism generates lactate and other organic acids. The profound hypoxemia triggers multiple organ dysfunction, including cardiac arrhythmias, renal failure, hepatic dysfunction, and activation of coagulation cascades, creating irreversible multi-organ failure. Death typically results from intractable hypoxemia unresponsive to maximal mechanical ventilation and supplemental oxygen, often occurring after several days of futile intensive care support when the family and medical team determine that further aggressive intervention is not beneficial and comfort-focused care is initiated.[28][49]
Cellular Signaling Pathways Dysregulated in Achondrogenesis Type II
The BMP-SMAD1 Signaling Pathway and Loss of Chondrocyte Homeostasis
Recent molecular discoveries have revealed that type II collagen functions as an extracellular signaling molecule that actively suppresses chondrocyte hypertrophy through inhibition of bone morphogenetic protein (BMP) signaling, specifically the BMP-SMAD1 pathway, a finding with major implications for understanding how COL2A1 mutations disrupt normal skeletal development.[7][20][21][23][39][41][42] The BMP signaling pathway is initiated when BMP ligands bind to type I and type II BMP receptors on the chondrocyte surface, triggering receptor autophosphorylation and leading to phosphorylation of intracellular receptor-regulated SMADs (particularly SMAD1/5), which then form complexes with the common mediator SMAD (SMAD4), translocate to the nucleus, and activate transcription of BMP target genes involved in chondrocyte hypertrophy and osteoblast differentiation.[21][24][39] This BMP-SMAD1 signaling pathway normally promotes chondrocyte hypertrophy, mineralization of cartilage matrix, and progression toward endochondral ossification, making it essential for normal bone development.[21][24][39][42]
However, type II collagen actively suppresses this BMP-SMAD1 pathway through a sophisticated mechanism involving the major type II collagen receptor integrin β1 (ITGB1), which upon binding to type II collagen competes with BMP receptors for binding to SMAD1, thereby preventing SMAD1 phosphorylation and nuclear translocation.[7][23] Additionally, type II collagen activation of ITGB1 triggers ERK1/2 phosphorylation through downstream kinase cascades, and ERK1/2 phosphorylation further represses BMP-SMAD1 pathway activation through direct interaction with SMAD1.[7][23] The net result is that normal type II collagen in properly assembled fibrils acts as a potent suppressor of BMP-SMAD1 signaling, maintaining chondrocytes in a proliferative state and preventing premature hypertrophy.[7][23][41] This regulatory mechanism appears critical for proper growth plate structure and function, as conditional deletion of ITGB1 in chondrocytes results in accelerated chondrocyte hypertrophy and growth plate defects similar to those observed in type II collagenopathies.[7][23]
In achondrogenesis type II, the failure of mutant type II collagen to assemble into proper fibrils and to be present in adequate quantities in the extracellular matrix means that chondrocytes are deprived of the suppressive signals normally provided by type II collagen-integrin β1 interactions.[7][12][23][41] The consequence is that BMP-SMAD1 pathway signaling becomes abnormally elevated in ACG2 chondrocytes, leading to accelerated and aberrant hypertrophic differentiation, increased expression of hypertrophic markers, enhanced osteogenic gene expression, and increased chondrocyte apoptosis.[7][12][23][41] This loss of type II collagen-mediated suppression of BMP signaling represents a major mechanism through which ACG2 mutations disrupt normal endochondral ossification: rather than chondrocytes proceeding through a carefully orchestrated program of proliferation, pre-hypertrophic transition, hypertrophic differentiation, and then either apoptosis or osteogenic transformation, instead they undergo dysregulated hypertrophic differentiation driven by unopposed BMP-SMAD1 signaling. The aberrant hypertrophy, combined with the abnormal extracellular matrix, results in disorganized growth plates incapable of directing normal bone elongation.
