ADGRG1-related Bilateral Frontoparietal Polymicrogyria
Bilateral frontoparietal polymicrogyria (BFPP) is an autosomal recessive malformation of cortical development caused by loss-of-function mutations in ADGRG1 (formerly GPR56), which encodes an adhesion G protein-coupled receptor. GPR56 is expressed in cortical radial glial progenitors and their basal endfeet, where it binds its extracellular-matrix ligand collagen III (COL3A1) to maintain the integrity of the pial basement membrane and to restrain neuronal migration through Galpha-12/13-RhoA signaling. Loss of GPR56 function breaches the pial basement membrane and permits ectopic overmigration of neurons beyond the pial limit, producing a frontoparietally predominant polymicrogyria that overlaps with the cobblestone (type II lissencephaly) malformation spectrum. Affected individuals present with severe intellectual disability, motor delay, drug-resistant seizures, cerebellar dysplasia and white-matter abnormalities. Because GPR56 is expressed in a regionally restricted, promoter-controlled pattern, the malformation is anatomically patterned (frontoparietal in classic BFPP; perisylvian when a non-coding regulatory element is disrupted), rather than a global cortical defect. The disorder is mechanistically distinct from generic polymicrogyria and is better represented by the ADGRG1/COL3A1 pial basement-membrane / radial glial endfoot skeleton; biallelic COL3A1 (ligand-side) disease produces a closely related cobblestone-like phenotype and is curated separately.
Ask OpenScientist
Ask a research question about ADGRG1-related Bilateral Frontoparietal Polymicrogyria. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Discussions and Knowledge Gaps
1Show evidence (2 references)
Pathophysiology
6Show evidence (3 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Pathograph
Phenotypes
6Nervous System 5
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Other 1
Show evidence (1 reference)
Genetic Associations
1Show evidence (2 references)
Medical Actions
2Source YAML
click to showname: ADGRG1-related Bilateral Frontoparietal Polymicrogyria
creation_date: "2026-06-10T00:00:00Z"
category: Mendelian
disease_term:
preferred_term: bilateral frontoparietal polymicrogyria
term:
id: MONDO:0011738
label: bilateral frontoparietal polymicrogyria
description: >-
Bilateral frontoparietal polymicrogyria (BFPP) is an autosomal recessive
malformation of cortical development caused by loss-of-function mutations in
ADGRG1 (formerly GPR56), which encodes an adhesion G protein-coupled receptor.
GPR56 is expressed in cortical radial glial progenitors and their basal
endfeet, where it binds its extracellular-matrix ligand collagen III (COL3A1)
to maintain the integrity of the pial basement membrane and to restrain
neuronal migration through Galpha-12/13-RhoA signaling. Loss of GPR56 function
breaches the pial basement membrane and permits ectopic overmigration of
neurons beyond the pial limit, producing a frontoparietally predominant
polymicrogyria that overlaps with the cobblestone (type II lissencephaly)
malformation spectrum. Affected individuals present with severe intellectual
disability, motor delay, drug-resistant seizures, cerebellar dysplasia and
white-matter abnormalities. Because GPR56 is expressed in a regionally
restricted, promoter-controlled pattern, the malformation is anatomically
patterned (frontoparietal in classic BFPP; perisylvian when a non-coding
regulatory element is disrupted), rather than a global cortical defect. The
disorder is mechanistically distinct from generic polymicrogyria and is
better represented by the ADGRG1/COL3A1 pial basement-membrane / radial glial
endfoot skeleton; biallelic COL3A1 (ligand-side) disease produces a closely
related cobblestone-like phenotype and is curated separately.
parents:
- congenital nervous system disorder
- disorder of development or morphogenesis
- hereditary neurological disease
references:
- reference: PMID:15044805
title: "G protein-coupled receptor-dependent development of human frontal cortex."
