ADan amyloidosis
ADan amyloidosis is an autosomal dominant cerebral amyloidosis caused by pathogenic ITM2B variants that generate the amyloidogenic ADan peptide. The disease was first recognized as familial Danish dementia and is characterized by cataracts, hearing loss, progressive ataxia, and dementia together with widespread cerebral amyloid angiopathy and neurofibrillary degeneration.
Ask OpenScientist
Ask a research question about ADan amyloidosis. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (1 reference)
Mechanistic Hypotheses
2Show evidence (1 reference)
Show evidence (2 references)
Pathophysiology
7Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (3 references)
Histopathology
2Show evidence (2 references)
Show evidence (1 reference)
Pathograph
Phenotypes
8Ear 1
Show evidence (1 reference)
Eye 1
Show evidence (1 reference)
Nervous System 5
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Other 1
Show evidence (1 reference)
Genetic Associations
1Show evidence (1 reference)
Medical Actions
5Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Biochemical Markers
1Show evidence (1 reference)
Show evidence (1 reference)
Differential Diagnoses
2Conditions with similar clinical presentations that must be differentiated from ADan amyloidosis:
Show evidence (1 reference)
- ABri rather than ADan amyloid peptide
- Prominent spastic tetraparesis or spastic paraparesis in FBD
- FDD has earlier ocular and hearing manifestations and lacks the spastic paraparesis pattern emphasized for FBD
Show evidence (2 references)
Experimental Models
1Show evidence (1 reference)
Source YAML
click to showname: ADan amyloidosis
creation_date: '2026-04-14T12:05:00Z'
updated_date: '2026-04-15T01:00:00Z'
category: Mendelian
synonyms:
- familial Danish dementia
- Danish dementia
- heredopathia ophthalmo-oto-encephalica
description: >-
ADan amyloidosis is an autosomal dominant cerebral amyloidosis caused by
pathogenic ITM2B variants that generate the amyloidogenic ADan peptide. The
disease was first recognized as familial Danish dementia and is characterized
by cataracts, hearing loss, progressive ataxia, and dementia together with
widespread cerebral amyloid angiopathy and neurofibrillary degeneration.
disease_term:
preferred_term: ADan amyloidosis
term:
id: MONDO:0007297
label: ADan amyloidosis
parents:
- hereditary disease
- amyloidosis
inheritance:
- name: Autosomal dominant inheritance
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
description: >-
ADan amyloidosis/familial Danish dementia segregates as an autosomal
dominant ITM2B-related disorder.
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD), also known as heredopathia ophthalmo-oto-encephalica, is an autosomal dominant disorder characterized by cataracts, deafness, progressive ataxia, and dementia.
explanation: >-
This directly establishes autosomal dominant inheritance for familial
Danish dementia/ADan amyloidosis.
progression:
- phase: Natural history
age_range: third to sixth decade
duration: Progressive over decades
notes: >-
The published Danish kindred shows staged progression from cataracts in the
third decade to hearing impairment, cerebellar signs, dementia in later
adulthood, and death in the fifth or sixth decade.
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Clinically, FDD is characterized by the development and progression of cataracts during the third decade of life (Strömgren et al. 1970). Hearing impairment was reported to appear 10–20 years following the development of ocular problems. Severe to total perceptive hearing loss developed by the fifth decade of life, with vestibular reflex disturbances, slurred speech, and a swaying gait (Strömgren et al. 1970; Strömgren 1981). Patients with FDD developed cerebellar ataxia with intention tremor in their fourth decade of life, however, unlike FBD cases, did not develop spastic para-paresis. Progressive dementia probably begins in the sixth decade and may be associated with paranoid reactions and temporal disturbances of consciousness. The majority of patients succumb within the fifth or sixth decade of life.
explanation: >-
This review summarizes the staged natural history of the Danish kindred.
mechanistic_hypotheses:
- hypothesis_group_id: canonical_adan_amyloid_angiopathy_tau_model
hypothesis_label: Canonical ADan Amyloid Angiopathy and Tau Model
status: CANONICAL
description: >-
The human disease mechanism centers on the ITM2B Danish mutation generating
ADan peptide, with widespread cerebral amyloid angiopathy, parenchymal ADan
protein deposits, and severe neurofibrillary degeneration.
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD) is pathologically characterized by widespread cerebral amyloid angiopathy (CAA), parenchymal protein deposits, and neurofibrillary degeneration.
explanation: >-
Human neuropathology establishes the canonical ADan amyloid angiopathy,
parenchymal-deposit, and tau-degeneration axis.
- hypothesis_group_id: bri2_synaptic_loss_app_model
hypothesis_label: BRI2 Synaptic Loss and APP-Processing Model
status: EMERGING
description: >-
Model-organism and limited human-brain evidence support a superimposed
mature-BRI2 loss-of-function mechanism with impaired glutamatergic
transmission and increased APP processing; this is separated from the
canonical human ADan deposition pathology.
notes: >-
Classified as emerging because the synaptic-transmission evidence is mainly
from knock-in mice, and the human APP-processing study used very limited
postmortem FDD material.
evidence:
- reference: PMID:33172889
reference_title: Danish and British dementia ITM2b/BRI2 mutations reduce BRI2 protein stability and impair glutamatergic synaptic transmission.
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Collectively, the data show that FDD and FBD mutations cause a reduction of BRI2 levels and function at synapses, which results in reduced glutamatergic transmission.
explanation: >-
Knock-in mouse data support a distinct synaptic loss-of-function model
downstream of pathogenic ITM2B mutations.
- reference: PMID:21841249
reference_title: Increased AbetaPP processing in familial Danish dementia patients.
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
The levels of soluble Aβ42 were increased in FDD and AD brains compared to aging brains (FDD versus Con., 11-fold increase; AD1 versus Con., 4-fold increase; AD2 versus Con., 14-fold increase, Fig. 3A).
explanation: >-
Limited human FDD brain data support increased APP/Abeta processing, but
the evidence is not as mature as the canonical ADan pathology model.
pathophysiology:
- name: ITM2B frameshift generates the ADan amyloidogenic peptide
description: >-
A decamer duplication near the normal stop codon of ITM2B creates an
extended precursor from which the amyloidogenic ADan peptide is released.
genes:
- preferred_term: ITM2B
term:
id: hgnc:6174
label: ITM2B
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The decamer duplication mutation produces a frame-shift in the BRI sequence generating a larger-than-normal precursor protein, of which the amyloid subunit (designated ADan) comprises the last 34 C-terminal amino acids.
explanation: >-
This directly supports generation of the ADan peptide as the initiating
molecular lesion.
downstream:
- target: Cerebral amyloid deposition
description: Amyloidogenic ADan peptide is deposited in brain vessels and parenchyma.
causal_link_type: DIRECT
hypothesis_groups:
- canonical_adan_amyloid_angiopathy_tau_model
- target: Synaptic BRI2 loss
description: The same ITM2B mutation also reduces availability of functional mature BRI2 at synapses.
causal_link_type: DIRECT
hypothesis_groups:
- bri2_synaptic_loss_app_model
- target: Cataract
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
description: The ITM2B frameshift syndrome includes early ocular involvement with cataracts, but the tissue-level ocular mechanism is not yet separated in this pathograph.
- target: Sensorineural hearing impairment
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
description: The same ITM2B frameshift syndrome includes progressive otologic involvement with perceptive hearing loss, although the cochlear mechanism remains unresolved here.
- name: Cerebral amyloid deposition
description: >-
ADan accumulates in leptomeninges, vessel walls, and brain parenchyma,
producing cerebral amyloid angiopathy and parenchymal deposits; Abeta can
co-deposit in a subset of vascular and parenchymal lesions.
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD) is pathologically characterized by widespread cerebral amyloid angiopathy (CAA), parenchymal protein deposits, and neurofibrillary degeneration.
explanation: >-
This directly supports the core tissue pathology of ADan amyloidosis.
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Abeta was also present in a proportion of both vascular and parenchymal lesions.
explanation: >-
This supports Falcon's emphasis that ADan deposition may be accompanied by
Abeta co-deposition in affected brain lesions.
downstream:
- target: Cerebral amyloid angiopathy
description: ADan deposition in leptomeningeal and cerebral vessel walls produces the cerebral amyloid angiopathy tissue phenotype.
causal_link_type: DIRECT
hypothesis_groups:
- canonical_adan_amyloid_angiopathy_tau_model
- target: Neurofibrillary degeneration
description: Amyloid deposition is accompanied by severe downstream tau pathology and neurodegeneration.
hypothesis_groups:
- canonical_adan_amyloid_angiopathy_tau_model
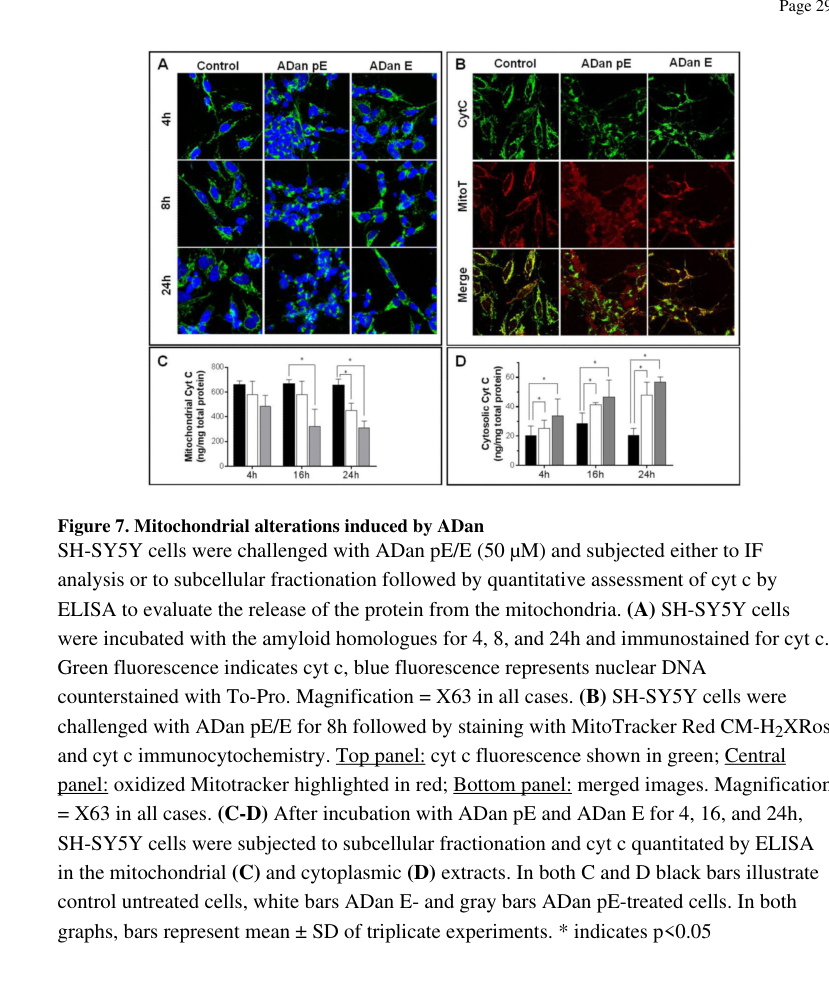
- target: ADan-induced mitochondrial oxidative apoptosis
description: >-
Amyloidogenic ADan species can trigger neuronal-cell oxidative stress,
mitochondrial cytochrome c release, mitochondrial membrane-potential loss,
and caspase-dependent apoptosis in vitro.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
evidence:
- reference: PMID:26459115
reference_title: Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial Danish dementia ADan amyloid.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
The data presented herein demonstrates that ADan neurotoxicity takes place through a mechanism involving many components of intrinsic apoptosis, including high levels of ROS generation, cyt c release into the cytoplasm, disruption of mitochondrial membrane potential, and final activation of terminal caspase-3.
explanation: >-
Differentiated SH-SY5Y cell data support a downstream toxic pathway from
ADan amyloid species to oxidative mitochondrial apoptosis.
- name: Neurofibrillary degeneration
description: >-
Amyloid deposition is accompanied by severe downstream tau pathology and
progressive neurodegeneration.
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD) is pathologically characterized by widespread cerebral amyloid angiopathy (CAA), parenchymal protein deposits, and neurofibrillary degeneration.
explanation: >-
This directly supports neurofibrillary degeneration as a downstream
tissue-level consequence of ADan deposition.
downstream:
- target: Dementia
description: Progressive tau-associated neurodegeneration contributes to dementia.
causal_link_type: DIRECT
- target: Ataxia
description: Widespread neurodegeneration contributes to the progressive ataxic phenotype.
causal_link_type: DIRECT
- target: Intention tremor
description: Cerebellar neurodegeneration in familial Danish dementia produces the intention tremor component of the adult ataxic syndrome.
causal_link_type: DIRECT
- target: Dysarthria
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- cerebellar and vestibular neurologic involvement
description: Progressive neurodegeneration contributes to slurred speech and dysarthria during the neurologic phase.
- target: Psychosis
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- progressive dementia
- temporal disturbances of consciousness
description: Late neurodegeneration and dementia can be accompanied by paranoid reactions captured as psychosis.
- name: Synaptic BRI2 loss
description: >-
Danish-dementia ITM2B mutations reduce mature BRI2 abundance at synapses.
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
evidence:
- reference: PMID:33172889
reference_title: Danish and British dementia ITM2b/BRI2 mutations reduce BRI2 protein stability and impair glutamatergic synaptic transmission.
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
In vivo and in vitro studies show that both pathogenic mutations alter maturation of BRI2 resulting in reduced levels of functional mature BRI2 protein at synapses.
explanation: >-
Knock-in mouse and cellular data support a distinct synaptic-loss-of-function
mechanism downstream of pathogenic ITM2B mutations.
downstream:
- target: Impaired glutamatergic transmission
description: Reduced mature synaptic BRI2 impairs excitatory neurotransmission.
causal_link_type: DIRECT
hypothesis_groups:
- bri2_synaptic_loss_app_model
- target: Increased APP processing and Abeta burden
description: >-
BRI2 normally inhibits amyloid-beta precursor protein processing; reduced
mature BRI2 in FDD models and human brain is associated with increased
APP-derived metabolites including Abeta.
causal_link_type: DIRECT
hypothesis_groups:
- bri2_synaptic_loss_app_model
evidence:
- reference: PMID:21841249
reference_title: Increased AbetaPP processing in familial Danish dementia patients.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We find that the levels of several AβPP metabolites, including Aβ, are significantly increased in the brain sample derived from an FDD patient.
explanation: >-
Human FDD brain data support increased APP/Abeta processing downstream
of reduced BRI2 function.
- name: Impaired glutamatergic transmission
description: >-
Loss of functional mature BRI2 at synapses impairs excitatory
glutamatergic neurotransmission.
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: synaptic transmission, glutamatergic
term:
id: GO:0035249
label: synaptic transmission, glutamatergic
modifier: DECREASED
evidence:
- reference: PMID:33172889
reference_title: Danish and British dementia ITM2b/BRI2 mutations reduce BRI2 protein stability and impair glutamatergic synaptic transmission.
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Collectively, the data show that FDD and FBD mutations cause a reduction of BRI2 levels and function at synapses, which results in reduced glutamatergic transmission.
explanation: >-
This directly links mutant BRI2 loss at synapses to impaired
glutamatergic transmission.
downstream:
- target: Dementia
description: >-
Impaired excitatory neurotransmission likely contributes to the
progressive cognitive decline of familial Danish dementia.
causal_link_type: DIRECT
hypothesis_groups:
- bri2_synaptic_loss_app_model
- name: Increased APP processing and Abeta burden
description: >-
Loss of mature BRI2 function increases amyloid-beta precursor protein
processing and Abeta metabolite burden in FDD model systems and a human FDD
brain sample, providing a mechanistic bridge between ITM2B-related dementia
and Alzheimer-like amyloid biology.
biological_processes:
- preferred_term: amyloid precursor protein metabolic process
term:
id: GO:0042982
label: amyloid precursor protein metabolic process
modifier: INCREASED
evidence:
- reference: PMID:21841249
reference_title: Increased AbetaPP processing in familial Danish dementia patients.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The levels of soluble Aβ42 were increased in FDD and AD brains compared to aging brains (FDD versus Con., 11-fold increase; AD1 versus Con., 4-fold increase; AD2 versus Con., 14-fold increase, Fig. 3A).
explanation: >-
This human brain analysis quantifies increased soluble Abeta42 in FDD.
- reference: PMID:21841249
reference_title: Increased AbetaPP processing in familial Danish dementia patients.
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Strikingly, AβPP haplodeficiency prevents memory and synaptic dysfunctions in FDDKI mice, supporting the claim that Danish dementia is mediated, like FAD, through toxic AβPP products [17].
explanation: >-
Mouse genetic-modifier data support APP-derived products as mediators of
synaptic and memory phenotypes in FDD models.
downstream:
- target: Cerebral amyloid deposition
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
hypothesis_groups:
- bri2_synaptic_loss_app_model
description: >-
Increased APP processing provides Abeta that co-deposits with ADan in
some vascular and parenchymal lesions.
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Abeta was also present in a proportion of both vascular and parenchymal lesions.
explanation: >-
This links increased Abeta biology to observed Abeta co-deposition in FDD
lesions.
- name: ADan-induced mitochondrial oxidative apoptosis
description: >-
Pyroglutamate-modified ADan is neurotoxic in differentiated SH-SY5Y cells,
inducing reactive oxygen species, cytochrome c release, mitochondrial
membrane-potential loss, and caspase-3 activation.
biological_processes:
- preferred_term: response to oxidative stress
term:
id: GO:0006979
label: response to oxidative stress
modifier: INCREASED
- preferred_term: intrinsic apoptotic signaling pathway
term:
id: GO:0097193
label: intrinsic apoptotic signaling pathway
modifier: INCREASED
evidence:
- reference: PMID:26459115
reference_title: Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial Danish dementia ADan amyloid.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
ADan pE provoked DNA fragmentation after only 8 hour peptide challenge, with increased and statistically significant levels of nucleosome formation observed following 24h treatment.
explanation: >-
This supports apoptotic toxicity from pyroglutamate-modified ADan in a
neuronal cell model.