Effects on Chondrocyte Proliferation and Metabolic Function
Beyond the effects on hypertrophy regulation, loss of type II collagen also impairs chondrocyte proliferation through multiple mechanisms. Normal chondrocyte proliferation is regulated by a complex interplay of positive signals (including Indian hedgehog/Ihh signaling, parathyroid hormone-related peptide/PTHrP signaling, FGF signaling in specific contexts, and BMP signaling in specific doses) and negative signals (including high-dose FGF signaling and type II collagen-mediated suppression of excess BMP signaling).[39][42] When type II collagen is absent or abnormally structured, chondrocytes lose a critical brake on BMP signaling that would normally constrain excessive proliferation and ensure that chondrocyte division occurs at appropriate rates matched to the developmental stage.[7][23][39][41][42] Additionally, the endoplasmic reticulum stress triggered by accumulation of misfolded procollagen directly reduces proliferation rates at the growth plates through mechanisms involving activation of the unfolded protein response and phosphorylation of eIF2α, leading to global translation attenuation and cell cycle arrest.[2][12][41]
The consequence is a growth plate where chondrocytes fail to proliferate normally despite having growth-promoting signals, creating a population bottleneck that severely limits the number of cells available to undergo differentiation and contribute to bone elongation. The reduction in chondrocyte number, combined with the impaired progression through normal differentiation stages, results in severely shortened long bones and overall dwarfism.[2][12][41]
Disrupted Indian Hedgehog and Parathyroid Hormone-Related Peptide Signaling
The Indian hedgehog/parathyroid hormone-related peptide (Ihh/PTHrP) negative feedback loop represents another critical regulatory system disrupted in achondrogenesis type II.[2][39][41][42] In normal growth plates, Indian hedgehog is secreted by pre-hypertrophic and hypertrophic chondrocytes and acts to stimulate expression of parathyroid hormone-related peptide in perichondrial cells and in the resting zone chondrocytes, and parathyroid hormone-related peptide then diffuses back into the growth plate where it suppresses further Ihh expression and maintains a proliferative chondrocyte phenotype, preventing premature hypertrophy.[2][39][41][42] This feedback loop is essential for maintaining the normal thickness and organized structure of the growth plate's proliferative zone and for ensuring that chondrocytes do not undergo hypertrophic differentiation prematurely.[39][42]
The endoplasmic reticulum stress and abnormal chondrocyte differentiation occurring in ACG2 appear to severely disrupt the normal expression and spatial organization of Ihh and PTHrP, as documented by marked reduction or complete absence of Ihh mRNA expression in growth plates of affected individuals.[2][12][41] The loss of Ihh expression disrupts the feedback loop that normally maintains proliferative chondrocytes and prevents premature hypertrophy, leading to disorganization of the proliferative zone and inappropriate hypertrophic differentiation of chondrocytes that should remain proliferative.[2][12][41] This disruption of the Ihh/PTHrP feedback loop represents an additional mechanism through which ACG2 mutations produce the characteristic disorganized growth plate phenotype with loss of normal columnar architecture and impaired bone elongation.
Genotype-Phenotype Correlations in Achondrogenesis Type II
Spectrum of COL2A1 Mutations and Their Clinical Consequences
Achondrogenesis Type II demonstrates remarkable genotypic diversity, with more than 570 different mutations documented in the COL2A1 gene in scientific literature and public databases, and this genetic diversity correlates imperfectly with clinical phenotype severity.[2][3][12][26][33][43] Understanding the genotype-phenotype correlation is critical for explaining why some fetuses with ACG2 mutations might be stillborn with severe hydrops while others survive the immediate neonatal period with intensive support, though all uniformly have lethal outcomes eventually.[33] The most common mutations (over 70% of cases) are missense mutations, particularly those resulting in glycine substitutions in the Gly-X-Y repeat of the triple-helical domain, and these glycine substitutions produce the most severe phenotypes through dominant-negative mechanisms.[2][12][26][33]
Among missense mutations causing glycine substitutions, glycine-to-serine substitutions appear to produce slightly milder skeletal phenotypes compared with glycine-to-aspartate, glycine-to-arginine, or glycine-to-valine substitutions, suggesting that the specific chemical properties of the substituted amino acid influence the degree of triple-helix destabilization and thus the severity of the skeletal dysplasia.[2][12][26][31][33][34] For example, in one comparative analysis, glycine-to-serine substitutions resulted in alternating zones producing severer and milder skeletal phenotypes, whereas glycine-to-nonserine residue substitutions exclusively created more severe phenotypes without such alternating severity zones.[26] However, even the "milder" glycine-to-serine mutations in ACG2 still produce lethal skeletal dysplasia indistinguishable from other ACG2 mutations in terms of lethality and clinical course, suggesting that phenotypic variation among ACG2 cases is more about variation in survival time and degree of organ dysfunction rather than fundamental differences in skeletal dysplasia severity.[26][33][43]