- reference: PMID:21768377
title: "G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination."
pathophysiology:
- name: Loss of GPR56 Adhesion GPCR Function
description: >-
Recessive loss-of-function mutations in ADGRG1/GPR56, an adhesion-family G
protein-coupled receptor expressed in cortical radial glial progenitors and
their basal endfeet, abolish receptor signaling. Disease-associated missense
variants cluster in the large extracellular region (ectodomain, GPS
autoproteolysis site, and extracellular loops) and impair surface
expression, autoproteolysis, ligand interaction and downstream signaling,
establishing GPR56 loss of function as the initiating molecular lesion of
BFPP.
genes:
- preferred_term: ADGRG1
term:
id: hgnc:4512
label: ADGRG1
cell_types:
- preferred_term: cortical radial glial cell
term:
id: CL:0013000
label: forebrain radial glial cell
biological_processes:
- preferred_term: G protein-coupled receptor signaling pathway
term:
id: GO:0007186
label: G protein-coupled receptor signaling pathway
modifier: DECREASED
evidence:
- reference: PMID:15044805
reference_title: "G protein-coupled receptor-dependent development of human frontal cortex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
we show that mutations in GPR56, which encodes an orphan G
protein-coupled receptor (GPCR) with a large extracellular domain, cause a
human brain cortical malformation called bilateral frontoparietal
polymicrogyria (BFPP)
explanation: >-
Establishes GPR56/ADGRG1 mutations as the cause of BFPP and identifies the
gene product as a GPCR with a large extracellular domain.
- reference: PMID:21349848
reference_title: Disease-associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
Loss-of-function mutations in the gene encoding G protein-coupled receptor
56 (GPR56) lead to bilateral frontoparietal polymicrogyria (BFPP), an
autosomal recessive disorder affecting brain development.
explanation: >-
Confirms BFPP as a recessive disorder driven by loss-of-function GPR56
mutations.
- reference: PMID:21349848
reference_title: Disease-associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
individual GPR56 mutants most likely cause BFPP via different combination
of multiple mechanisms. These include reduced surface receptor
expression, loss of GPS proteolysis, reduced receptor shedding, inability
to interact with a novel protein ligand
explanation: >-
Details the convergent molecular consequences of disease-associated
extracellular-region mutations that abolish GPR56 function.
downstream:
- target: Disrupted GPR56-Collagen III Signaling
description: >-
Loss of functional GPR56 abolishes ligand binding and the downstream
G protein signaling that normally restrains neuronal migration.
- name: Disrupted GPR56-Collagen III Signaling
description: >-
Collagen III (COL3A1), a major constituent of the pial basement membrane, is
the extracellular-matrix ligand of GPR56. Engagement of GPR56 by collagen
III couples to the Galpha-12/13 family of G proteins and activates the RhoA
pathway, which inhibits neuronal migration. Loss of receptor or ligand
removes this brake on migration and destabilizes cortical lamination.
conforms_to: pial_basement_membrane_radial_glial_endfoot_failure#GPR56-COL3A1 Pial ECM Signaling Failure
cell_types:
- preferred_term: cortical radial glial cell
term:
id: CL:0013000
label: forebrain radial glial cell
biological_processes:
- preferred_term: cell-matrix adhesion
term:
id: GO:0007160
label: cell-matrix adhesion
modifier: DECREASED
- preferred_term: Rho protein signal transduction
term:
id: GO:0007266
label: Rho protein signal transduction
modifier: DECREASED
evidence:
- reference: PMID:21768377
reference_title: "G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
As for intracellular signaling, GPR56 couples to the Gα(12/13) family of
G proteins and activates RhoA pathway upon ligand binding.
explanation: >-
Identifies the GPR56 intracellular signaling output (Galpha-12/13 to RhoA)
activated by ligand binding.
- reference: PMID:21768377
reference_title: "G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Functional studies suggest that the interaction of collagen III with its
receptor GPR56 inhibits neural migration in vitro.
explanation: >-
Establishes that the collagen III-GPR56 interaction normally restrains
neuronal migration, the brake lost in disease.
downstream:
- target: Pial Basement Membrane Breach
description: >-
Loss of collagen III-GPR56 signaling destabilizes the pial basement
membrane and removes the migratory brake, permitting overmigration.