- reference: PMID:26459115
reference_title: Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial Danish dementia ADan amyloid.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
ADan pE challenge resulted, in addition to the release of cytochrome c to the cytoplasm, in a concurrent loss of mitochondrial membrane potential, indicated by the diffuse MitoTracker staining and poor co-localization of both signals (Figure 7B, bottom panel).
explanation: >-
This supports mitochondrial cytochrome c release and membrane-potential
loss as part of ADan toxicity.
- reference: PMID:26459115
reference_title: Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial Danish dementia ADan amyloid.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
In cells challenged with either ADan pE or ADan E, the ROS-scavenging ability of Trolox almost completely abolished caspase-3 activation, an indicator of its protective effect from ADan-induced toxicity.
explanation: >-
Trolox rescue supports oxidative stress upstream of caspase activation in
this in vitro mechanism.
phenotypes:
- name: Cataract
category: Ophthalmologic
frequency: FREQUENT
description: >-
Early cataracts are part of the classic familial Danish dementia syndrome.
phenotype_term:
preferred_term: Cataract
term:
id: HP:0000518
label: Cataract
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD), also known as heredopathia ophthalmo-oto-encephalica, is an autosomal dominant disorder characterized by cataracts, deafness, progressive ataxia, and dementia.
explanation: >-
This directly lists cataracts as a defining clinical manifestation.
- name: Sensorineural hearing impairment
category: Otologic
frequency: FREQUENT
description: >-
Progressive hearing loss is part of the core syndromic presentation.
phenotype_term:
preferred_term: Sensorineural hearing impairment
term:
id: HP:0000407
label: Sensorineural hearing impairment
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD), also known as heredopathia ophthalmo-oto-encephalica, is an autosomal dominant disorder characterized by cataracts, deafness, progressive ataxia, and dementia.
explanation: >-
This directly supports hearing loss as part of the syndrome phenotype.
- name: Ataxia
category: Neurologic
frequency: FREQUENT
description: >-
Progressive ataxia reflects cerebellar and broader neurodegenerative
involvement.
phenotype_term:
preferred_term: Ataxia
term:
id: HP:0001251
label: Ataxia
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD), also known as heredopathia ophthalmo-oto-encephalica, is an autosomal dominant disorder characterized by cataracts, deafness, progressive ataxia, and dementia.
explanation: >-
This directly identifies progressive ataxia as a core clinical feature.
- name: Intention tremor
category: Neurologic
description: >-
Intention tremor accompanies the adult cerebellar syndrome in familial
Danish dementia.
phenotype_term:
preferred_term: Intention tremor
term:
id: HP:0002080
label: Intention tremor
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Patients with FDD developed cerebellar ataxia with intention tremor in their fourth decade of life, however, unlike FBD cases, did not develop spastic para-paresis.
explanation: >-
This directly supports intention tremor as part of the FDD cerebellar
phenotype.
- name: Dysarthria
category: Neurologic
description: >-
Slurred speech is reported during progression after ocular and hearing
manifestations.
phenotype_term:
preferred_term: Dysarthria
term:
id: HP:0001260
label: Dysarthria
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Severe to total perceptive hearing loss developed by the fifth decade of life, with vestibular reflex disturbances, slurred speech, and a swaying gait (Strömgren et al. 1970; Strömgren 1981).
explanation: >-
The review reports slurred speech in the FDD natural history, supporting a
dysarthria phenotype annotation.
- name: Dementia
category: Neurologic
frequency: FREQUENT
description: >-
Progressive cognitive decline is a defining late neurodegenerative
manifestation.
phenotype_term:
preferred_term: Dementia
term:
id: HP:0000726
label: Dementia
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD), also known as heredopathia ophthalmo-oto-encephalica, is an autosomal dominant disorder characterized by cataracts, deafness, progressive ataxia, and dementia.
explanation: >-
This directly supports dementia as one of the defining clinical outcomes.
- name: Psychosis
category: Psychiatric
description: >-
Paranoid reactions may accompany late progressive dementia in familial
Danish dementia.
phenotype_term:
preferred_term: Psychosis
term:
id: HP:0000709
label: Psychosis
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Progressive dementia probably begins in the sixth decade and may be associated with paranoid reactions and temporal disturbances of consciousness.
explanation: >-
The review reports paranoid reactions during the dementia stage,
supporting a psychosis-related phenotype annotation.
- name: Cerebral amyloid angiopathy
category: Neurologic
frequency: FREQUENT
description: >-
Vascular amyloid deposition is a defining ADan amyloidosis tissue phenotype
and overlaps with the broader cerebral amyloid angiopathy spectrum.
phenotype_term:
preferred_term: Cerebral amyloid angiopathy
term:
id: HP:0011970
label: Cerebral amyloid angiopathy
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD) is pathologically characterized by widespread cerebral amyloid angiopathy (CAA), parenchymal protein deposits, and neurofibrillary degeneration.
explanation: >-
This directly supports cerebral amyloid angiopathy as part of the ADan
amyloidosis phenotype/pathology spectrum.
histopathology:
- name: Cerebral Amyloid Angiopathy and Parenchymal ADan Deposits
finding_term:
preferred_term: Parenchymal and vascular amyloid deposition
term:
id: NCIT:C54018
label: Amyloid Deposition
description: >-
Widespread ADan deposition involves leptomeninges, cerebral blood vessels,
and brain parenchyma, producing a cerebral amyloid angiopathy-dominant
pathology with parenchymal pre-amyloid lesions.
diagnostic: true
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We showed that ADan is widely distributed in the central nervous system (CNS) in the leptomeninges, blood vessels, and parenchyma.
explanation: >-
This supports the tissue distribution of ADan deposits in FDD.
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
A predominance of parenchymal pre-amyloid (non-fibrillary) lesions was found.
explanation: >-
This distinguishes the parenchymal lesion type observed in FDD brain.
- name: Neurofibrillary Degeneration with PHF-like Tau
description: >-
Severe tau neurofibrillary pathology accompanies ADan/Abeta deposition and
has a paired-helical-filament tau immunoblot pattern similar to Alzheimer
disease.
diagnostic: false
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
There was severe neurofibrillary pathology, and tau immunoblotting revealed a triplet electrophoretic migration pattern comparable with PHF-tau.
explanation: >-
This supports severe neurofibrillary degeneration and PHF-like tau
biochemistry as part of FDD neuropathology.
biochemical:
- name: ADan amyloid peptide deposition
presence: PRESENT
context: >-
ADan peptide in CNS leptomeninges, vessel walls, and parenchyma is the
defining tissue biomarker of familial Danish dementia/ADan amyloidosis.
readouts:
- target: Cerebral amyloid deposition
relationship: READOUT_OF
direction: PRESENT_ABSENT
endpoint_context: DIAGNOSTIC
interpretation: >-
Detection of ADan in CNS vessels and parenchyma reports the defining ADan
amyloid deposition mechanism.
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We showed that ADan is widely distributed in the central nervous system (CNS) in the leptomeninges, blood vessels, and parenchyma.
explanation: >-
Human neuropathology supports ADan peptide distribution as a diagnostic
readout of the cerebral deposition mechanism.
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We showed that ADan is widely distributed in the central nervous system (CNS) in the leptomeninges, blood vessels, and parenchyma.
explanation: >-
This supports ADan peptide deposition as the defining biochemical/tissue
marker of ADan amyloidosis.
genetic:
- name: ITM2B
association: Gain-of-toxic-fragment / loss-of-function
gene_term:
preferred_term: ITM2B
term:
id: hgnc:6174
label: ITM2B
notes: >-
The Danish mutation in ITM2B creates the amyloidogenic ADan peptide and
also reduces normal mature BRI2 availability.
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Molecular genetic analysis of the BRI gene in the Danish kindred showed a different defect, namely the presence of a 10-nt duplication (795-796insTTTAATTTGT) between codons 265 and 266, one codon before the normal stop codon 267.
explanation: >-
This directly identifies the pathogenic ITM2B/BRI mutation in familial
Danish dementia.
environmental: []
treatments:
- name: Genetic counseling and cascade testing
description: >-
Genetic counseling and family testing are relevant because ADan amyloidosis
is an autosomal dominant, gene-defined ITM2B disorder.
treatment_term:
preferred_term: genetic counseling
term:
id: MAXO:0000079
label: genetic counseling
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Familial Danish dementia (FDD), also known as heredopathia ophthalmo-oto-encephalica, is an autosomal dominant disorder characterized by cataracts, deafness, progressive ataxia, and dementia.
explanation: >-
Autosomal dominant inheritance and molecular diagnosis support genetic
counseling and cascade testing in affected families.
- name: Supportive multidisciplinary care
description: >-
No disease-modifying treatment is established; care is supportive and may
involve ophthalmology, audiology, neurology, rehabilitation, and dementia
management matched to the staged manifestations.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
target_phenotypes:
- preferred_term: Cataract
term:
id: HP:0000518
label: Cataract
- preferred_term: Sensorineural hearing impairment
term:
id: HP:0000407
label: Sensorineural hearing impairment
- preferred_term: Ataxia
term:
id: HP:0001251
label: Ataxia
- preferred_term: Dementia
term:
id: HP:0000726
label: Dementia
- preferred_term: Psychosis
term:
id: HP:0000709
label: Psychosis
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
Clinically, FDD is characterized by the development and progression of cataracts during the third decade of life (Strömgren et al. 1970). Hearing impairment was reported to appear 10–20 years following the development of ocular problems. Severe to total perceptive hearing loss developed by the fifth decade of life, with vestibular reflex disturbances, slurred speech, and a swaying gait (Strömgren et al. 1970; Strömgren 1981).
explanation: >-
The staged visual, hearing, speech, and gait manifestations support the
need for multidisciplinary supportive management, though this is not a
disease-specific treatment trial.
- name: Ophthalmology-directed cataract care
description: >-
Ophthalmology evaluation and cataract-directed care are clinically relevant
because cataracts are an early, progressive feature of familial Danish
dementia.
treatment_term:
preferred_term: ophthalmologist evaluation
term:
id: MAXO:0000703
label: ophthalmologist evaluation
target_phenotypes:
- preferred_term: Cataract
term:
id: HP:0000518
label: Cataract
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
Clinically, FDD is characterized by the development and progression of cataracts during the third decade of life (Strömgren et al. 1970).
explanation: >-
Early progressive cataracts support ophthalmology-directed supportive
management, although the citation is natural-history rather than
treatment-efficacy evidence.
- name: Audiology evaluation and hearing support
description: >-
Audiology evaluation and hearing-support planning are relevant because
hearing loss follows the ocular manifestations and may become severe by mid
adulthood.
treatment_term:
preferred_term: audiologist evaluation
term:
id: MAXO:0000734
label: audiologist evaluation
target_phenotypes:
- preferred_term: Sensorineural hearing impairment
term:
id: HP:0000407
label: Sensorineural hearing impairment
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
Hearing impairment was reported to appear 10–20 years following the development of ocular problems.
explanation: >-
Progressive hearing impairment supports audiology-directed supportive
management, although this is natural-history rather than treatment-efficacy
evidence.
- name: Physical therapy for gait and balance impairment
description: >-
Physical therapy and gait/balance rehabilitation may be used
symptomatically for progressive ataxia and motor dysfunction.
treatment_term:
preferred_term: physical therapy
term:
id: MAXO:0000011
label: physical therapy
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
Patients with FDD developed cerebellar ataxia with intention tremor in their fourth decade of life, however, unlike FBD cases, did not develop spastic para-paresis.
explanation: >-
The clinical motor phenotype supports rehabilitation as symptomatic care;
the citation does not establish disease-specific treatment efficacy.
diagnosis:
- name: ITM2B genetic testing
diagnosis_term:
preferred_term: genetic testing
term:
id: MAXO:0000127
label: genetic testing
description: >-
Molecular diagnosis is established by identifying the pathogenic ITM2B
duplication associated with familial Danish dementia.
results: Pathogenic ITM2B variant supports the diagnosis of ADan amyloidosis.
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Molecular genetic analysis of the BRI gene in the Danish kindred showed a different defect, namely the presence of a 10-nt duplication (795-796insTTTAATTTGT) between codons 265 and 266, one codon before the normal stop codon 267.
explanation: >-
This supports targeted ITM2B/BRI genetic testing for the pathogenic
Danish duplication.
- name: Eye examination for cataract
diagnosis_term:
preferred_term: eye examination
term:
id: MAXO:0001155
label: eye examination
description: >-
Ocular assessment can identify the early cataracts that characterize the
staged familial Danish dementia phenotype.
results: Cataracts support the clinical syndrome in an at-risk ITM2B family.
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Clinically, FDD is characterized by the development and progression of cataracts during the third decade of life (Strömgren et al. 1970).
explanation: >-
Early progressive cataracts support eye examination as part of clinical
evaluation.
- name: Hearing examination
diagnosis_term:
preferred_term: hearing examination
term:
id: MAXO:0000873
label: hearing examination
description: >-
Hearing assessment helps document the progressive auditory component of the
ophthalmo-oto-encephalopathic syndrome.
results: Progressive perceptive hearing loss supports the clinical syndrome.
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hearing impairment was reported to appear 10–20 years following the development of ocular problems.
explanation: >-
The staged natural history supports hearing examination as part of
diagnostic evaluation.
- name: Neuropsychological assessment
diagnosis_term:
preferred_term: neuropsychological assessment
term:
id: MAXO:0009018
label: neuropsychological assessment
description: >-
Neuropsychological assessment can document progressive dementia and
accompanying neuropsychiatric features during later disease stages.
results: Progressive dementia with paranoid reactions supports late-stage clinical involvement.
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Progressive dementia probably begins in the sixth decade and may be associated with paranoid reactions and temporal disturbances of consciousness.
explanation: >-
The natural-history description supports cognitive and neuropsychiatric
assessment during later stages.
- name: Neuropathologic confirmation of ADan amyloid pathology
description: >-
Neuropathologic assessment can confirm the defining pattern of ADan
distribution, cerebral amyloid angiopathy, parenchymal lesions, Abeta
co-deposition, and PHF-like tau pathology.
markers: ADan peptide, cerebral amyloid angiopathy, parenchymal protein deposits, Abeta co-deposition, PHF-like tau
results: ADan-positive vascular and parenchymal pathology supports definitive disease classification.
evidence:
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We showed that ADan is widely distributed in the central nervous system (CNS) in the leptomeninges, blood vessels, and parenchyma.
explanation: >-
ADan distribution in CNS vessels and parenchyma supports neuropathologic
confirmation of the disease.
- reference: PMID:11895040
reference_title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta"
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
There was severe neurofibrillary pathology, and tau immunoblotting revealed a triplet electrophoretic migration pattern comparable with PHF-tau.
explanation: >-
PHF-like tau pathology supports the Alzheimer-like neurofibrillary
component of the diagnostic neuropathology.
differential_diagnoses:
- name: Alzheimer disease
disease_term:
preferred_term: Alzheimer disease
term:
id: MONDO:0004975
label: Alzheimer disease
description: >-
ADan amyloidosis overlaps with Alzheimer disease through dementia,
amyloid deposition, and neurofibrillary pathology.
evidence:
- reference: PMID:10781099
reference_title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Neuropathological findings include severe widespread cerebral amyloid angiopathy, hippocampal plaques, and neurofibrillary tangles, similar to Alzheimer's disease.
explanation: >-
This directly supports Alzheimer disease as an important neuropathologic
differential.
- name: Familial British dementia
description: >-
Familial British dementia is another chromosome 13/ITM2B amyloidosis and
may resemble ADan amyloidosis through progressive dementia, ataxia, cerebral
amyloid angiopathy, and Alzheimer-like tau pathology, but it generates ABri
rather than ADan and has prominent spastic tetraparesis.
distinguishing_features:
- ABri rather than ADan amyloid peptide
- Prominent spastic tetraparesis or spastic paraparesis in FBD
- FDD has earlier ocular and hearing manifestations and lacks the spastic paraparesis pattern emphasized for FBD
evidence:
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Clinical symptoms in patients with FBD appear during the fifth decade of life and include progressive dementia, spastic tetraparesis, and cerebellar ataxia.
explanation: >-
This supports FBD as a clinically overlapping dementia/ataxia differential
and identifies spastic tetraparesis as a distinguishing feature.
- reference: PMID:19779737
reference_title: Modeling familial British and Danish dementia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The 34 amino acid carboxy-terminal (C-terminal) peptide of the mutant protein was isolated from amyloid deposits from FBD patients and was referred to as British amyloid (ABri) (Vidal et al. 1999, 2000b).
explanation: >-
ABri peptide generation distinguishes familial British dementia from the
ADan peptide mechanism in familial Danish dementia.
animal_models:
- species: Rattus norvegicus
genotype: FDD-KI Itm2b Danish knock-in with humanized App
description: >-
FDD-KI rats harbor the endogenous Itm2b Danish mutation with a humanized
App allele and model vascular ADan/Aβ deposition, fibrinogen leakage,
neuroinflammation, demyelination, axonal injury, and age-accelerated motor
and gait deficits.
evidence:
- reference: PMID:41354963
reference_title: "Pathological mechanisms of motor dysfunction in familial Danish dementia: insights from a knock-in rat model."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
In this study, we investigate the pathological mechanisms linking CAA, white matter damage, and motor dysfunction using a recently developed FDD knock-in (FDD-KI) rat model.
explanation: >-
This establishes the FDD knock-in rat model and its use for motor and
white-matter pathophysiology.
- reference: PMID:41354963
reference_title: "Pathological mechanisms of motor dysfunction in familial Danish dementia: insights from a knock-in rat model."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Motor function assessments in FDD-KI rats demonstrated age-accelerated motor deficits and gait abnormalities, mirroring the clinical characteristics of FDD patients.
explanation: >-
The rat model recapitulates motor and gait abnormalities relevant to the
human disorder.
- reference: PMID:41354963
reference_title: "Pathological mechanisms of motor dysfunction in familial Danish dementia: insights from a knock-in rat model."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
These findings suggest that CAA and fibrinogen leakage in FDD may drive neuroinflammation, demyelination, and axonal damage in the cerebellum, potentially contributing to the motor and gait impairments observed in FDD.
explanation: >-
This supports the model's vascular leakage, neuroinflammation, and white
matter injury mechanism for motor dysfunction.