Missense mutations affecting positions within the C-propeptide domain of type II collagen (the C-terminal propeptide region) produce distinctive skeletal phenotypes that differ from triple-helical domain mutations, characterized prominently by brachydactyly (abnormally short digits) and metaphyseal involvement in addition to spinal abnormalities, reflecting the distinct role of the C-propeptide in procollagen assembly and possible specific signaling functions of this domain.[2][9][33][36] The C-propeptide also known as chondrocalcin appears to play a role in growth plate development and potentially in regulation of chondrocyte proliferation, and mutations disrupting this region produce phenotypes with more prominent short-digit abnormalities than typical triple-helical mutations.[2][9]
Clinical Variability Among ACG2 Cases
Despite the severe and uniformly lethal nature of achondrogenesis type II, some degree of clinical variability exists in the severity of skeletal dysplasia, the degree of organ involvement (particularly respiratory), and the timing and rapidity of progression to respiratory failure.[33][43][49] Some ACG2 cases present with more severe in-utero manifestations including very early-onset hydrops fetalis and fetal demise in mid-pregnancy, while others allow survival to term or near-term with delivery of a liveborn but severely compromised infant.[3][25][28][49] Some fetuses present with mild to absent hydrops and normal skull ossification but severe limb shortening and vertebral non-ossification, while others develop prominent hydrops with cystic hygroma and generalized edema.[3][25][49] Once born, some ACG2 infants survive only hours due to immediate respiratory failure refractory to all interventions, while others survive for days or even up to several weeks with intensive respiratory support before ultimately succumbing to respiratory failure or multi-organ dysfunction.[3][25][28][49]
This clinical variability likely reflects inter-individual differences in the specific COL2A1 mutation and its precise effects on collagen structure, differences in the timing of mutation occurrence during germ cell or early embryonic development (affecting the degree of mosaicism if present), and potentially differences in genetic background factors affecting modifier gene expression and physiological resilience.[33][37][40][43] One documented case of achondrogenesis type II in a liveborn infant survived for 25 days with intensive support despite having a novel mutation, illustrating that rare infants with severe ACG2 can survive beyond the immediate neonatal period with dedicated intensive care.[3][25] However, survival beyond a few weeks appears extraordinarily rare, and no documented cases of long-term survival into childhood exist, making ACG2 uniformly fatal in the short term despite variation in the exact timing and pace of decline.[28][49]
Conclusion: Integrating Mechanisms from Genomics to Clinical Phenotype
Achondrogenesis Type II represents one of the most severe skeletal dysplasias, caused by heterozygous COL2A1 gene mutations that disrupt type II collagen structure and function through dominant-negative mechanisms.[1][2][3][4] The fundamental pathophysiology involves the production of structurally abnormal type II collagen molecules that fail to form stable triple helices, become retained in the endoplasmic reticulum triggering ER stress, and fail to assemble into functional fibrillar networks in the extracellular matrix.[2][12][41] This primary molecular defect cascades into profound disruption of multiple interconnected cellular and developmental processes: chondrocytes experience ER stress that reduces proliferation and triggers apoptosis; abnormal chondrocyte differentiation disrupts normal growth plate architecture; dysregulated BMP-SMAD1 signaling due to loss of type II collagen-mediated suppression causes aberrant hypertrophy; disruption of Indian hedgehog and PTHrP signaling further disorganizes the growth plate; and the gelatinous, poorly organized extracellular matrix fails to provide proper mechanical support or present growth factors in normal spatial distributions.[2][7][12][23][39][41][42]
The consequence of these dysregulated molecular and cellular processes is severe micromelic dwarfism, narrow thorax with underdeveloped lungs, incomplete ossification of vertebral bodies and pelvis, and characteristic facial features including frontal bossing and micrognathia.[1][3][4][13][25][28] Most critically from a clinical perspective, the narrow thorax and pulmonary hypoplasia create a fatal combination in which the severely restricted ribcage physically prevents adequate lung expansion, leading to respiratory failure that is uniformly fatal within hours to days of birth in most cases, with rare exceptions allowing survival for several weeks with intensive respiratory support but inevitable progression to fatal respiratory failure.[3][28][49][53] The identification of novel COL2A1 mutations through next-generation sequencing continues to expand our understanding of the genotype-phenotype correlations in this condition and increasingly provides opportunities for molecular diagnosis during pregnancy, enabling informed genetic counseling and family planning despite the universally lethal nature of the condition.[3][43][49]