- name: Pial Basement Membrane Breach
description: >-
GPR56 signaling at radial glial basal endfeet maintains the integrity of the
pial basement membrane in the developing forebrain and rostral cerebellum.
Loss of GPR56 (or its collagen III ligand) dysregulates maintenance of the
pial basement membrane, breaching the glia limitans and creating gaps
through which neurons can escape the cortical plate.
conforms_to: pial_basement_membrane_radial_glial_endfoot_failure#Pial Basement Membrane Breach
cell_types:
- preferred_term: cortical radial glial cell
term:
id: CL:0013000
label: forebrain radial glial cell
biological_processes:
- preferred_term: basement membrane organization
term:
id: GO:0071711
label: basement membrane organization
modifier: ABNORMAL
evidence:
- reference: PMID:20929962
reference_title: "GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
loss of GPR56 leads to a dysregulation of the maintenance of the pial
basement membrane integrity in the forebrain and the rostral cerebellum
explanation: >-
Identifies pial basement membrane integrity failure as the consequence of
GPR56 loss, the structural lesion enabling overmigration.
- reference: PMID:21768377
reference_title: "G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Mutations in the GPR56 gene cause a malformed cerebral cortex in both
humans and mice that resembles cobblestone lissencephaly, which is
characterized by overmigration of neurons beyond the pial basement
membrane.
explanation: >-
Links GPR56 mutation to a cobblestone-like cortex defined by neuronal
overmigration beyond the breached pial basement membrane.
downstream:
- target: Radial-Glial Basal Endfoot Detachment
description: >-
Pial basement membrane breach destabilizes radial glial basal endfeet at
the cortical surface.
- target: Cerebellar Dysplasia
description: >-
GPR56-dependent pial basement membrane failure also affects the rostral
cerebellum, producing cerebellar dysplasia with cysts.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- rostral cerebellar pial basement membrane failure
- abnormal cerebellar cortical development
- name: Radial-Glial Basal Endfoot Detachment
description: >-
GPR56 is enriched in radial glial endfeet, where the GPR56-COL3A1 axis helps
maintain the pial basement membrane and radial-glial scaffold. Loss of this
endfoot anchoring function removes the boundary constraint that normally
prevents overmigration beyond the pial surface.
conforms_to: pial_basement_membrane_radial_glial_endfoot_failure#Radial-Glial Basal Endfoot Detachment
cell_types:
- preferred_term: cortical radial glial cell
term:
id: CL:0013000
label: forebrain radial glial cell
biological_processes:
- preferred_term: formation of radial glial scaffolds
term:
id: GO:0021943
label: formation of radial glial scaffolds
modifier: DECREASED
evidence:
- reference: PMID:18509043
reference_title: "GPR56 regulates pial basement membrane integrity and cortical lamination."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
We show further that GPR56 is present in abundance in radial glial
endfeet.
explanation: >-
Localizes GPR56 to radial glial endfeet, supporting this as the cellular
site of the pial-boundary failure.
- reference: PMID:18509043
reference_title: "GPR56 regulates pial basement membrane integrity and cortical lamination."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
There are four crucial events in the development of cobblestone cortex,
namely defective pial basement membrane (BM), abnormal anchorage of radial
glial endfeet, mislocalized Cajal-Retzius cells, and neuronal
overmigration.
explanation: >-
Identifies abnormal radial-glial endfoot anchorage as part of the shared
GPR56/pial-boundary overmigration skeleton.
downstream:
- target: Neuronal Overmigration and Cortical Dyslamination
description: >-
Endfoot detachment and pial boundary failure permit neurons to overmigrate
beyond the pial limit, disrupting cortical lamination.