- species: Drosophila melanogaster
genotype: UAS-ADan transgenic expression
description: >-
Site-directed Drosophila UAS lines expressing ADan model amyloid-peptide
neurotoxicity, with ADan showing the strongest eye and CNS toxicity among
the tested peptides and early age-dependent climbing impairment.
evidence:
- reference: DOI:10.1186/1750-1326-9-5
reference_title: Amyloid peptides ABri and ADan show differential neurotoxicity in transgenic Drosophila models of familial British and Danish dementia
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
The highest toxicity was seen for ADan, followed by Aβ42 and ABri.
explanation: >-
This supports ADan neurotoxicity in the Drosophila eye model.
- reference: DOI:10.1186/1750-1326-9-5
reference_title: Amyloid peptides ABri and ADan show differential neurotoxicity in transgenic Drosophila models of familial British and Danish dementia
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
This effect was stronger for ADan, detected at 7 days post-eclosion, and followed by ABri and Aβ42, whose toxicity became evident after 15 and 21 days, respectively.
explanation: >-
This supports age-dependent ADan-associated motor dysfunction in the
Drosophila CNS model.
experimental_models:
- name: ADan pE-challenged SH-SY5Y neuronal cell model
description: >-
Differentiated human SH-SY5Y cells treated with pyroglutamate-modified ADan
model ADan-driven oxidative stress, mitochondrial cytochrome c release, and
caspase-mediated apoptosis.
experimental_model_type: CELL_LINE
organism:
preferred_term: human
term:
id: NCBITaxon:9606
label: Homo sapiens
cell_source: Human SH-SY5Y neuroblastoma cell line differentiated with retinoic acid
culture_system: Two-dimensional peptide-challenge culture with ADan pE or control peptides
modeled_mechanisms:
- target: ADan-induced mitochondrial oxidative apoptosis
description: >-
Measures ADan-driven oxidative stress, mitochondrial dysfunction, and
intrinsic apoptosis in a neuronal cell-line peptide-challenge model.
findings:
- statement: ADan pE induces mitochondrial cytochrome c release and loss of mitochondrial membrane potential.
supporting_text: >-
ADan pE challenge resulted, in addition to the release of cytochrome c to the cytoplasm, in a concurrent loss of mitochondrial membrane potential, indicated by the diffuse MitoTracker staining and poor co-localization of both signals (Figure 7B, bottom panel).
evidence:
- reference: PMID:26459115
reference_title: Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial Danish dementia ADan amyloid.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
ADan pE challenge resulted, in addition to the release of cytochrome c to the cytoplasm, in a concurrent loss of mitochondrial membrane potential, indicated by the diffuse MitoTracker staining and poor co-localization of both signals (Figure 7B, bottom panel).
explanation: >-
This supports the SH-SY5Y peptide-challenge model for mitochondrial ADan
toxicity.
clinical_trials: []
datasets: []
references:
- reference: PMID:10781099
title: A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: FDD is an autosomal dominant syndrome caused by a 10-nt BRI/ITM2B duplication that generates the ADan amyloid peptide.
supporting_text: >-
The decamer duplication mutation produces a frame-shift in the BRI sequence generating a larger-than-normal precursor protein, of which the amyloid subunit (designated ADan) comprises the last 34 C-terminal amino acids.
- reference: PMID:11895040
title: "Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta."
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: FDD neuropathology includes widespread cerebral amyloid angiopathy, parenchymal deposits, neurofibrillary degeneration, and Abeta co-deposition.
supporting_text: >-
Familial Danish dementia (FDD) is pathologically characterized by widespread cerebral amyloid angiopathy (CAA), parenchymal protein deposits, and neurofibrillary degeneration.
- reference: PMID:19779737
title: Modeling familial British and Danish dementia.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: FDD progresses from cataracts in the third decade to hearing impairment, cerebellar ataxia with intention tremor, later dementia, and death in the fifth or sixth decade.
supporting_text: >-
Clinically, FDD is characterized by the development and progression of cataracts during the third decade of life (Strömgren et al. 1970).
- reference: PMID:15968464
title: Chromosome 13 dementias.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: FBD and FDD share Alzheimer-like pathology including neurofibrillary tangles, amyloid deposits, cerebral amyloid angiopathy, inflammatory components, and de novo ABri/ADan peptide generation.
supporting_text: >-
Two non-Abeta cerebral amyloidoses, familial British and Danish dementias (FBD and FDD), share many aspects of Alzheimer's disease, including the presence of neurofibrillary tangles, parenchymal preamyloid and amyloid deposits, cerebral amyloid angiopathy and a variety of amyloid-associated proteins and inflammatory components.
- reference: DOI:10.1007/s00018-005-5092-5
title: Chromosome 13 dementias.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
- reference: DOI:10.1007/s00429-009-0221-9
title: Modeling familial British and Danish dementia.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
- reference: DOI:10.3233/JAD-2011-110785
title: Increased AβPP Processing in Familial Danish Dementia Patients.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
- reference: DOI:10.1016/j.nbd.2015.10.003
title: Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial Danish dementia ADan amyloid.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
- reference: PMID:21841249
title: Increased AbetaPP processing in familial Danish dementia patients.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: APP-derived metabolites, including Abeta, are increased in an FDD brain sample, supporting altered APP processing in Danish dementia.
supporting_text: >-
We find that the levels of several AβPP metabolites, including Aβ, are significantly increased in the brain sample derived from an FDD patient.
- reference: PMID:33172889
title: Danish and British dementia ITM2b/BRI2 mutations reduce BRI2 protein stability and impair glutamatergic synaptic transmission.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: Danish and British ITM2B mutations reduce mature BRI2 at synapses and impair glutamatergic transmission.
supporting_text: >-
Collectively, the data show that FDD and FBD mutations cause a reduction of BRI2 levels and function at synapses, which results in reduced glutamatergic transmission.
- reference: DOI:10.1074/jbc.ra120.015679
title: Danish and British dementia ITM2b/BRI2 mutations reduce BRI2 protein stability and impair glutamatergic synaptic transmission.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
- reference: PMID:26459115
title: Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial Danish dementia ADan amyloid.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: ADan neurotoxicity involves ROS generation, cytochrome c release, mitochondrial membrane-potential disruption, and caspase-3 activation.
supporting_text: >-
The data presented herein demonstrates that ADan neurotoxicity takes place through a mechanism involving many components of intrinsic apoptosis, including high levels of ROS generation, cyt c release into the cytoplasm, disruption of mitochondrial membrane potential, and final activation of terminal caspase-3.
- reference: DOI:10.1186/s12974-025-03537-w
title: "Pathological mechanisms of motor dysfunction in familial Danish dementia: insights from a knock-in rat model"
found_in:
- ADan_amyloidosis-deep-research-falcon.md
- reference: PMID:41354963
title: "Pathological mechanisms of motor dysfunction in familial Danish dementia: insights from a knock-in rat model."
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: A FDD-KI rat model links vascular amyloid deposition, fibrinogen leakage, neuroinflammation, white-matter injury, and motor dysfunction.
supporting_text: >-
These findings suggest that CAA and fibrinogen leakage in FDD may drive neuroinflammation, demyelination, and axonal damage in the cerebellum, potentially contributing to the motor and gait impairments observed in FDD.
- reference: DOI:10.1038/s44319-024-00077-x
title: "Functional BRI2-TREM2 interactions in microglia: implications for Alzheimer's and related dementias"
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: BRI2 interacts functionally with TREM2 in microglia, influences TREM2 processing, and affects phagocytosis.
supporting_text: >-
Lastly, Bri2 deletion reduces phagocytosis similarly to a pathogenic TREM2 variant that enhances processing.
- reference: DOI:10.1007/s00401-024-02820-z
title: Microglia contribute to the production of the amyloidogenic ABri peptide in familial British dementia
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: ITM2B/BRI2 is enriched in iPSC-derived microglia, which can produce amyloidogenic ABri peptide in familial British dementia.
supporting_text: >-
Using patient-derived induced pluripotent stem cells, we show that expression of ITM2B/BRI2 is 34-fold higher in microglia than neurons and 15-fold higher in microglia compared with astrocytes.
- reference: DOI:10.1101/2023.06.27.546552
title: Microglia produce the amyloidogenic ABri peptide in familial British dementia.
found_in:
- ADan_amyloidosis-deep-research-falcon.md
- reference: DOI:10.1101/2023.09.15.557952
title: "Amyloids at the border: deep mutagenesis and random sequence extension reveal an incomplete amyloid-forming motif in Bri2 that turns amyloidogenic upon C-terminal extension"
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: Deep mutagenesis maps an ADan amyloid core and supports C-terminal extension as a mechanism that can generate de novo amyloid motifs.
supporting_text: >-
To systematically explore the consequences of Bri2 C-terminal extension, here, we measure amyloid formation for 676 ADan substitutions and identify the region that forms the putative amyloid core of ADan fibrils, located between positions 20 and 26, where stop-loss occurs.
- reference: DOI:10.1186/1750-1326-9-5
title: Amyloid peptides ABri and ADan show differential neurotoxicity in transgenic Drosophila models of familial British and Danish dementia
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: ADan is neurotoxic in Drosophila eye and CNS models and causes age-dependent climbing impairment earlier than ABri or Abeta42.
supporting_text: >-
This effect was stronger for ADan, detected at 7 days post-eclosion, and followed by ABri and Aβ42, whose toxicity became evident after 15 and 21 days, respectively.
- reference: DOI:10.1101/2021.06.24.449787
title: Familial Danish dementia young Knock-in rats expressing humanized APP and human Abeta show impaired pre and postsynaptic glutamatergic transmission
found_in:
- ADan_amyloidosis-deep-research-falcon.md
findings:
- statement: Young Itm2b Danish knock-in rats expressing humanized App show early glutamatergic synaptic abnormalities.
supporting_text: >-
Collectively, the data show that the pathogenic Danish mutation alters the physiological function of BRI2 at glutamatergic synapses; these functional alterations are detected across species and occur early in life.
References & Deep Research
References
20Deep Research
2Asta Literature Retrieval: Pathophysiology and clinical mechanisms of ADan amyloidosis. Core disease mechanisms, molecular and cellular pathways...
This report is retrieval-only and is generated directly from Asta results.
- Papers retrieved: 20
- Snippets retrieved: 20
Relevant Papers
[1] Common immunopathogenesis of central nervous system diseases: the protein-homeostasis-system hypothesis
- Authors: Kyung-Yil Lee
- Year: 2022
- Venue: Cell & Bioscience
- URL: https://www.semanticscholar.org/paper/2984270ae67451b93007040848d9694d19714c9f
- DOI: 10.1186/s13578-022-00920-5
- PMID: 36384812
- PMCID: 9668226
- Citations: 9
- Influential citations: 1
- Summary: This article proposes a common immunopathogenesis of CNS diseases, including prion diseases, Alzheimer’s disease, and genetic diseases, through the PHS hypothesis, which proposes that the immune systems in the host control those substances according to the size and biochemical properties of the substances.
- Evidence snippets:
- Snippet 1 (score: 0.480) > They are usually fibrillar when visualized by electronic microscopy and can be detected by the Congo red stain method. The pathophysiology of amyloidosis remains unknown, but the associated diseases include those with genetic traits, such as ATTRv and AFib, and those with underlying diseases that are accompanied by abnormal immune reactions, such as AL and AA amyloidosis [91]. Furthermore, the clinical manifestations and pathologic findings, such as the components of the deposited amyloidogenic proteins and peptides, are different in each disease, whereas the structure and properties of the protein aggregates, such as their insolubility, protease resistance, birefringent staining with Congo red, and high beta-sheet content, are similar across diseases, as observed in the prion diseases. These findings suggest that a common immunopathogenesis is involved in prion diseases and non-prion amyloid diseases [92]. > Certain chronic progressive CNS diseases, including AD, PD, and HD, are associated with genetic traits, similar to familial CJG and GSS in prion diseases. The pathological hallmarks of these diseases include intracellular inclusion bodies, extracellular amyloid deposits with various components, and neuronal loss. Well-studied amyloid proteins are amyloid beta (Aβ) and phosphorylated tau in AD and α-synuclein in PD. Inclusion bodies and extracellular amyloid proteins, such as Lewy bodies in PD and neurofibrillary tangles (tau protein) in AD, are considered to play important etiopathogenetic roles in neurodegenerative diseases. However, some affected patients have no intracellular inclusion lesions, and the degree of inclusion-body involvement in the pathology findings is not related to the clinical severity of the diseases. Furthermore, multiple risk factors are related to the diseases [93]. There are genetic forms of these diseases, and experimental studies of gene-null mice for the tau protein or α-synuclein show that those genes do not play a significant role in disease phenotype, pathologic lesions, or disease progression [94,95].
[2] Molecular Mechanism of Pathogenesis and Treatment Strategies for AL Amyloidosis
- Authors: Hidehiko Ikura, J. Endo, Hiroki Kitakata, Hidenori Moriyama, M. Sano et al.
- Year: 2022
- Venue: International Journal of Molecular Sciences
- URL: https://www.semanticscholar.org/paper/1450c3006397005e391453bb1d59585e7ecef7d9
- DOI: 10.3390/ijms23116336
- PMID: 35683015
- PMCID: 9181426
- Citations: 23
- Influential citations: 1
- Summary: The pathogenesis and treatment strategies for AL amyloidosis with respect to its molecular mechanisms are outlined and the mechanism of cellular and tissue damage, the mass effect due to amyloids deposition, as well as the toxicity of pre-fibrillar LC, is gradually being elucidated.
- Evidence snippets:
- Snippet 1 (score: 0.461) > In amyloid light-chain (AL) amyloidosis, small B-cell clones (mostly plasma cell clones) present in the bone marrow proliferate and secrete unstable monoclonal free light chains (FLCs), which form amyloid fibrils that deposit in the interstitial tissue, resulting in organ injury and dysfunction. AL amyloidosis progresses much faster than other types of amyloidosis, with a slight delay in diagnosis leading to a marked exacerbation of cardiomyopathy. In some cases, the resulting heart failure is so severe that chemotherapy cannot be administered, and death sometimes occurs within a few months. To date, many clinical studies have focused on therapeutics, especially chemotherapy, to treat this disease. Because it is necessary to promptly lower FLC, the causative protein of amyloid, to achieve a hematological response, various anticancer agents targeting neoplastic plasma cells are used for the treatment of this disease. In addition, many basic studies using human specimens to elucidate the pathophysiology of AL have been conducted. Gene mutations associated with AL, the characteristics of amyloidogenic LC, and the structural specificity of amyloid fibrils have been clarified. Regarding the mechanism of cellular and tissue damage, the mass effect due to amyloid deposition, as well as the toxicity of pre-fibrillar LC, is gradually being elucidated. This review outlines the pathogenesis and treatment strategies for AL amyloidosis with respect to its molecular mechanisms.
[3] New treatment strategies for Alzheimer's disease: is there a hope?
- Authors: I. Aprahamian, F. Stella, O. Forlenza
- Year: 2013
- Venue: The Indian Journal of Medical Research
- URL: https://www.semanticscholar.org/paper/ea073be2ee0f8291a441455f49ba13689e1440a9
- PMID: 24434253
- PMCID: 3868059
- Citations: 71
- Influential citations: 8
- Summary: The available evidence on the new therapeutic approaches that target amyloid and Tau pathology in AD are summarized, focusing on pharmaceutical compounds undergoing phase 2 and phase 3 randomized controlled trials.
- Evidence snippets:
- Snippet 1 (score: 0.450) > The recognition of core and secondary pathophysiological mechanisms in AD has led to the identification of molecular targets for the development of specific drugs. In fact, more than 200 pharmaceutical compounds are currently undergoing phase 2 and 3 trials 11 . These compounds can be grossly divided into anti-amyloid agents and drugs that target other pathological pathways. Anti-amyloid compounds can be subdivided into drugs designed to block or inhibit the overproduction or aggregation of Aβ, or to favour its clearance from the brain (Table I; Fig. 2) 20 , whereas the latter group can be subdivided according to predominant mechanism of action of the drug, i.e., neurotransmitter and cell-signalling agents, glial cell modulators, neuroprotective agents, and Tau-based therapies (Table II). Studies involving stem-cell and gene therapy are also under way, but at more incipient stages of experimental validation. > In AD, pathophysiological mechanisms change soluble Aβ peptides into fibrillary oligomers and insoluble fibrils, which accumulate extracellularly in the neural tissue and also in the intima of brain and systemic vessels 34 . Extracellular Aβ oligomers and fibrillary forms cause synaptic dysfunction, affect axons and dendritic spines, and eventually lead to neuronal loss in AD 35 . Toxic Aβ species also trigger secondary pathological mechanisms (e.g., oxidative stress and inflammation), which hasten neuronal dysfunction and death 36 . Therefore, pharmacological compounds that favour the clearance of Aβ from the brain, or prevent its aggregation, may represent a strategy to delay the progression of the pathological process in AD. Intracerebral amyloidosis may start in the brain of individuals with AD many years before the onset of clinical symptoms 37,38 . Evidence of this pathological process can be depicted at prodromal or even at preclinical stages of the disease by the analysis of cerebrospinal fluid (CSF) and molecular neuroimaging biomarkers [37][38][39] .
[4] Transthyretin mutagenesis: impact on amyloidogenesis and disease
- Authors: Zaida L. Almeida, D. Vaz, Rui M. M. Brito
- Year: 2024
- Venue: Critical Reviews in Clinical Laboratory Sciences
- URL: https://www.semanticscholar.org/paper/543c86839cb0c86a5ba2ea68fa8cc897ab376b55
- DOI: 10.1080/10408363.2024.2350379
- PMID: 38850014
- Citations: 16
- Influential citations: 1
- Summary: This article reviews and discusses TTR mutagenesis and amyloidogenesis, and their implications in disease onset, and compiles the various in vitro TTR aggregation protocols currently in use for research and drug development purposes.
- Evidence snippets:
- Snippet 1 (score: 0.447) > Protein aggregation and amyloid formation contribute to several debilitating diseases collectively known as Amyloidosis [1]. To date, more than fifty amyloid diseases have been identified, including localized amyloidosis found in neurodegenerative conditions like Alzheimer's and Parkinson's diseases, and systemic amyloidosis such as transthyretin amyloidosis and lysozyme amyloidosis [1]. These pathologies result from mutations, post-translational modifications, or partial proteolysis, and by abnormal folding or unfolding events affecting approximately fifty different peptides/ proteins. These end up adopting non-native, misfolded conformations prone to aggregate into highly ordered soluble oligomers and insoluble fibrils with a characteristic cross-β structure -the amyloid substance. Generally, amyloid diseases are not a consequence of the loss of function of the native protein but result from the cytotoxic nature of the amyloid aggregates and/or the action of amyloid fibrils on inter-cellular communication and tissue physiology. Although most amyloids are found extracellularly, amyloid-like deposits are also found inside cells [1]. > Transthyretin amyloid disorders include sporadic age-related wild-type TTR amyloidosis (ATTRwt), hereditary TTR amyloidosis polyneuropathy (ATTRv-PN), hereditary TTR amyloidosis cardiomyopathy (ATTRv-CM), hereditary leptomeningeal TTR amyloidosis (ATTRv-LM) and hereditary ocular TTR amyloidosis (ATTRv-OC). Although some of the ATTR clinical manifestations have unmet medical needs, in the last decade several disease-modifying therapies have contributed to slowing down disease progression and, in some cases, have ameliorated disease symptoms. The continuing efforts to better understand the molecular mechanisms of disease progression and tissue specificity are critical for the rational development of new and improved therapies for the treatment of TTR amyloidoses.
[5] Modulation of the Mechanisms Driving Transthyretin Amyloidosis
- Authors: Filipa Bezerra, M. Saraiva, M. R. Almeida
- Year: 2020
- Venue: Frontiers in Molecular Neuroscience
- URL: https://www.semanticscholar.org/paper/c1e8e4cf5970a0ac939aeffc6db8d5dd475f9611
- DOI: 10.3389/fnmol.2020.592644
- PMID: 33362465
- PMCID: 7759661
- Citations: 38
- Influential citations: 1
- Summary: Transthyretin (TTR) amyloidoses are systemic diseases associated with TTR aggregation and extracellular deposition in tissues as amyloid. The most frequent and severe forms of the disease are hereditary and associated with amino acid substitutions in the protein due to single point mutations in the TTR gene (ATTRv amyloidosis). However, the wild type TTR (TTR wt) has an intrinsic amyloidogenic potential that, in particular altered physiologic conditions and aging, leads to TTR aggregation in...
- Evidence snippets:
- Snippet 1 (score: 0.433) > ATTR amyloidosis is an under-recognized disease which is characterized by extracellular deposition of TTR aggregates in several organs, being polyneuropathy and cardiomyopathy the major clinical manifestations. The mechanism by which the tetramer disassembles and aggregates into amyloid fibrils has been considered the main driver of the disease. However, TTR proteolysis, namely occurring in the cardiac tissue, as well as its modulation have been increasingly documented as fundamental for understanding the development and progression of ATTR amyloidosis. > Many therapeutic approaches have been suggested for the treatment of ATTR amyloidosis targeting different steps of the pathology. Those therapies include interventions from the synthesis of the TTR variants through liver transplant or gene silencing therapies, to TTR stabilization, inhibition of aggregation, disruption of amyloid fibrils and clearance of amyloid deposits. The main targets for intervention on TTR amyloid formation are summarized in Figure 2. Although some the available therapies are more efficient than others, it becomes increasingly evident that combination of different therapies may improve the therapeutic outcome. In this sense, it would be interesting to test TTR gene silencing therapies in combination with protein stabilizers or disruptors of pre-existing amyloid deposits. It is also important to obtain more efficient and targeted therapies specific to organ and tissues with limited drug access as is the case of the eye and brain, that are particularly relevant in some forms of the disease. Moreover, it is crucial to continue with studies that can contribute to a better understanding of the mechanisms involved in the disease, in particular, TTR proteolysis, which has been mainly valued in the case of ATTR-CM and, also at the extracellular level involving either interactions with components of the extracellular matrix or with molecular and chemical chaperones acting as disease modulators. > Overall, detailed knowledge of the mechanisms of amyloid formation and the availability of different approaches allows directed and personalized interventions aiming higher specificity and efficacy of chosen therapeutic solutions.
[6] Neuroprotective Function of Non-Proteolytic Amyloid-β Chaperones in Alzheimer’s Disease
- Authors: Bhargy Sharma, K. Pervushin
- Year: 2019
- Venue: Amyloid Diseases
- URL: https://www.semanticscholar.org/paper/966e3e024e09b5c9bf4c6967acd39639d1a542fc
- DOI: 10.5772/INTECHOPEN.84238
- Citations: 2
- Summary: This chapter focuses on structural and morphological changes during aggregation of amyloids which have been identified using Nuclear magnetic resonance spectroscopy, X-ray crystallography, Electron microscopy, Atomic force microscopy and other biophysical techniques as well as interactions between chaperone proteins and amyloid moieties.
- Evidence snippets:
- Snippet 1 (score: 0.425) > Abnormal deposition of amyloids or "Amyloidosis" is hallmark of several chronic cerebrovascular diseases including neurodegeneration culminating into dementia. Efforts to develop targeted drugs against amyloids have been hindered since there is no universal mechanism that leads to protein misfolding or aggregation, and the aggregates usually do not correspond directly to clinical symptoms of the diseases. A clearer understanding of molecular interactions of amyloids can drive the ongoing therapeutic efforts to prevent aggregation of nascent amyloids into pathological species and to design timely interventions. In addition to aging, precursor mutations in genes and proteins, gene multiplication, expansion of amyloidogenic sequences, and xenobiotics such as air pollutants are risk factors usually associated with amyloidosis disorders [1]. There are different amyloidogenic species causing a variety of neuropathic diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), Poly-glutamine disorders like Huntington disease (HD), Prion diseases including Creutzfeldt-Jakob disease, Lewy body disease, Amyotrophic Lateral Sclerosis (ALS) as well as metabolic diseases such as type II diabetes (T2D) and corneal dystrophy to name a few [2]. AD is a pandemic form of dementia caused due to improper aggregation of amyloidogenic proteins-amyloid-β (Aβ), which is a cleavage product of amyloid precursor protein (APP) and tau, stabilizes microtubules in neurons [3]. Prognosis is also closely associated with aggregation of α-Synuclein (αS) protein into Lewy bodies usually concentrated in presynaptic terminals [4]. Human islet amyloid polypeptide (IAPP), also known as amylin, is secreted along with insulin from pancreatic β-islet cells. IAPP rich amyloid plaques, facilitated by insulin resistance, are hallmarks of T2D [5]. β-sheeted infectious isoform of cellular prion protein (PrP) causes transmissible spongiform encephalopathy (TSE), broadly known as prion disease [2]. > In this chapter, we review various structures and conformations attained by Aβ peptides during the process
[7] Arrhythmic Manifestations of Cardiac Amyloidosis: Challenges in Risk Stratification and Clinical Management
- Authors: N. Laptseva, V. Rossi, I. Sudano, Rahel Schwotzer, F. Ruschitzka et al.
- Year: 2023
- Venue: Journal of Clinical Medicine
- URL: https://www.semanticscholar.org/paper/bfbf1950ad68a4aff16d3b2163345380608c3ddf
- DOI: 10.3390/jcm12072581
- PMID: 37048664
- PMCID: 10095126
- Citations: 19
- Influential citations: 3
- Summary: A deepened insight is given into the arrhythmic features of cardiac amyloidosis by discussing the pathogenesis of these arrhythmias, addressing the challenges in risk stratification and strategies for management in these patients.
- Evidence snippets:
- Snippet 1 (score: 0.424) > Amyloidosis is a systemic disease that is characterized by extracellular deposits of an insoluble fibrillar protein called amyloid within various tissues and organs, mainly the heart, kidneys and the peripheral nervous system [1]. There are more than 30 amyloidogenic precursor proteins in humans. The clinical phenotype depends on the nature of the amyloid and stage of disease, and can vary considerably in different patients. Cardiac amyloidosis is a frequent feature of the disease (in up to 60% of patients), causing a progressive, restrictive type of cardiomyopathy, and is associated with adverse clinical outcomes [2,3]. > There are two main forms of amyloidosis that significantly affect the heart: light chain amyloidosis (AL) and transthyretin amyloidosis (ATTR) [4,5]. The two forms have a completely different pathophysiology and their treatment differs considerably [6,7]. AL type is caused by overproduction and misfolding of antibody light chain fragments by an underlying clonal plasma cell in the bone marrow. Other vital organs may also be involved in AL amyloidosis, such as kidneys, liver, peripheral and autonomic nervous system. The majority of the patients are male and over the age of 50, although the disease may also occur in younger patients. Early diagnosis of AL cardiac amyloidosis is very important since the disease, if untreated, typically progresses rapidly and causes severe heart failure (HF), life-threatening arrhythmias, and results in death. ATTR, which occurs due to deposits of misfolded monomers of transthyretin (a small molecule mainly produced by the liver) can be observed either as a genetic disease (variant ATTR, or vATTR) or more commonly as a nonhereditary disease, called wild-type ATTR (wtATTR). In general, progression of ATTR cardiac amyloidosis (especially wtATTR) is slower as compared to that observed in AL cardiac amyloidosis, and untreated affected individuals may live many years after initial manifestation of disease. > The typical clinical presentation in patients with cardiac amyloidosis is HF with preserved ejection fraction (HFpEF).
[8] Alzheimer's disease mechanisms in peripheral cells: Promises and challenges
- Authors: E. Trushina
- Year: 2019
- Venue: Alzheimer's & Dementia : Translational Research & Clinical Interventions
- URL: https://www.semanticscholar.org/paper/43207b6874701048ebc4e950e37122fa446f16a5
- DOI: 10.1016/j.trci.2019.06.008
- PMID: 31720366
- PMCID: 6838468
- Citations: 29
- Influential citations: 3
- Summary: Development of efficacious therapeutic interventions for Alzheimer's disease is hampered by the lack of understanding early disease mechanisms, biomarkers, and models that mimic complex pathophysiology of human disease.
- Evidence snippets:
- Snippet 1 (score: 0.422) > Alzheimer's disease (AD) is the leading form of dementia where underlying molecular mechanisms are poorly understood. Therapeutic strategies designed to reduce levels of amyloid beta (Ab) plaques or hyperphosphorylated tau (ptau) containing tangles, two hallmarks of AD, have failed in clinical trials [1][2][3]. Factors contributing to this failure include limited understanding of early disease mechanisms and associated biomarkers, and poor translation of preclinical research conducted in model organisms [4,5]. Familial AD (FAD) accounts for~5% of all cases and is linked to mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes [6]. Most AD cases are sporadic late-onset AD (LOAD) with age being the greatest risk factor [6]. Recent clinical investigations using systems biology approaches and imaging techniques suggest that AD is a complex disorder where changes in multiple pathways occur years before the onset of clinical symptoms [7]. Moreover, the disease differentially affects males and females presenting additional challenges for biomarker and drug discovery [8]. Animal models currently used for preclinical therapeutic development do not recapitulate the complexity of sporadic AD. Thus, there is an urgent need to identify translational models that better represent AD mechanisms and could complement existing animal models to test novel therapeutic approaches and develop panels of disease stage-and sex-specific diagnostic and prognostic biomarkers. > This article aims to test the hypothesis that AD is a systemic disorder where peripheral cells recapitulate major molecular mechanisms affected in the brain. The hypothesis predicts that alterations in pathways shown fundamentally important in the etiology of AD including inflammation, abnormal calcium signaling, amyloid precursor protein processing, Ab and p-tau accumulation, altered oxidative metabolism, mitochondrial dysfunction, and abnormal cellular energetics will be detected in peripheral cells and biofluids of AD patients providing a unique opportunity to study/ manipulate these mechanisms in a contest of the individuals' genetic, epigenetic, and metabolic background. Here, we will (1) provide the rationale for this hypothesis reviewing evidence that peripheral cells and biofluids recapitulate mechanisms affected in the brain; (2) discuss opportunities
[9] The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure
- Authors: Christian Haupt
- Year: 2021
- Venue: Hemato
- URL: https://www.semanticscholar.org/paper/4d1c46c8467ff75e1f0e5ce51f2d67b5cc6c3824
- DOI: 10.3390/hemato2030032
- Citations: 7
- Summary: Recent biochemical and biophysical studies that have expanded knowledge on how versatile the structure of AL fibrils in patients is are reviewed and highlighted their implications for the molecular mechanism of fibril formation in AL amyloidosis are highlighted.
- Evidence snippets:
- Snippet 1 (score: 0.415) > The formation and deposition of fibrils derived from immunglobulin light chains is a hallmark of systemic AL amyloidosis. A particularly remarkable feature of the disease is the diversity and complexity in pathophysiology and clinical manifestations. This is related to the variability of immunoglobulins, as virtually every patient has a variety of mutations resulting in their own unique AL protein and thus a unique fibril deposited in the body. Here, I review recent biochemical and biophysical studies that have expanded our knowledge on how versatile the structure of AL fibrils in patients is and highlight their implications for the molecular mechanism of fibril formation in AL amyloidosis.
[10] Are we creating a new phenotype? Physiological barriers and ethical considerations in the treatment of hereditary transthyretin-amyloidosis
- Authors: Maike F. Dohrn, J. Medina, K. Olaciregui Dague, E. Hund
- Year: 2021
- Venue: Neurological Research and Practice
- URL: https://www.semanticscholar.org/paper/74e5dd6052df6b49203e21d721efd832f467d2c8
- DOI: 10.1186/s42466-021-00155-8
- PMID: 34719408
- PMCID: 8559355
- Citations: 18
- Summary: The current advances in treating ATTRv amyloidosis have become a meaningful example for mechanism-based treatment and will still have to face new challenges including shifts in the phenotype spectrum and the ongoing need for improved treatment precision.
- Evidence snippets:
- Snippet 1 (score: 0.414) > Hereditary transthyretin (TTR) amyloidosis (ATTRv) is an autosomal dominant, systemic disease transmitted by amyloidogenic mutations in the TTR gene. To prevent the otherwise fatal disease course, TTR stabilizers and mRNA silencing antisense drugs are currently approved treatment options. With 90% of the amyloidogenic protein produced by the liver, disease progression including polyneuropathy and cardiomyopathy, the two most prominent manifestations, can successfully be halted by hepatic drug targeting or—formerly—liver transplantation. Certain TTR variants, however, favor disease manifestations in the central nervous system (CNS) or eyes, which is mostly associated with TTR production in the choroid plexus and retina. These compartments cannot be sufficiently reached by any of the approved medications. From liver-transplanted patients, we have learned that with longer lifespans, such CNS manifestations become more relevant over time, even if the underlying TTR mutation is not primarily associated with such. Are we therefore creating a new phenotype? Prolonging life will most likely lead to a shift in the phenotypic spectrum, enabling manifestations like blindness, dementia, and cerebral hemorrhage to come out of the disease background. To overcome the first therapeutic limitation, the blood–brain barrier, we might be able to learn from other antisense drugs currently being used in research or even being approved for primary neurodegenerative CNS diseases like spinal muscular atrophy or Alzheimer’s disease. But what effects will unselective CNS TTR knock-down have considering its role in neuroprotection? A potential approach to overcome this second limitiation might be allele-specific targeting, which is, however, still far from clinical trials. Ethical standpoints underline the need for seamless data collection to enable more evidence-based decisions and for thoughtful consenting in research and clinical practice. We conclude that the current advances in treating ATTRv amyloidosis have become a meaningful example for mechanism-based treatment. With its great success in improving patient life spans, we will still have to face new challenges including shifts in the phenotype spectrum and the ongoing need for improved treatment precision. Further investigation is needed to address these closed barriers and open questions.
[11] Tissue Characterization in Cardiac Amyloidosis
- Authors: V. Musetti, F. Greco, V. Castiglione, A. Aimo, C. Palmieri et al.
- Year: 2022
- Venue: Biomedicines
- URL: https://www.semanticscholar.org/paper/e42701c0e211d214f44aa633a34d8719838c3109
- DOI: 10.3390/biomedicines10123054
- PMID: 36551810
- PMCID: 9775200
- Citations: 17
- Influential citations: 2
- Summary: The present review focuses on the status of tissue characterization in cardiac amyloidosis, from histochemistry to immunohistochemistry and mass spectrometry, as well as on its future directions.
- Evidence snippets:
- Snippet 1 (score: 0.412) > Amyloidoses are characterized by the extra-cellular deposition of amyloidogenic proteins in a cross-β-sheet structure that confers a typical green birefringence by Congo red staining under polarized light microscopy [1]. The tissue deposition of amyloid may lead to organ failure [1,2]. Cardiac amyloidosis (CA) has long been considered a rare disease [1,3]; however, recent advances in non-invasive diagnostic tools have led to a reconsideration of the epidemiology of the disease. Furthermore, the new therapeutic strategies available have increased the clinical impact of CA diagnosis [4][5][6][7][8]. > More than 30 amyloidogenic proteins have been identified so far, with immunoglobulin light chains and transthyretin (TTR) being the most common amyloidogenic precursors of CA, respectively reasonable for AL and ATTR amyloidosis [9,10]. AL amyloidosis is a systemic disease involving almost every organ and has a poor prognosis, particularly when the heart is affected [11]. Two major forms of ATTR amyloidosis are recognized: wild-type ATTR (ATTRwt) and variant ATTR (ATTRv; due to point mutations in the TTR gene) amyloidosis [12,13]. In CA, the myocardial damage and the functional impairment are related to the type of the amyloidogenic protein as well as to the pattern and the extension of the amyloid deposits [11]. Recent investigations have also identified other mechanisms, including fibrosis and inflammatory pathways, which may play a significant role in myocardial dysfunction [14,15]. Amyloid typing is crucial for the clinical management of affected patients, since the misidentification of the amyloidogenic protein can lead to inappropriate treatments [7,16]. In some cases, particularly in ATTR amyloidosis, a non-biopsy diagnosis can be obtained; nevertheless, tissue characterization is always needed when a plasma cell dyscrasia is present and/or when nuclear medicine findings are inconclusive [17][18][19].