- name: Neuronal Overmigration and Cortical Dyslamination
description: >-
With the pial basement membrane breached and the GPR56-RhoA migratory brake
lost, postmitotic neurons overmigrate beyond their normal pial stopping
point, producing a disorganized, abnormally laminated cortex. The result is
a frontoparietally predominant polymicrogyria that, at its severe end,
forms a cobblestone-like cortex with ectopic neuronal overmigration,
forming a phenotypic continuum from BFPP to cobblestone-like lissencephaly.
conforms_to: pial_basement_membrane_radial_glial_endfoot_failure#Neuronal Overmigration Across the Pial Boundary
cell_types:
- preferred_term: cerebral cortex neuron
term:
id: CL:0010012
label: cerebral cortex neuron
biological_processes:
- preferred_term: neuron migration
term:
id: GO:0001764
label: neuron migration
modifier: INCREASED
- preferred_term: cerebral cortex radial glia-guided migration
term:
id: GO:0021801
label: cerebral cortex radial glia-guided migration
modifier: DYSREGULATED
- preferred_term: layer formation in cerebral cortex
term:
id: GO:0021819
label: layer formation in cerebral cortex
modifier: ABNORMAL
evidence:
- reference: PMID:20929962
reference_title: "GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
showed a cobblestone-like lissencephaly with a succession of normal,
polymicrogyric and 'cobblestone-like' cortex with ectopic neuronal
overmigration
explanation: >-
Documents ectopic neuronal overmigration and the polymicrogyria-to-
cobblestone cortical continuum in a fetopathological BFPP case.
- reference: PMID:15044805
reference_title: "G protein-coupled receptor-dependent development of human frontal cortex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
BFPP is characterized by disorganized cortical lamination that is most
severe in frontal cortex.
explanation: >-
Establishes disordered cortical lamination with frontal predominance as
the defining cortical pathology of BFPP.
downstream:
- target: Bilateral Frontoparietal Polymicrogyria
description: >-
Neuronal overmigration through a breached pial boundary directly produces
the frontoparietal polymicrogyria/cobblestone cortical malformation.
causal_link_type: DIRECT
- target: White Matter Abnormalities
description: >-
The same developmental cortical malformation is accompanied by patchy to
diffuse white-matter/myelination abnormalities in the ADGRG1 cohort.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Delayed Myelination
description: >-
Early severe hypomyelination and later patchy white-matter lesions are
downstream developmental imaging manifestations of the malformation
spectrum.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
- target: Severe Intellectual Disability
description: >-
Severe disruption of cortical lamination and associated white-matter
development produces profound neurodevelopmental impairment.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- disorganized cortical lamination
- impaired cortical network development
- target: Seizures
description: Disorganized cortical lamination creates an epileptogenic cortical substrate.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- cortical dyslamination
- epileptogenic network formation
- name: Regional Cortical Patterning Dependence on GPR56 Expression
description: >-
GPR56 is expressed in cortical progenitor cells in a regionally restricted,
promoter-controlled pattern, and its expression levels regulate progenitor
proliferation. This regional control explains why the malformation is
anatomically patterned rather than global: classic protein-coding mutations
produce frontoparietal disease, whereas a non-coding deletion that disrupts
a region-specific regulatory element produces a perisylvian-predominant
polymicrogyria by selectively abolishing lateral cortical GPR56 expression.
cell_types:
- preferred_term: cortical progenitor cell
term:
id: CL:0000047
label: neural stem cell
biological_processes:
- preferred_term: cerebral cortex regionalization
term:
id: GO:0021796
label: cerebral cortex regionalization
modifier: ABNORMAL
evidence:
- reference: PMID:24531968
reference_title: Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
we describe a 15-base pair deletion mutation in a regulatory element of
GPR56 that selectively disrupts human cortex surrounding the Sylvian
fissure bilaterally including "Broca's area," the primary language area,
by disrupting regional GPR56 expression and blocking RFX transcription
factor binding
explanation: >-
Shows that a non-coding regulatory mutation produces a regionally
restricted (perisylvian) malformation by disrupting regional GPR56
expression, demonstrating promoter-level regional patterning.