[12] Gelsolin pathogenic Gly167Arg mutation promotes domain-swap dimerization of the protein
- Authors: F. Bonì, M. Milani, A. Barbiroli, L. Diomede, E. Mastrangelo et al.
- Year: 2017
- Venue: Human Molecular Genetics
- URL: https://www.semanticscholar.org/paper/02e035ff678ddb449cb65f97c28e695ddc1a77a5
- DOI: 10.1093/hmg/ddx383
- PMID: 29069428
- PMCID: 5886171
- Citations: 18
- Summary: Structural and biophysical characterizations reveal that the Gly167Arg mutation alone is responsible for the monomer to dimer transition and that, even in the context of the full-length protein, the pathogenic variant is prone to form dimers.
- Evidence snippets:
- Snippet 1 (score: 0.408) > examples in (16)(17)(18)], in kindred lacking Finnish ancestors, suggesting that this disease, owing to its neglected state and complex and variable clinical picture, has often been un/ misdiagnosed. > Two novel pathological variants of gelsolin have recently been described to be associated with renal amyloidosis containing the following mutations: Asn184 to Lys and Gly167 to Arg (C633 to A and G580 to A, respectively) (19)(20)(21). In addition, a sporadic form of AGel with marked wild-type (wt) gelsolin deposits surrounding a sellar glioma of the hypophysis has also recently been discovered (22). This finding is particularly relevant since, in several clinical reports of pituicytoma-associated amyloidosis (23)(24)(25), it is the first one in which the etiological agent, i.e. the main constituent of the aggregates, was identified. Therefore, AGel can be classified into three different forms according to the nature of the protein, mutants or wt, in addition to the organ(s) involved in fibril deposition: (i) systemic (or Finnish-type), (ii) kidney localized and (iii) sporadic. > All AGel types share the lack of apt pharmacological therapies that cure the disease, targeting the source of toxicity, rather than only acting as palliative, symptomatic treatments. However, the use of nanobodies raised against mutated gelsolin recently showed great potential as a novel strategy against AGel, both in vitro and in vivo exploiting adeno-associated viruses (26)(27)(28). Dissection of the molecular bases that lead to wt and mutant gelsolin misfolding, and knowledge of the mechanisms underlying each AGel form, are crucial to identify pharmacologically relevant targets and to develop effective therapeutic strategies. > With the exception of our recent description of the underlying molecular bases of Asn184Lys amyloidosis (29), little is known about the pathological mechanisms that lead to renal or sporadic forms. On the contrary, the amyloidogenic pathway of the systemic Asp187
[13] Oxidative Stress, Chronic Inflammation, and Amyloidoses
- Authors: A. Orzechowski, A. Cywińska, A. Rostagno, F. Rizzi
- Year: 2019
- Venue: Oxidative Medicine and Cellular Longevity
- URL: https://www.semanticscholar.org/paper/b1edc328b4cf156216b002e11396f08538bd4fb2
- DOI: 10.1155/2019/6024975
- PMID: 31612076
- PMCID: 6755281
- Citations: 11
- Summary: Department of Physiological Sciences, Warsaw University of Life Sciences (SGGW), Nowoursynowska 159, 02-776 Warsaw, Poland Department of Pathology and Veterinary Diagnostics, Warsaw Universitas degli Studi di Parma, Via Volturno 39/E, Parma.
- Evidence snippets:
- Snippet 1 (score: 0.408) > Amyloid diseases are part of an emerging complex group of chronic and progressive entities collectively known as disorders of protein folding. For reasons still under active investigation, soluble proteins normally found in interstitial or biological fluids change their conformation and form poorly soluble molecular assemblies that accumulate as extracellular fibrillar aggregates within the parenchyma and blood vessels of different organs. Once formed, these amyloid fibrils that typically exhibit a β-sheet-rich secondary structure are highly resistant to proteolytic degradation, a characteristic that impairs their effective physiologic removal and leads to their tissue accumulation inducing local hypoxia and cellular damage with overall organ dysfunction and, eventually, death. > The molecular pathogenic mechanisms associated with amyloid diseases are complex and involve cross-talk among different cell populations and cellular pathways. Mounting evidence points out to inflammation-related mechanisms and oxidative stress as associated burden for amyloidosis. In spite of multiple studies, it still remains to be elucidated whether oxidative stress is a key contributor to the disease pathogenesis and progression or whether-on the contrary-it is a mere consequence of the cellular responses elicited by the presence of amyloid deposits and the concomitant inflammatory processes affecting injured tissue. This special issue provides an updated view on the role of oxidative stress and inflammation-related cellular pathways to the etiopathogenesis, progression, and treatment strategies of amyloid diseases. > One of the papers of this special issue, "Hypoxia and Inflammation as a Consequence of β-fibril Accumulation: A Perspective View for New Potential Therapeutic Targets," addresses the link between local hypoxia and the induction of chronic long-lasting inflammation in the context of the tissue accumulation of the β-sheet-rich amyloid deposits. The authors propose that the induction of cell death mechanisms in conjunction with local tissue hypoxia associated with the amyloid accumulation constitutes important events for the pathogenesis and progression of amyloidosis. The authors hypothesize that molecules released by necrotic cells activate inflammatory responses through Damage-Associated Molecular Patterns (DAMPs) and evoke activation of HIF-1α/NF-κB-dependent pathways suggesting that modulation of these cellular paths may constitute new therapeutic strategies. Another manuscript of the special issue focuses on
[14] Unusual Pain Disorders – What Can Be Learned from Them?
- Authors: J. Sachau, D. Kersebaum, R. Baron, A. Dickenson
- Year: 2021
- Venue: Journal of Pain Research
- URL: https://www.semanticscholar.org/paper/11a1bceba8d0d4afec7cc632571f8f6d014c648d
- DOI: 10.2147/JPR.S287603
- PMID: 33758536
- PMCID: 7980038
- Citations: 3
- Influential citations: 1
- Summary: Pain therapists and researchers should be aware of these rare and unusual pain disorders as they offer the unique opportunity to study mechanisms, identify new druggable targets and finally because early diagnosis might save many patient lives.
- Evidence snippets:
- Snippet 1 (score: 0.408) > In the past, systemic transthyretin (ATTR) amyloidosis was classified as a poorly or completely untreatable, rapidly progressive disease.Painful polyneuropathy is an early symptom that significantly reduces the quality of life in the affected patients.Extensive research within the last years has led to the identification of new -also non-invasive -diagnostic tools and promising therapeutic options to improve the overall treatment of patients, making it critical and relevant to raise awareness of this disease. > Systemic ATTR amyloidosis results from 1) a mutation causing a change in the TTR protein structure, ie hereditary ATTR (ATTRv) amyloidosis or 2) a process during ageing, ie wild-type ATTR (ATTwt) amyloidosis.Physiologically, transthyretin (TTR) acts as a carrier protein to transport thyroxin and retinol (vitamin A) via associating with its binding protein and is mostly synthesized by the liver.In ATTR amyloidosis, the TTR protein loses its stable tetrameric conformation, dissociates into monomers that are misfolded, aggregate and finally deposit as amyloid into tissues and organs (Figure 1). 43Since amyloid deposition can theoretically affect every organ, the clinical manifestation is very heterogenous.This clinical chameleon complicates the correct diagnosis resulting in delayed start of therapy and hence rapid disease progression. > Pain is a common clinical symptom in ATTRv amyloidosis and led to the first description of the disease by Corino Andrade in 1952 who observed an increased incidence of a rapid progressive painful foot disease in families from north of Portugal, that was associated with symptoms of a peripheral neuropathy and a high mortality rate. 44In a study by Lozeron et al, pain was present in more than half of the patients with demyelinating ATTRv amyloid polyneuropathy. 45Different pathophysiological mechanisms have been proposed that lead to nerve fiber damage in ATTRv amyloidosis. 46
[15] Iatrogenic Dementia: Providing Insight into Transmissible Subtype of Alzheimer’s Disease, Creutzfeldt–Jakob Disease and Cerebral Amyloid Angiopathy
- Authors: Stella Karatzetzou, Serafeim Ioannidis, E. Konstantinopoulou, D. Parisis, Theodora Afrantou et al.
- Year: 2025
- Venue: Biomolecules
- URL: https://www.semanticscholar.org/paper/24ab859dfee1209eeecb40472a4c291945fcadee
- DOI: 10.3390/biom15040522
- PMID: 40305264
- PMCID: 12025122
- Summary: The present review aims to explore the distinct features of acquired dementia encompassing a history of potential exposure and relatively early age of onset, highlighting transmission potential with a rather prion-like pattern.
- Evidence snippets:
- Snippet 1 (score: 0.406) > An extensive literature review was conducted in order to elucidate the transmission potential of dementia in a clinical setting of a neurodegenerative disease. Original articles dealing with the underlying pathomechanisms and the distinct clinical phenotype of acquired type of cognitive decline were identified and reviewed. > Proteinopathies represent a group of diseases characterized by the deposition of misfolded protein aggregates. These abnormally accumulated proteinaceous structures in the brain can trigger the pathological cascade that leads to neurologic dysfunction and subsequently to clinically evident neurodegeneration with manifestations within the dementia spectrum. Prion proteins, Aβ and tau being among them, stand for the pathological hallmarks of various neurodegenerative diseases, reflecting the main underlying pathomechanisms [62]. > In an attempt to explore the underlying molecular pathways involved in iatrogenically triggered amyloidogenesis, Bonilauri [71] proposed a classification of relevant cases according to the nature of medical intervention and subsequent impact on amyloid proteins. As far as neurodegenerative disorders including AD, CAA and CJD are concerned, the amyloidogenic cascade can be initiated by the transmission of amyloid oligomers and fibrils through different medical procedures. Already found in a pre-existing amyloid state in cases of organ/tissue transplantation or contaminated instruments, amyloid seeds may enhance and ultimately accelerate the formation of insoluble mature amyloid fibrils, thus disrupting normal tissue functionality and leading to a wide range of clinical manifestations. According to growing experimental evidence, the ability of misfolded proteins to continuously induce the conversion of similar physiological proteins into a pathological form through seeding is a characteristic feature not only of prion protein but also Aβ, tau and a-synuclein [10]. A common molecular mechanism seems to be responsible for the replication and spread of these different misfolded protein aggregates within the central nervous system [3]. > It is of great interest that a-synuclein might exhibit at least some of the misfolded protein properties, including the generation of a-synuclein aggregates that act as a template for the formation of seeds, propagation between cells and spread across certain anatomical pathways.
[16] Targeting Hepatic Stellate Cells for the Prevention and Treatment of Liver Cirrhosis and Hepatocellular Carcinoma: Strategies and Clinical Translation
- Authors: Hao Xiong, Jinsheng Guo
- Year: 2025
- Venue: Pharmaceuticals
- URL: https://www.semanticscholar.org/paper/76e92127053136900f7e3f10e2c9278251ced5d2
- DOI: 10.3390/ph18040507
- PMID: 40283943
- PMCID: 12030350
- Citations: 8
- Summary: HSC-targeted approaches using specific surface markers and receptors may enable the selective delivery of drugs, oligonucleotides, and therapeutic peptides that exert optimized anti-fibrotic and anti-HCC effects.
- Evidence snippets:
- Snippet 1 (score: 0.402) > Significant progress has been made in elucidating the cellular and molecular mechanisms of liver fibrosis; however, only a few findings have been successfully translated into clinical applications. Firstly, the high cost of drug development and target validation necessitates prolonged timelines and substantial financial investment. Secondly, as regulatory requirements become more stringent, there is an increasing demand for drugs with well-defined clinical efficacy and safety profiles. Moreover, the efficacy observed in animal models often fails to fully translate to clinical settings due to differences in pharmacokinetics, extracellular matrix (ECM) cross-linking, and disease pathophysiology. Despite advancements in anti-fibrotic drug development, accurately identifying ideal noninvasive biomarkers for fibrotic activity and establishing consensus on optimal clinical endpoints remain significant challenges [113,114]. > Currently, addressing the underlying cause remains the only proven strategy to halt or reverse liver fibrosis progression, while the development of effective anti-fibrotic therapies continues to pose a major challenge in liver disease management. Over the past few decades, substantial progress has been made in elucidating the cellular and molecular mechanisms underlying liver fibrosis. Liver fibrosis is a complex pathological change involving multiple cells, factors, and pathways, and the study of the cellular and molecular mechanisms of its occurrence and development provides an important theoretical basis and therapeutic target for clinical drug development. It is anticipated that improved animal models and well-designed clinical trials will facilitate the successful translation of anti-fibrotic research into effective clinical treatments in the near future.
[17] Structural Basis for Vital Function and Malfunction of Serum Amyloid A: an Acute-Phase Protein that Wears Hydrophobicity on Its Sleeve
- Authors: O. Gursky
- Year: 2020
- Venue: Current Atherosclerosis Reports
- URL: https://www.semanticscholar.org/paper/1c041f2c08905510171a02b26816bb26f11bd435
- DOI: 10.1007/s11883-020-00888-y
- PMID: 32968930
- PMCID: 7511256
- Citations: 28
- Summary: Recent advances in understanding of the structure-function relationship of SAA are summarized, its newly emerging beneficial roles in lipid transport and inflammation control are outlined, and factors that critically influence its misfolding in AA amyloidosis are discussed.
- Evidence snippets:
- Snippet 1 (score: 0.402) > The major therapies for AA amyloidosis target the underlying inflammatory disease and include antibacterial, anti-inflammatory, and immunosuppressive drugs; dialysis and kidney transplant are used to treat advanced renal damage. The drugs reduce SAA levels, which reduces the amyloid load and reverses the disease. However, factors other than amyloid load can also contribute to clinical symptoms [91], and the existing treatments are not always efficient [92]. For example, antiinflammatory approaches are unsuited for patients with idiopathic AA amyloidosis without the underlying chronic inflammation [3•, 93], necessitating the development of new therapies. > Some therapeutic approaches target the protein misfolding pathway. This includes interactions with GAGs, particularly heparan sulfate, that dissociate SAA from HDL and accelerate fibrillogenesis [83,94]. Sulfonated or sulfated GAG mimetics were proposed to block SAA-GAG interactions. Unfortunately, a sulfonated small molecule drug eprodisate failed to show efficiency in phase-3 clinical trials and was discontinued in 2016 (ClinicalTrials.gov Identifier: NCT01215747). > Other approaches propose to block amyloidogenic segments of SAA by using complementary peptides [95] or lipids. In particular, decreasing triglyceride levels in the core of HDL helps retain SAA and other apolipoproteins on the HDL surface and thereby retard fibrillogenesis [96]. If so, existing triglyceride-lowering therapies hold promise for treating AA amyloidosis, including cases without the underlying inflammation. Conversely, increased plasma triglycerides are expected to augment AA amyloidosis. This idea is consistent with clinical studies reporting a direct correlation between idiopathic AA amyloidosis and obesity, a condition wherein plasma triglycerides are elevated [93,97]. > Another proposed approach uses a cell-based system to identify nontoxic inhibitors of SAA fibrillogenesis regardless of specific mechanisms of action [98]. This approach targets the protein quality control cellular machinery and has a high screening capability, which helps select candidates for future animal model studies.
[18] Familial Mediterranean Fever: Assessing the Overall Clinical Impact and Formulating Treatment Plans
- Authors: D. Rigante, R. Manna
- Year: 2019
- Venue: Mediterranean Journal of Hematology and Infectious Diseases
- URL: https://www.semanticscholar.org/paper/3675c7536975c9ceacb1d96eb667a053c06d0fc7
- DOI: 10.4084/MJHID.2019.027
- PMID: 31205631
- PMCID: 6548206
- Citations: 50
- Influential citations: 2
- Summary: Colchicine, a tricyclic alkaloid with anti-microtubule and anti-inflammatory properties, is the bedrock of FMF management: daily administration of colchicines prevents the recurrence ofFMF attacks and the development of secondary AA amyloidosis.
- Evidence snippets:
- Snippet 1 (score: 0.396) > 112 Diagnosis of FMF is mainly made on the basis of the typical clinical findings in association with the peculiar ethnicity, family history, and response to colchicine.113,114 Despite the great steps in our understanding of FMF, we still have a number of hanging questions: for instance, what is the exact role of additional genes in the definition of the final FMF phenotype, what is the pathophysiology of the disease in patients with only one MEFV mutation or in those without any MEFV mutation, which are further unexplored inflammatory pathways which might be involved in the disease expression or progression to amyloidosis, and so on.The progress in the knowledge of genetic determinants of FMF could constitute a significant step towards the understanding of the human genome power and general mechanisms of inflammation with future relevant therapeutic implications. Wier awareness of FMF will probably reduce the diagnostic delay in recognition of the disease and positively affect the quality of life of patients who will have a lower risk of long-term morbidity and complications.
[19] Human Dermal Fibroblast: A Promising Cellular Model to Study Biological Mechanisms of Major Depression and Antidepressant Drug Response
- Authors: P. Mesdom, R. Colle, É. Lebigot, S. Trabado, Eric Deflesselle et al.
- Year: 2020
- Venue: Current Neuropharmacology
- URL: https://www.semanticscholar.org/paper/79368e365458486de96794333613c12a6063bf54
- DOI: 10.2174/1570159X17666191021141057
- PMID: 31631822
- PMCID: 7327943
- Citations: 12
- Summary: This review highlights the great and still underused potential of HDF, which stands out as a very promising tool in the understanding of MDD and AD mechanisms of action.
- Evidence snippets:
- Snippet 1 (score: 0.391) > Background: Human dermal fibroblasts (HDF) can be used as a cellular model relatively easily and without genetic engineering. Therefore, HDF represent an interesting tool to study several human diseases including psychiatric disorders. Despite major depressive disorder (MDD) being the second cause of disability in the world, the efficacy of antidepressant drug (AD) treatment is not sufficient and the underlying mechanisms of MDD and the mechanisms of action of AD are poorly understood. Objective The aim of this review is to highlight the potential of HDF in the study of cellular mechanisms involved in MDD pathophysiology and in the action of AD response. Methods The first part is a systematic review following PRISMA guidelines on the use of HDF in MDD research. The second part reports the mechanisms and molecules both present in HDF and relevant regarding MDD pathophysiology and AD mechanisms of action. Results HDFs from MDD patients have been investigated in a relatively small number of works and most of them focused on the adrenergic pathway and metabolism-related gene expression as compared to HDF from healthy controls. The second part listed an important number of papers demonstrating the presence of many molecular processes in HDF, involved in MDD and AD mechanisms of action. Conclusion The imbalance in the number of papers between the two parts highlights the great and still underused potential of HDF, which stands out as a very promising tool in our understanding of MDD and AD mechanisms of action
[20] New therapeutic targets in rare genetic skeletal diseases
- Authors: M. Briggs, Peter A. Bell, M. Wright, K. A. Pirog
- Year: 2015
- Venue: Expert Opinion on Orphan Drugs
- URL: https://www.semanticscholar.org/paper/1363107f71ae6d2d60abca471cddf3da5d13644b
- DOI: 10.1517/21678707.2015.1083853
- PMID: 26635999
- PMCID: 4643203
- Citations: 37
- Influential citations: 1
- Summary: An overview of disease mechanisms that are shared amongst groups of different GSDs and potential therapeutic approaches that are under investigation are described to generate critical mass for the identification and validation of novel therapeutic targets and biomarkers.
- Evidence snippets:
- Snippet 1 (score: 0.390) > proteins of the cartilage ECM such as type II collagen [50]. However, emerging knowledge suggests that the primary genetic defect may be less important than the cells' response to the expression of the mutant gene product [107]. Moreover, the largely overlooked response of a cell (i.e. chondrocyte) to the abnormal extracellular environment is also important for disease progression as illustrated by several GSDs discussed in this review. > It is important that 'omics'-based approaches and technologies are systematically applied to the study of rare GSDs so that definitive reference profiles and disease signatures are generated for each phenotype. These can then be used in a Systems Biology approach to identify both common and dissimilar pathological signatures and disease mechanisms. This approach is entirely dependent upon relevant in vitro and in vivo models (and also novel 'disease-mechanism phenocopies' [107]) for testing new diagnostic and prognostic tools and for determining the molecular mechanisms that underpin the pathophysiology so that effective therapeutic treatments can be developed and validated. This approach will eventually lead to personalized treatments and care strategies centred on shared disease mechanisms with the use of relevant biomarkers to monitor the efficacy of treatment and disease progression. > It is vital that all relevant stakeholders are involved from the outset in defining the appropriate outcomes of any potential therapeutic regime. The perceptions of a successful therapy can differ widely between the clinical academic community and the relevant patient-support groups and it is vital that there is engagement on all these issues. > In summary, the identification of causative genes and mutations for GSDs over the last 20 years, coupled with the generation and in-depth analysis of a plethora of relevant cell and mouse models, has derived new knowledge on disease mechanisms and suggested potential therapeutic targets. The fast-evolving hypothesis that clinically disparate diseases can share common disease mechanisms is a powerful concept that will generate critical mass for the identification and validation of novel therapeutic targets and biomarkers.
Notes
- This provider combines
search_papers_by_relevancewithsnippet_search. - No synthesis or second-stage model call is performed.
ADan Amyloidosis (Familial Danish Dementia) — Comprehensive Disease Characteristics Report
Executive summary
ADan amyloidosis is a rare, autosomal dominant, Mendelian neurodegenerative disorder historically described in a Danish pedigree and commonly referred to as familial Danish dementia (FDD) or heredopathia ophthalmo‑oto‑encephalica. It is caused by a pathogenic stop-region duplication in ITM2B (BRI2) that creates an extended precursor protein (277 aa) and a de novo 34–amino‑acid amyloidogenic peptide (ADan) produced by furin-like processing. The clinical syndrome classically begins with early cataracts, followed by progressive hearing loss, then cerebellar ataxia/tremor and later progressive dementia, accompanied by prominent cerebral and retinal amyloid angiopathy and tau neurofibrillary pathology. Recent (2023–2024) work has strengthened links between ITM2B/BRI2 biology and microglial pathways (including TREM2 processing/phagocytosis) and has quantitatively mapped sequence determinants of ADan amyloid nucleation. (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, yin2024functionalbri2trem2interactions pages 1-2, arber2024microgliacontributeto pages 1-2, martin2024amyloids“atthe pages 3-5)
1. Disease information
1.1 What is the disease?
Familial Danish dementia (FDD) is a hereditary neurodegenerative disorder characterized by early ophthalmologic disease (cataracts), progressive hearing loss, cerebellar ataxia with tremor, and later progressive dementia. Neuropathology includes cerebral and retinal amyloid angiopathy with deposition of the ADan amyloid peptide (and often co-deposition of Aβ) and neurofibrillary tangles resembling Alzheimer-like tau pathology. (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11)
1.2 Key identifiers (with availability constraints)
A curated disease mapping from OpenTargets identifies ADan amyloidosis as MONDO_0007297 and connects it to ITM2B with supporting literature PMIDs (e.g., PMID: 10391242; PMID: 10781099) in the association evidence. (OpenTargets Search: familial Danish dementia,ADan amyloidosis-ITM2B)
Other identifiers (OMIM, Orphanet, MeSH, ICD-10/ICD-11) were not directly retrieved in the current tool context; they are flagged as missing rather than inferred. (OpenTargets Search: familial Danish dementia,ADan amyloidosis-ITM2B)
| Identifier type | Value | Synonyms / alternate names | Primary supporting citation IDs |
|---|---|---|---|
| MONDO | MONDO_0007297 | ADan amyloidosis; familial Danish dementia (FDD); heredopathia ophthalmo-oto-encephalica | (OpenTargets Search: familial Danish dementia,ADan amyloidosis-ITM2B, garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) |
| OMIM | Not retrieved in current tool context | familial Danish dementia (FDD); heredopathia ophthalmo-oto-encephalica | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) |
| Orphanet | Not retrieved in current tool context | ADan amyloidosis; familial Danish dementia (FDD) | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) |
| MeSH | Not retrieved in current tool context | familial Danish dementia; chromosome 13 dementia | (rostagno2005chromosome13dementias pages 1-2, rostagno2005chromosome13dementias pages 4-6) |
| ICD | Not retrieved in current tool context | familial Danish dementia; ADan amyloidosis | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) |
Table: This table summarizes the key identifiers and accepted names for ADan amyloidosis based only on evidence retrieved in the current tool context. It is useful for mapping the disease entity across nomenclature systems while clearly flagging identifiers not yet directly retrieved.
1.3 Synonyms / alternative names
Common synonyms include: - Familial Danish dementia (FDD) (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) - Heredopathia ophthalmo‑oto‑encephalica (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) - “Chromosome 13 dementias” (category including familial British and Danish dementias) (rostagno2005chromosome13dementias pages 1-2)
1.4 Evidence provenance
Much clinical characterization derives from aggregated disease-level descriptions anchored in a multigenerational pedigree/case series (e.g., 13 cases across five generations) and from neuropathologic analyses of post-mortem tissue; mechanistic insights largely derive from animal models, iPSC-derived cell types, and in vitro systems. (rostagno2005chromosome13dementias pages 4-6, yin2024functionalbri2trem2interactions pages 1-2, todd2016oxidativestressand pages 11-13)
2. Etiology
2.1 Disease causal factors
Primary cause (genetic): FDD is caused by a 10-nucleotide duplication near the ITM2B/BRI2 stop codon (reported as 787_796dupTTTAATTTGT) producing a mutant 277-aa precursor (often described as p.Ser266PhefsTer11 / p.Ser266PhefsX11). Furin-like processing at the BRI2 C-terminus releases ADan, a 34-aa amyloidogenic peptide that deposits in brain and vessel walls. (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 2-4)
Two mechanistic classes are repeatedly discussed: 1) Toxic gain-of-function due to aggregation/accumulation of ADan (and ADan-associated CAA). (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) 2) Loss-of-function due to reduced levels of functional mature BRI2 (mBRI2), impairing physiological roles at synapses and altering APP processing. (matsuda2011increasedaβppprocessing pages 4-5, yin2021danishandbritish pages 1-2)
| Gene (symbol, name) | Inheritance | Key pathogenic variant | Variant type / mechanism | Pathogenic product | Processing enzyme / cleavage | Key evidence / PMID |
|---|---|---|---|---|---|---|
| ITM2B (integral membrane protein 2B; also BRI2) | Autosomal dominant | NM_021999.4(ITM2B):c.787_796dup (reported in review as 787_796dupTTTAATTTGT); protein consequence p.Ser266PhefsTer11 / p.Ser266PhefsX11 (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) | 10-nt duplication immediately before the normal stop codon; creates a frameshift/stop-loss–like C-terminal extension, producing a 277-aa mutant precursor instead of the normal 266-aa protein (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 1-2) | Mutant precursor ADanPP / mutant BRI2-277; proteolytic release of ADan, a 34-aa amyloidogenic peptide; deposits in parenchyma and vasculature (CAA), often with Aβ co-deposition (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11) | Furin-like convertase cleavage at Arg243–Glu244 / ...KGIQKR243↓E244... releases the C-terminal peptide that becomes ADan (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 2-4) | Primary review evidence cites original disease-gene papers including PMID: 10391242 and PMID: 10781099 via curated disease-target association (OpenTargets Search: familial Danish dementia,ADan amyloidosis-ITM2B); supporting mechanistic reviews/details (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 2-4) |
| ITM2B (integral membrane protein 2B; BRI2) | Autosomal dominant | Same Danish mutation above; human FDD brain data also support pathogenic effect through reduced mature BRI2 levels (matsuda2011increasedaβppprocessing pages 4-5) | Dual mechanism proposed: toxic gain of function from amyloidogenic ADan generation plus loss of normal BRI2 function, including impaired inhibition of APP processing and altered synaptic physiology (matsuda2011increasedaβppprocessing pages 4-5, yin2021danishandbritish pages 1-2, yin2024functionalbri2trem2interactions pages 1-2) | Increased APP-derived metabolites/Aβ in human FDD brain; ADan/Aβ co-accumulation; reduced functional mature BRI2 at synapses in KI models (matsuda2011increasedaβppprocessing pages 4-5, yin2021danishandbritish pages 1-2) | BRI2 normally undergoes furin cleavage; disease mutation alters downstream peptide composition and is also associated with reduced mature BRI2 stability/maturation (rostagno2005chromosome13dementias pages 2-4, yin2021danishandbritish pages 1-2) | Human FDD brain: soluble Aβ42 11-fold, insoluble Aβ42 125-fold, soluble Aβ40 27-fold, insoluble Aβ40 38-fold vs control in one analyzed case (matsuda2011increasedaβppprocessing pages 4-5); synaptic LOF model support (yin2021danishandbritish pages 1-2) |
Table: This table summarizes the currently supported genetic cause and pathogenic mechanism of ADan amyloidosis/familial Danish dementia from the retrieved evidence. It highlights the ITM2B Danish duplication, its processing into ADan, and the evidence for both toxic peptide accumulation and BRI2 loss-of-function.
2.2 Risk factors
Genetic: The causal ITM2B duplication is the principal risk factor; the disorder is autosomal dominant. (rostagno2005chromosome13dementias pages 4-6, choudhury2025pathologicalmechanismsof pages 1-2)
Environmental: No environment-specific risk factors were retrieved in this evidence set.
2.3 Protective factors
No definitive protective genetic or environmental factors were retrieved. However, mechanistic genetic modification in models (APP haplodeficiency) prevents synaptic/memory deficits, supporting APP-derived products as mediators of some phenotypes (this is not a population protective factor but a mechanistic modifier). (matsuda2011increasedaβppprocessing pages 4-5)
2.4 Gene–environment interactions
No gene–environment interaction evidence was retrieved.
3. Phenotypes
3.1 Core phenotype spectrum and characteristics
Clinical course described in reviews: - Cataracts often in the third decade (median visual symptom onset 27 years in a 13-case series). (rostagno2005chromosome13dementias pages 4-6) - Hearing loss typically 10–20 years after ocular symptoms, often severe by the fifth decade. (garringer2010modelingfamilialbritish pages 3-4) - Cerebellar ataxia with intention tremor in adulthood, progressive. (garringer2010modelingfamilialbritish pages 3-4, choudhury2025pathologicalmechanismsof pages 1-2) - Dementia probably beginning in the sixth decade, progressive and fatal. (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) - Other reported features include psychosis and spastic paralysis/spasticity in review literature. (rostagno2005chromosome13dementias pages 10-11)
| Clinical feature | Phenotype type (symptom/sign/lab/pathology) | Typical timing (with any quantitative age data) | Notes (frequency if available) | Suggested HPO term(s) | Supporting citation IDs |
|---|---|---|---|---|---|
| Early cataracts / visual impairment | Sign / symptom | Usually first manifestation; often before age 30; median visual symptom onset 27 years | Described as early and prominent in the original Danish pedigree; ophthalmic disease precedes neurologic decline by years to decades | HP:0000518 Cataract; HP:0000505 Visual impairment | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11) |
| Progressive hearing loss / deafness | Symptom / sign | Typically appears 10–20 years after ocular problems; often severe by the 5th decade | Major non-cognitive feature; contributes substantially to disability | HP:0000365 Hearing impairment; HP:0000407 Sensorineural hearing impairment | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11) |
| Cerebellar ataxia | Sign | Develops in adulthood after sensory manifestations; progressive | Core neurologic phenotype in reviews and case descriptions | HP:0001251 Ataxia; HP:0002060 Cerebellar ataxia | (garringer2010modelingfamilialbritish pages 3-4, choudhury2025pathologicalmechanismsof pages 1-2, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11) |
| Intention tremor | Sign | Adult onset; accompanies cerebellar syndrome | Reported as part of the cerebellar phenotype | HP:0002080 Intention tremor; HP:0001337 Tremor | (garringer2010modelingfamilialbritish pages 3-4) |
| Progressive dementia / cognitive deterioration | Symptom / syndrome | Probably begins in the 6th decade; chronic progressive course | Central late manifestation; disease is neurodegenerative and usually fatal | HP:0000726 Dementia; HP:0100543 Cognitive impairment | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 1-2, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11) |
| Psychosis | Symptom / behavioral change | Adult onset; timing not well quantified in retrieved evidence | Reported in reviews, but frequency not available in current evidence set | HP:0000709 Psychosis | (rostagno2005chromosome13dementias pages 10-11) |
| Spasticity / spastic paralysis | Sign | Adult onset; progressive in advanced disease | Mentioned in review literature; severity likely increases with disease progression | HP:0001257 Spasticity; HP:0007020 Spastic paraplegia | (rostagno2005chromosome13dementias pages 10-11) |
| Gait abnormality / wide-based gait | Sign | Adult to later-stage disease; progressive | Seen clinically and recapitulated in transgenic/rat models; likely related to cerebellar and white-matter pathology | HP:0001288 Gait disturbance; HP:0002136 Wide-based gait | (choudhury2025pathologicalmechanismsof pages 1-2, garringer2010modelingfamilialbritish pages 4-6, garringer2010modelingfamilialbritish pages 6-7) |
| Motor dysfunction | Symptom / sign | Progressive with age; later-stage manifestation in both humans and models | Rat model links this to cerebellar ADan-CAA, demyelination, axonal loss, and fibrinogen leakage | HP:0004305 Motor delay/abnormality (general); HP:0001270 Motor impairment | (choudhury2025pathologicalmechanismsof pages 1-2, choudhury2025pathologicalmechanismsof pages 15-16) |
| White-matter abnormalities on neuroimaging | Imaging / pathology | Reported in symptomatic adults; timing not well quantified | Related imaging findings include periventricular white-matter changes and ventricular dilatation | HP:0002500 Abnormal cerebral white matter morphology; HP:0002119 Ventriculomegaly | (rostagno2005chromosome13dementias pages 4-6) |
| Cerebral amyloid angiopathy (CAA), including retinal and cerebral vascular amyloid | Pathology | Progressive age-related accumulation over disease course | Major histopathologic hallmark; deposits are predominantly vascular, including retina and small cerebral vessels/capillaries | HP:0031630 Cerebral amyloid angiopathy; HP:0100651 Retinal vascular abnormality | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11) |
| Parenchymal ADan amyloid deposition | Pathology | Progressive; present in affected brain, though compact plaques may be absent | ADan is a 34-aa peptide derived from mutant ITM2B/BRI2; some reports emphasize pre-amyloid/non-compact deposits rather than classic plaques | HP:0011972 Amyloidosis; HP:0012759 Abnormality of CNS physiology/pathology | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, garringer2010modelingfamilialbritish pages 10-12) |
| Co-deposition of Aβ with ADan | Pathology / biochemical | Detected in affected brain during established disease | Important mechanistic feature linking FDD to Alzheimer-like amyloid biology | HP:0011972 Amyloidosis | (garringer2010modelingfamilialbritish pages 3-4, garringer2010modelingfamilialbritish pages 9-10, matsuda2011increasedaβppprocessing pages 4-5, garringer2010modelingfamilialbritish pages 10-12) |
| Neurofibrillary tangles / tau pathology | Pathology | Later-stage neurodegenerative pathology | Prominent in hippocampus and limbic structures; paired helical filaments reported | HP:0002185 Neurofibrillary tangles | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 1-2, rostagno2005chromosome13dementias pages 4-6) |
| Death / shortened survival | Outcome | Median age at death 58 years in a series of 13 cases across 5 generations | Indicates substantial mortality burden; no modern survival curves retrieved | HP:0003819 Premature death | (rostagno2005chromosome13dementias pages 4-6) |
Table: This table summarizes the major clinical and pathologic features reported for familial Danish dementia/ADan amyloidosis, including approximate timing and suggested HPO mappings. It is useful for disease knowledge-base curation and phenotype annotation.
3.2 Quality-of-life impact
While formal QoL instruments were not retrieved, the phenotype timing suggests a substantial, staged disability burden driven by early visual impairment, progressive deafness, gait/motor dysfunction, and eventual dementia requiring comprehensive supportive care. (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6)
4. Genetic / molecular information
4.1 Causal gene
- ITM2B (BRI2) encodes a type II transmembrane glycoprotein with a BRICHOS domain and undergoes furin processing at a C-terminal site (…KGIQKR243↓E244…). (rostagno2005chromosome13dementias pages 2-4)
4.2 Pathogenic variant and functional consequence
- Danish FDD duplication creates an extended precursor and generates ADan (34 aa). (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6)
- In knock-in mouse models of Danish/British dementias, pathogenic ITM2B mutations reduce BRI2 protein stability/maturation, lowering functional mature BRI2 at synapses and producing synaptic physiology abnormalities consistent with loss of function. (yin2021danishandbritish pages 1-2)
4.3 Modifier pathways and intersecting biology
APP processing / Aβ: In one human FDD brain compared with one control, APP/Aβ metabolites were markedly increased (e.g., soluble Aβ42 11×, insoluble Aβ42 125×). These data support a mechanistic link between BRI2 loss-of-function and increased APP processing/Aβ burden, though human sample size was extremely limited in the retrieved evidence. (matsuda2011increasedaβppprocessing pages 4-5)
Microglia / TREM2 (recent development, 2024): BRI2 interacts with TREM2 and inhibits TREM2 processing. In Bri2 loss-of-function contexts, TREM2-CTF and soluble sTREM2 are elevated, and microglial phagocytosis is reduced—supporting BRI2 as a microglial regulatory node relevant to AD and ITM2B-associated dementias. (yin2024functionalbri2trem2interactions pages 1-2)
5. Environmental information
No specific environmental, lifestyle, toxin, or infectious triggers were identified in the retrieved evidence.
6. Mechanism / pathophysiology
6.1 Causal chain (integrated current understanding)
1) ITM2B Danish duplication → extended BRI2 precursor (277 aa) (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) 2) Furin-like cleavage near residue 243/244 → release of ADan 34-mer (rostagno2005chromosome13dementias pages 2-4, rostagno2005chromosome13dementias pages 4-6) 3) ADan misfolding/aggregation and deposition predominantly in vessels (CAA, including retinal vasculature) with frequent co-deposition of Aβ (rostagno2005chromosome13dementias pages 4-6, matsuda2011increasedaβppprocessing pages 4-5) 4) Downstream neurodegenerative cascades: vascular dysfunction/BBB leakage and neuroinflammation (rat KI model), oxidative stress/mitochondria-mediated apoptosis (cell models), synaptic dysfunction and altered neurotransmission (KI models), and tau neurofibrillary pathology. (choudhury2025pathologicalmechanismsof pages 1-2, todd2016oxidativestressand pages 11-13, yin2021danishandbritish pages 1-2, garringer2010modelingfamilialbritish pages 3-4)
6.2 Oxidative stress and mitochondria-mediated apoptosis (cell models)
In differentiated SH-SY5Y cells, pyroglutamate-modified ADan (ADan pE) triggers rapid ROS generation and intrinsic apoptosis markers including cytochrome c release, loss of mitochondrial membrane potential, and caspase-3 activation. An abstract-supported mechanistic statement from the paper includes that “ADan pE provoked DNA fragmentation after only 8 hour peptide challenge” and that “ADan pE treatment caused the release of cyt c…” with loss of mitochondrial membrane potential. (todd2016oxidativestressand pages 11-13, todd2016oxidativestressand pages 13-14)
Intervention evidence in vitro: the antioxidant Trolox reduced ROS-linked caspase-3 activation, and an ADan C-terminal monoclonal antibody (1B7) “completely neutralize[d] ADan-mediated cytotoxicity,” supporting oxidative stress and ADan sequence-specific toxicity as targetable processes (preclinical). (todd2016oxidativestressand pages 13-14, todd2016oxidativestressand pages 11-13, todd2016oxidativestressand pages 14-16)
Visual evidence (figures): Todd et al. show cytochrome c redistribution and Trolox-sensitive caspase-3 activation in figure panels. (todd2016oxidativestressand media f315265f, todd2016oxidativestressand media d7873ce6)
6.3 Synaptic dysfunction and glutamatergic transmission (KI models)
In Danish and British dementia knock-in mice, spontaneous glutamate release and AMPAR-mediated responses are decreased while short-term synaptic facilitation is increased, resembling Itm2b knockout phenotypes and supporting a BRI2 loss-of-function contribution. (yin2021danishandbritish pages 1-2)
6.4 Microglia, innate immunity, and TREM2 (2023–2024)
Recent work highlights cell-type specificity: - ITM2B expression is ~34-fold higher in microglia than neurons and ~15-fold higher than astrocytes in iPSC-derived models, and ABri peptide is detectable in patient iPSC-microglia lysates/conditioned media (British dementia), suggesting microglia can be a major cellular source of ITM2B-derived amyloid peptides; the mechanism is relevant to ADan generation because both ABri and ADan derive from ITM2B stop-region mutations and furin cleavage. (arber2024microgliacontributeto pages 1-2, arber2023microgliaproducethe pages 1-4) - In 2024 EMBO Reports, BRI2 directly interacts with TREM2 and inhibits its processing; Bri2 deletion reduces microglial phagocytosis, suggesting innate immune dysfunction as a convergent mechanism relevant to ITM2B dementias and AD. (yin2024functionalbri2trem2interactions pages 1-2)
6.5 Sequence-level determinants of ADan amyloid nucleation (2024)
A 2024 deep mutagenesis/random extension study provides quantitative mapping of the ADan amyloid core: ADan showed strong nucleation in a yeast-based assay (25.14 ± 3.41% growth; ~2× Aβ42), while ABri was essentially non-nucleating in that assay. The authors map a putative core region between residues 20–26 (L20–F26) and quantify mutation effects on nucleation, supporting the concept that stop-loss extensions can generate de novo amyloid motifs. (martin2024amyloids“atthe pages 3-5)
Suggested ontology terms (examples)
- GO biological processes: amyloid fibril formation; regulation of APP processing; oxidative stress response; mitochondrial outer membrane permeabilization; intrinsic apoptotic signaling; microglial phagocytosis; regulation of TREM2 processing. (matsuda2011increasedaβppprocessing pages 4-5, yin2024functionalbri2trem2interactions pages 1-2, todd2016oxidativestressand pages 13-14)
- CL cell types: microglia (CL:0000129); neurons; astrocytes; vascular smooth muscle cells; endothelial cells. (rostagno2005chromosome13dementias pages 1-2, arber2024microgliacontributeto pages 1-2)
7. Anatomical structures affected
7.1 Organ/tissue systems
- Eye (lens/retina): early cataracts and retinal amyloid angiopathy. (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6)
- Central nervous system: cerebellum (ataxia; subpial/leptomeningeal vascular pathology in models), hippocampus (tangles; cognitive impairment), widespread cerebral vasculature (CAA). (rostagno2005chromosome13dementias pages 4-6, choudhury2025pathologicalmechanismsof pages 1-2, garringer2010modelingfamilialbritish pages 3-4)
7.2 Cell/tissue pathology
- Vascular walls and capillaries are prominent sites of amyloid deposition (CAA), with neuroinflammatory responses including activated microglia in proximity to deposits in models. (rostagno2005chromosome13dementias pages 4-6, garringer2010modelingfamilialbritish pages 6-7)
UBERON suggestions (examples): UBERON:0000955 brain; UBERON:0002037 cerebellum; UBERON:0002421 hippocampus; UBERON:0000970 retina; UBERON:0000965 lens. (rostagno2005chromosome13dementias pages 4-6, choudhury2025pathologicalmechanismsof pages 1-2)
8. Temporal development
- Onset pattern: insidious, chronic, progressive. Sensory manifestations precede neurodegeneration (cataracts → hearing loss → ataxia → dementia). (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6)
- Progression: progressive worsening over decades; median death in one pedigree series at 58 years. (rostagno2005chromosome13dementias pages 4-6)
9. Inheritance and population
9.1 Inheritance
Autosomal dominant inheritance is consistently reported in reviews and mechanistic summaries. (choudhury2025pathologicalmechanismsof pages 1-2, rostagno2005chromosome13dementias pages 4-6)
9.2 Epidemiology
No population prevalence/incidence estimates were retrieved in the current evidence set. The most concrete human aggregation in the retrieved evidence is a 13-case, five-generation pedigree-based series. (rostagno2005chromosome13dementias pages 4-6)
10. Diagnostics
10.1 Practical diagnostic approach (current evidence)
- Genetic confirmation: identify the ITM2B Danish duplication (c.787_796dupTTTAATTTGT) in the setting of compatible phenotype and family history. (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6)
- Neuropathology: predominance of cerebral and retinal amyloid angiopathy with ADan deposits, frequent co-deposition of Aβ, and hippocampal neurofibrillary tangles. (rostagno2005chromosome13dementias pages 4-6, garringer2010modelingfamilialbritish pages 3-4)
- Neuroimaging: ventricular dilatation and periventricular white-matter changes have been reported (nonspecific). (rostagno2005chromosome13dementias pages 4-6)
| Domain | Test or intervention | Purpose | Notes | Suggested ontology term(s) | Evidence/citation IDs |
|---|---|---|---|---|---|
| genetic | Targeted ITM2B/BRI2 testing for the Danish duplication (c.787_796dupTTTAATTTGT, p.Ser266PhefsTer11 / p.Ser266PhefsX11) | Confirm molecular diagnosis in symptomatic individuals and enable family testing | Core disease-defining test; the mutation is an autosomal dominant 10-nt duplication near the stop codon that generates the 277-aa precursor processed to ADan | Ontology: not assigned | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 1-2) |
| diagnostic | Family-history assessment and pedigree analysis | Identify at-risk relatives and support suspicion of autosomal dominant inherited amyloidosis | Especially useful because published human disease derives from a multigenerational Danish pedigree/case series | Ontology: not assigned | (rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 1-2) |
| imaging | Brain neuroimaging (reported findings: ventricular dilatation, periventricular white-matter changes) | Support clinical workup and assess CNS structural involvement | Imaging findings are nonspecific but compatible with disease burden; no disease-specific imaging guideline retrieved | Ontology: not assigned | (rostagno2005chromosome13dementias pages 4-6) |
| pathology | Neuropathologic examination of brain/retina/vessels for ADan amyloid and CAA | Establish tissue-level diagnosis and characterize disease severity/distribution | Hallmarks include widespread cerebral and retinal amyloid angiopathy, predominantly vascular deposits, and often absence of compact parenchymal plaques | SNOMED/LOINC: not assigned | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11) |
| pathology | Histochemistry for amyloid (e.g., Congo red / thioflavin-based methods in reported studies) | Detect amyloid deposition in tissue | Some FDD hippocampal deposits were reported Congo red/ThS negative in certain reports, so interpretation depends on lesion type/stage | SNOMED/LOINC: not assigned | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) |
| pathology | Assessment for tau pathology / neurofibrillary tangles | Document Alzheimer-like downstream neurodegeneration | Hippocampal neurofibrillary tangles and paired helical filaments are recurrent pathologic features | SNOMED/LOINC: not assigned | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 1-2, rostagno2005chromosome13dementias pages 4-6) |
| diagnostic | Ophthalmologic evaluation (including cataract assessment) | Detect early disease manifestations and functional impairment | Visual symptoms are often the first manifestation, with median onset around 27 years in one family series | Ontology: not assigned | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) |
| diagnostic | Audiologic evaluation | Characterize progressive hearing loss and disability burden | Hearing loss typically follows ocular disease by 10–20 years and may be severe by the fifth decade | Ontology: not assigned | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) |
| diagnostic | Neurologic and cognitive assessment | Monitor progression of ataxia, tremor, gait dysfunction, and dementia | Important for longitudinal care; no standardized disease-specific criteria retrieved | Ontology: not assigned | (garringer2010modelingfamilialbritish pages 3-4, choudhury2025pathologicalmechanismsof pages 1-2, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11) |
| supportive | Symptomatic multidisciplinary care (neurology, ophthalmology, audiology, rehabilitation, dementia care) | Maintain function and quality of life | No disease-modifying approved therapy was retrieved; current care is supportive and complication-focused | MAXO: supportive care procedure; rehabilitation intervention; dementia management (specific term not assigned) | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6, rostagno2005chromosome13dementias pages 10-11) |
| supportive | Physical therapy / gait and balance rehabilitation | Address ataxia, gait abnormality, and motor dysfunction | Supported by the prominence of cerebellar ataxia and progressive motor impairment; evidence is extrapolated from phenotype, not trial-based | MAXO: physical therapy | (choudhury2025pathologicalmechanismsof pages 1-2, rostagno2005chromosome13dementias pages 4-6) |
| supportive | Hearing and vision support | Mitigate sensory disability from deafness and cataracts | Includes standard-of-care assistive and ophthalmologic management; disease-specific outcomes data not retrieved | MAXO: assistive device intervention / ophthalmologic management (not assigned) | (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6) |
| experimental-preclinical | Anti-ADan monoclonal antibody 1B7 | Neutralize ADan-mediated cytotoxicity | In SH-SY5Y experiments, 1B7 completely neutralized ADan-mediated toxicity, supporting target-specific anti-amyloid strategies | MAXO: monoclonal antibody therapy (preclinical) | (todd2016oxidativestressand pages 11-13, todd2016oxidativestressand pages 14-16) |
| experimental-preclinical | Antioxidant Trolox | Reduce ADan-triggered oxidative stress and downstream apoptosis | Preclinical cell data show Trolox reduced ROS-linked caspase-3 activation, supporting oxidative-stress targeting | MAXO: antioxidant therapy (preclinical) | (todd2016oxidativestressand pages 26-32, todd2016oxidativestressand pages 13-14, todd2016oxidativestressand pages 14-16, todd2016oxidativestressand media f315265f) |
| experimental-preclinical | APP-lowering genetic modification (APP haplodeficiency in KI models) | Test whether APP-derived products mediate synaptic/memory deficits | In FDD models, APP reduction prevented synaptic plasticity and memory deficits, arguing that APP metabolites are mechanistically important | MAXO: gene dosage reduction / genetic interaction study (preclinical) | (matsuda2011increasedaβppprocessing pages 4-5, yin2021familialdanishdementia pages 9-11) |
| experimental-preclinical | Targeting microglial BRI2-TREM2 biology | Explore disease-modifying approaches via microglial processing/phagocytosis pathways | 2024 studies implicate microglial ITM2B enrichment, altered TREM2 processing, and reduced phagocytosis after BRI2 loss; therapeutic relevance remains investigational | MAXO: targeted molecular therapy (preclinical) | (yin2024functionalbri2trem2interactions pages 1-2, arber2024microgliacontributeto pages 1-2, arber2023microgliaproducethe pages 8-12) |
| experimental-preclinical | Clinical trials search for ADan/FDD-specific interventions | Assess real-world translational pipeline | No ADan/FDD-specific interventional trials were retrieved in the current tool context | Ontology: not assigned | (OpenTargets Search: familial Danish dementia,ADan amyloidosis-ITM2B) |
Table: This table summarizes currently supported diagnostic approaches, pathology findings, supportive care practices, and preclinical therapeutic leads for ADan amyloidosis/familial Danish dementia. It is useful for distinguishing established clinical implementation from experimental mechanisms-based interventions.
10.2 Differential diagnosis
Formal differential-diagnosis guidance was not retrieved. Based on pathology/phenotype overlap, differential considerations include other hereditary cerebral amyloid angiopathies, familial Alzheimer disease, and other inherited neurodegenerative syndromes with early cataracts/hearing loss/ataxia.
11. Outcome / prognosis
A multigenerational case series reported median age at death 58 years. No modern survival curves, prognostic models, or biomarker-based prognostic factors were retrieved. (rostagno2005chromosome13dementias pages 4-6)
12. Treatment
12.1 Current clinical management (real-world implementation)
No disease-modifying approved treatments were identified in the retrieved evidence. Current care is inferred to be supportive and multidisciplinary (vision, hearing, ataxia rehabilitation, cognitive/dementia care). (garringer2010modelingfamilialbritish pages 3-4, rostagno2005chromosome13dementias pages 4-6)
12.2 Experimental / preclinical therapeutic directions
- Anti-amyloid immunotherapy concept (preclinical): ADan-specific monoclonal antibody 1B7 completely neutralized ADan cytotoxicity in a neuronal cell model. (todd2016oxidativestressand pages 11-13, todd2016oxidativestressand pages 14-16)
- Anti-oxidant strategy (preclinical): Trolox reduced ADan-induced oxidative stress-linked caspase activation (figure-level evidence). (todd2016oxidativestressand pages 13-14, todd2016oxidativestressand media f315265f)
- Targeting APP metabolism (mechanistic modifier): human tissue and model data support altered APP processing in FDD and prevention of synaptic/memory phenotypes by APP haplodeficiency in models. (matsuda2011increasedaβppprocessing pages 4-5)
- Targeting microglial pathways: BRI2–TREM2 regulation and microglial phagocytosis effects suggest a possible therapeutic axis (early-stage hypothesis). (yin2024functionalbri2trem2interactions pages 1-2)
12.3 Clinical trials
A search of ClinicalTrials.gov within the current tool context retrieved no clearly relevant ADan/FDD interventional trials. (OpenTargets Search: familial Danish dementia,ADan amyloidosis-ITM2B)
13. Prevention
No primary-prevention strategies were retrieved. In practice, prevention is largely genetic counseling and cascade testing in affected families (concept supported by autosomal dominant inheritance and gene-defined diagnosis). (rostagno2005chromosome13dementias pages 4-6)
14. Other species / natural disease
No naturally occurring veterinary cases were retrieved in the current evidence set.
15. Model organisms
15.1 Mammalian models
- Transgenic FDD mice: age-dependent ADan deposition and cerebrovascular amyloid (CAA) and behavioral/motor abnormalities in some models, with onset of vascular pathology in early months in certain lines; crossing with tau P301S increases tau pathology. (garringer2010modelingfamilialbritish pages 4-6, garringer2010modelingfamilialbritish pages 6-7)
- Knock-in (KI) mice: some KI lines show synaptic/memory deficits with limited/no detectable amyloid deposition, underscoring mechanistic contributions beyond extracellular plaque burden and highlighting model limitations. (marcora2014amyloidpeptidesabri pages 1-2, garringer2010modelingfamilialbritish pages 6-7)
- Knock-in rats with humanized APP/Aβ: show vascular ADan deposition plus vascular Aβ, BBB leakage (fibrinogen extravasation), neuroinflammation, demyelination/axonal loss, and motor deficits—providing a platform for studying vascular/white-matter mechanisms. (choudhury2025pathologicalmechanismsof pages 1-2, choudhury2025pathologicalmechanismsof pages 15-16)
15.2 Invertebrate models
- Drosophila UAS models: ADan showed the highest age-dependent neurotoxicity among tested peptides, with functional motor decline detectable as early as 7 days post-eclosion in a geotaxis assay; deposits were thioflavin-S-negative, illustrating limitations in modeling fibrillar deposition. (marcora2014amyloidpeptidesabri pages 1-2)
15.3 In vitro / cellular models
- SH-SY5Y neuronal models show ADan-driven ROS and mitochondrial apoptosis, modifiable by Trolox and antibody neutralization. (todd2016oxidativestressand pages 11-13, todd2016oxidativestressand pages 13-14, todd2016oxidativestressand pages 14-16)
- iPSC-derived microglia studies (primarily FBD) show high ITM2B expression and peptide detection in microglial lysates/media, supporting microglial contributions to ITM2B-derived amyloid peptide biology. (arber2024microgliacontributeto pages 1-2)
Recent developments (prioritized 2023–2024)
1) Microglial production and enrichment of ITM2B/BRI2 (Acta Neuropathologica 2024): ITM2B expression is 34-fold higher in microglia vs neurons and 15-fold vs astrocytes, and patient iPSC-microglia contain detectable amyloidogenic peptide (ABri) in lysates and conditioned media; the same ITM2B processing logic applies to ADan generation in FDD. Publication date: Nov 2024; URL: https://doi.org/10.1007/s00401-024-02820-z (arber2024microgliacontributeto pages 1-2) 2) BRI2–TREM2 functional interaction (EMBO Reports 2024): demonstrates a direct BRI2–TREM2 ectodomain interaction, effects on TREM2 processing (TREM2-CTF/sTREM2), and reduced phagocytosis with Bri2 deletion. Publication date: Feb 2024; URL: https://doi.org/10.1038/s44319-024-00077-x (yin2024functionalbri2trem2interactions pages 1-2) 3) ADan sequence determinants and nucleation mapping (bioRxiv 2024): high-throughput mutagenesis maps an ADan amyloid core (L20–F26) and quantifies the distribution of nucleation-altering variants, providing a mechanistic framework for stop-loss amyloid diseases. Preprint posted 2024 (doi indicates 2023.09.15 submission); URL: https://doi.org/10.1101/2023.09.15.557952 (martin2024amyloids“atthe pages 3-5)
Key quantitative statistics (from retrieved studies)
- Natural history in a Danish pedigree series: 13 cases / 5 generations, median visual onset 27 years, median death 58 years. (rostagno2005chromosome13dementias pages 4-6)
- Human tissue (n=1 FDD vs n=1 control): soluble Aβ42 11×, insoluble Aβ42 125×, soluble Aβ40 27×, insoluble Aβ40 38×, sAPPβ 2.7×, sAPPα 5×. (matsuda2011increasedaβppprocessing pages 4-5)
- Cell-type expression (iPSC-derived): ITM2B/BRI2 expression 34× higher in microglia vs neurons and 15× higher vs astrocytes. (arber2024microgliacontributeto pages 1-2)
Evidence limitations and gaps
- Curated identifiers beyond MONDO (OMIM/Orphanet/MeSH/ICD) were not retrieved in the current context and should be filled by direct queries to those resources.
- Modern epidemiology (prevalence/incidence), penetrance estimates, and standardized diagnostic/treatment guidelines were not retrieved.
- Many therapeutic insights remain preclinical; no disease-specific interventional trial evidence was retrieved here.
Source list (URLs and dates available in retrieved texts)
- Rostagno et al., Cell Mol Life Sci (Jun 2005). https://doi.org/10.1007/s00018-005-5092-5 (rostagno2005chromosome13dementias pages 4-6)
- Garringer et al., Brain Struct Funct (Mar 2010). https://doi.org/10.1007/s00429-009-0221-9 (garringer2010modelingfamilialbritish pages 3-4)
- Matsuda et al., J Alzheimers Dis (Nov 2011). https://doi.org/10.3233/JAD-2011-110785 (matsuda2011increasedaβppprocessing pages 4-5)
- Todd et al., Neurobiol Dis (Jan 2016). https://doi.org/10.1016/j.nbd.2015.10.003 (todd2016oxidativestressand pages 11-13)
- Yin et al., EMBO Reports (Feb 2024). https://doi.org/10.1038/s44319-024-00077-x (yin2024functionalbri2trem2interactions pages 1-2)
- Arber et al., Acta Neuropathologica (Nov 2024). https://doi.org/10.1007/s00401-024-02820-z (arber2024microgliacontributeto pages 1-2)
- Martín & Bolognesi, bioRxiv (Aug 2024 posting/identifier). https://doi.org/10.1101/2023.09.15.557952 (martin2024amyloids“atthe pages 3-5)
References
-
(garringer2010modelingfamilialbritish pages 3-4): Holly J. Garringer, Jill Murrell, Luciano D’Adamio, Bernardino Ghetti, and Ruben Vidal. Modeling familial british and danish dementia. Brain Structure and Function, 214:235-244, Mar 2010. URL: https://doi.org/10.1007/s00429-009-0221-9, doi:10.1007/s00429-009-0221-9. This article has 55 citations and is from a peer-reviewed journal.
-
(rostagno2005chromosome13dementias pages 4-6): Agueda Rostagno, Y. Tomidokoro, T. Lashley, Douglas Ng, Gordon T. Plant, JL Holton, B. Frangione, Tamas Revesz, and J. Ghiso. Chromosome 13 dementias. Cellular and Molecular Life Sciences CMLS, 62:1814-1825, Jun 2005. URL: https://doi.org/10.1007/s00018-005-5092-5, doi:10.1007/s00018-005-5092-5. This article has 86 citations.
-
(yin2024functionalbri2trem2interactions pages 1-2): Tao Yin, Metin Yesiltepe, and Luciano D’Adamio. Functional bri2-trem2 interactions in microglia: implications for alzheimer’s and related dementias. EMBO Reports, 25(3):1326-1360, Feb 2024. URL: https://doi.org/10.1038/s44319-024-00077-x, doi:10.1038/s44319-024-00077-x. This article has 15 citations and is from a highest quality peer-reviewed journal.
-
(arber2024microgliacontributeto pages 1-2): Charles Arber, Jackie M. Casey, Samuel Crawford, Naiomi Rambarack, Umran Yaman, Sarah Wiethoff, Emma Augustin, Thomas M. Piers, Matthew Price, Agueda Rostagno, Jorge Ghiso, Patrick A. Lewis, Tamas Revesz, John Hardy, Jennifer M. Pocock, Henry Houlden, Jonathan M. Schott, Dervis A. Salih, Tammaryn Lashley, and Selina Wray. Microglia contribute to the production of the amyloidogenic abri peptide in familial british dementia. Acta Neuropathologica, Nov 2024. URL: https://doi.org/10.1007/s00401-024-02820-z, doi:10.1007/s00401-024-02820-z. This article has 6 citations and is from a highest quality peer-reviewed journal.
-
(martin2024amyloids“atthe pages 3-5): Mariano Martín and Benedetta Bolognesi. Amyloids “at the border”: deep mutagenesis and random sequence extension reveal an incomplete amyloid-forming motif in bri2 that turns amyloidogenic upon c-terminal extension. bioRxiv, Aug 2024. URL: https://doi.org/10.1101/2023.09.15.557952, doi:10.1101/2023.09.15.557952. This article has 3 citations.
-
(rostagno2005chromosome13dementias pages 10-11): Agueda Rostagno, Y. Tomidokoro, T. Lashley, Douglas Ng, Gordon T. Plant, JL Holton, B. Frangione, Tamas Revesz, and J. Ghiso. Chromosome 13 dementias. Cellular and Molecular Life Sciences CMLS, 62:1814-1825, Jun 2005. URL: https://doi.org/10.1007/s00018-005-5092-5, doi:10.1007/s00018-005-5092-5. This article has 86 citations.
-
(OpenTargets Search: familial Danish dementia,ADan amyloidosis-ITM2B): Open Targets Query (familial Danish dementia,ADan amyloidosis-ITM2B, 2 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(rostagno2005chromosome13dementias pages 1-2): Agueda Rostagno, Y. Tomidokoro, T. Lashley, Douglas Ng, Gordon T. Plant, JL Holton, B. Frangione, Tamas Revesz, and J. Ghiso. Chromosome 13 dementias. Cellular and Molecular Life Sciences CMLS, 62:1814-1825, Jun 2005. URL: https://doi.org/10.1007/s00018-005-5092-5, doi:10.1007/s00018-005-5092-5. This article has 86 citations.
-
(todd2016oxidativestressand pages 11-13): Krysti Todd, Jorge Ghiso, and Agueda Rostagno. Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial danish dementia adan amyloid. Neurobiology of Disease, 85:130-143, Jan 2016. URL: https://doi.org/10.1016/j.nbd.2015.10.003, doi:10.1016/j.nbd.2015.10.003. This article has 30 citations and is from a domain leading peer-reviewed journal.
-
(rostagno2005chromosome13dementias pages 2-4): Agueda Rostagno, Y. Tomidokoro, T. Lashley, Douglas Ng, Gordon T. Plant, JL Holton, B. Frangione, Tamas Revesz, and J. Ghiso. Chromosome 13 dementias. Cellular and Molecular Life Sciences CMLS, 62:1814-1825, Jun 2005. URL: https://doi.org/10.1007/s00018-005-5092-5, doi:10.1007/s00018-005-5092-5. This article has 86 citations.
-
(matsuda2011increasedaβppprocessing pages 4-5): Shuji Matsuda, Robert Tamayev, and Luciano D'Adamio. Increased aβpp processing in familial danish dementia patients. Journal of Alzheimer's Disease, 27:385-391, Nov 2011. URL: https://doi.org/10.3233/jad-2011-110785, doi:10.3233/jad-2011-110785. This article has 41 citations and is from a peer-reviewed journal.
-
(yin2021danishandbritish pages 1-2): Tao Yin, Wen Yao, Alexander D. Lemenze, and Luciano D’Adamio. Danish and british dementia itm2b/bri2 mutations reduce bri2 protein stability and impair glutamatergic synaptic transmission. Journal of Biological Chemistry, 296:100054, Jan 2021. URL: https://doi.org/10.1074/jbc.ra120.015679, doi:10.1074/jbc.ra120.015679. This article has 26 citations and is from a domain leading peer-reviewed journal.
-
(choudhury2025pathologicalmechanismsof pages 1-2): Arnab Choudhury, Metin Yesiltepe, Tammaryn Lashley, Vishal Singh, Veertegh Minhas, Tao Yin, Anllely Fernandez, Luciano D’Adamio, and Hyung Jin Ahn. Pathological mechanisms of motor dysfunction in familial danish dementia: insights from a knock-in rat model. Journal of Neuroinflammation, Dec 2025. URL: https://doi.org/10.1186/s12974-025-03537-w, doi:10.1186/s12974-025-03537-w. This article has 1 citations and is from a peer-reviewed journal.
-
(garringer2010modelingfamilialbritish pages 4-6): Holly J. Garringer, Jill Murrell, Luciano D’Adamio, Bernardino Ghetti, and Ruben Vidal. Modeling familial british and danish dementia. Brain Structure and Function, 214:235-244, Mar 2010. URL: https://doi.org/10.1007/s00429-009-0221-9, doi:10.1007/s00429-009-0221-9. This article has 55 citations and is from a peer-reviewed journal.
-
(garringer2010modelingfamilialbritish pages 6-7): Holly J. Garringer, Jill Murrell, Luciano D’Adamio, Bernardino Ghetti, and Ruben Vidal. Modeling familial british and danish dementia. Brain Structure and Function, 214:235-244, Mar 2010. URL: https://doi.org/10.1007/s00429-009-0221-9, doi:10.1007/s00429-009-0221-9. This article has 55 citations and is from a peer-reviewed journal.
-
(choudhury2025pathologicalmechanismsof pages 15-16): Arnab Choudhury, Metin Yesiltepe, Tammaryn Lashley, Vishal Singh, Veertegh Minhas, Tao Yin, Anllely Fernandez, Luciano D’Adamio, and Hyung Jin Ahn. Pathological mechanisms of motor dysfunction in familial danish dementia: insights from a knock-in rat model. Journal of Neuroinflammation, Dec 2025. URL: https://doi.org/10.1186/s12974-025-03537-w, doi:10.1186/s12974-025-03537-w. This article has 1 citations and is from a peer-reviewed journal.
-
(garringer2010modelingfamilialbritish pages 10-12): Holly J. Garringer, Jill Murrell, Luciano D’Adamio, Bernardino Ghetti, and Ruben Vidal. Modeling familial british and danish dementia. Brain Structure and Function, 214:235-244, Mar 2010. URL: https://doi.org/10.1007/s00429-009-0221-9, doi:10.1007/s00429-009-0221-9. This article has 55 citations and is from a peer-reviewed journal.
-
(garringer2010modelingfamilialbritish pages 9-10): Holly J. Garringer, Jill Murrell, Luciano D’Adamio, Bernardino Ghetti, and Ruben Vidal. Modeling familial british and danish dementia. Brain Structure and Function, 214:235-244, Mar 2010. URL: https://doi.org/10.1007/s00429-009-0221-9, doi:10.1007/s00429-009-0221-9. This article has 55 citations and is from a peer-reviewed journal.
-
(todd2016oxidativestressand pages 13-14): Krysti Todd, Jorge Ghiso, and Agueda Rostagno. Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial danish dementia adan amyloid. Neurobiology of Disease, 85:130-143, Jan 2016. URL: https://doi.org/10.1016/j.nbd.2015.10.003, doi:10.1016/j.nbd.2015.10.003. This article has 30 citations and is from a domain leading peer-reviewed journal.
-
(todd2016oxidativestressand pages 14-16): Krysti Todd, Jorge Ghiso, and Agueda Rostagno. Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial danish dementia adan amyloid. Neurobiology of Disease, 85:130-143, Jan 2016. URL: https://doi.org/10.1016/j.nbd.2015.10.003, doi:10.1016/j.nbd.2015.10.003. This article has 30 citations and is from a domain leading peer-reviewed journal.
-
(todd2016oxidativestressand media f315265f): Krysti Todd, Jorge Ghiso, and Agueda Rostagno. Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial danish dementia adan amyloid. Neurobiology of Disease, 85:130-143, Jan 2016. URL: https://doi.org/10.1016/j.nbd.2015.10.003, doi:10.1016/j.nbd.2015.10.003. This article has 30 citations and is from a domain leading peer-reviewed journal.
-
(todd2016oxidativestressand media d7873ce6): Krysti Todd, Jorge Ghiso, and Agueda Rostagno. Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial danish dementia adan amyloid. Neurobiology of Disease, 85:130-143, Jan 2016. URL: https://doi.org/10.1016/j.nbd.2015.10.003, doi:10.1016/j.nbd.2015.10.003. This article has 30 citations and is from a domain leading peer-reviewed journal.
-
(arber2023microgliaproducethe pages 1-4): Charles Arber, Jackie M. Casey, Samuel Crawford, Naiomi Rambarack, Umran Yaman, Sarah Wiethoff, Emma Augustin, Thomas M. Piers, Agueda Rostagno, Jorge Ghiso, Patrick A. Lewis, Tamas Revesz, John Hardy, Jennifer M. Pocock, Henry Houlden, Jonathan M. Schott, Dervis A. Salih, Tammaryn Lashley, and Selina Wray. Microglia produce the amyloidogenic abri peptide in familial british dementia. bioRxiv, Jun 2023. URL: https://doi.org/10.1101/2023.06.27.546552, doi:10.1101/2023.06.27.546552. This article has 2 citations.
-
(todd2016oxidativestressand pages 26-32): Krysti Todd, Jorge Ghiso, and Agueda Rostagno. Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial danish dementia adan amyloid. Neurobiology of Disease, 85:130-143, Jan 2016. URL: https://doi.org/10.1016/j.nbd.2015.10.003, doi:10.1016/j.nbd.2015.10.003. This article has 30 citations and is from a domain leading peer-reviewed journal.
-
(yin2021familialdanishdementia pages 9-11): Tao Yin, Wen Yao, Kelly A. Norris, and Luciano D’Adamio. Familial danish dementia young knock-in rats expressing humanized app and human aβ show impaired pre and postsynaptic glutamatergic transmission. bioRxiv, Jun 2021. URL: https://doi.org/10.1101/2021.06.24.449787, doi:10.1101/2021.06.24.449787. This article has 0 citations.
-
(arber2023microgliaproducethe pages 8-12): Charles Arber, Jackie M. Casey, Samuel Crawford, Naiomi Rambarack, Umran Yaman, Sarah Wiethoff, Emma Augustin, Thomas M. Piers, Agueda Rostagno, Jorge Ghiso, Patrick A. Lewis, Tamas Revesz, John Hardy, Jennifer M. Pocock, Henry Houlden, Jonathan M. Schott, Dervis A. Salih, Tammaryn Lashley, and Selina Wray. Microglia produce the amyloidogenic abri peptide in familial british dementia. bioRxiv, Jun 2023. URL: https://doi.org/10.1101/2023.06.27.546552, doi:10.1101/2023.06.27.546552. This article has 2 citations.
-
(marcora2014amyloidpeptidesabri pages 1-2): María S Marcora, Agata C Fernández-Gamba, Luz A Avendaño, Cecilia Rotondaro, Osvaldo L Podhajcer, Rubén Vidal, Laura Morelli, María F Ceriani, and Eduardo M Castaño. Amyloid peptides abri and adan show differential neurotoxicity in transgenic drosophila models of familial british and danish dementia. Molecular Neurodegeneration, 9:5-5, Jan 2014. URL: https://doi.org/10.1186/1750-1326-9-5, doi:10.1186/1750-1326-9-5. This article has 22 citations and is from a highest quality peer-reviewed journal.