- reference: PMID:24531968
reference_title: Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning.
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
GPR56 encodes a heterotrimeric guanine nucleotide-binding protein (G
protein)-coupled receptor required for normal cortical development and is
expressed in cortical progenitor cells. GPR56 expression levels regulate
progenitor proliferation.
explanation: >-
Establishes that GPR56 is expressed in cortical progenitors and that its
expression level controls progenitor proliferation and regional patterning.
phenotypes:
- name: Bilateral Frontoparietal Polymicrogyria
description: >-
The defining neuroradiological feature: bilateral, frontoparietally
predominant polymicrogyria with disorganized cortical lamination, reflecting
neuronal overmigration through the breached pial basement membrane.
phenotype_term:
preferred_term: Bilateral frontoparietal polymicrogyria
term:
id: HP:0002126
label: Polymicrogyria
evidence:
- reference: PMID:20929962
reference_title: "GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Neuroimaging demonstrated a common phenotype with bilateral
frontoparietally predominant polymicrogyria
explanation: >-
Documents bilateral frontoparietally predominant polymicrogyria as the
consistent imaging phenotype across the GPR56 patient cohort.
- name: Cerebellar Dysplasia
description: >-
Cerebellar dysplasia, frequently with cysts mainly affecting the superior
vermis, accompanies the cortical malformation and reflects pial basement
membrane failure in the rostral cerebellum.
phenotype_term:
preferred_term: Cerebellar dysplasia with cysts
term:
id: HP:0007033
label: Cerebellar dysplasia
evidence:
- reference: PMID:20929962
reference_title: "GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
cerebellar dysplasia with cysts mainly affecting the superior vermis
(11/13)
explanation: >-
Documents cerebellar dysplasia with cysts in the great majority of the
GPR56 cohort.
- name: White Matter Abnormalities
description: >-
Patchy to diffuse myelination abnormalities are a consistent feature,
evolving from severe hypomyelination in infancy to patchy lesions later in
childhood.
phenotype_term:
preferred_term: Abnormal cerebral white matter morphology
term:
id: HP:0002500
label: Abnormal cerebral white matter morphology
evidence:
- reference: PMID:20929962
reference_title: "GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
patchy to diffuse myelination abnormalities (13/13)
explanation: >-
Documents white-matter/myelination abnormalities in all imaged patients
of the GPR56 cohort.
- name: Delayed Myelination
description: >-
White-matter abnormalities begin as severe hypomyelination in infancy,
consistent with delayed myelination.
phenotype_term:
preferred_term: Delayed myelination
term:
id: HP:0012448
label: Delayed myelination
evidence:
- reference: PMID:20929962
reference_title: "GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
the white matter abnormalities showed a peculiar evolution from severe
hypomyelination at 4 months to patchy lesions later in childhood
explanation: >-
Documents early severe hypomyelination, supporting delayed myelination in
GPR56-related disease.
- name: Severe Intellectual Disability
description: >-
Affected individuals show a distinctive course beginning with pseudomyopathic
behaviour and evolving into severe mental and motor retardation.
phenotype_term:
preferred_term: Severe intellectual disability
term:
id: HP:0010864
label: Severe intellectual disability
evidence:
- reference: PMID:20929962
reference_title: "GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
a distinctive clinical course characterized by pseudomyopathic behaviour
at onset that subsequently evolved into severe mental and motor
retardation
explanation: >-
Documents the characteristic progression to severe mental and motor
impairment in GPR56-related BFPP.
- name: Seizures
description: >-
Generalized seizures occur in the great majority of patients, with onset in
childhood and characteristic anterior EEG bursts.
phenotype_term:
preferred_term: Seizure
term:
id: HP:0001250
label: Seizure
frequency: VERY_FREQUENT
evidence:
- reference: PMID:20929962
reference_title: "GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Generalized seizures (12/14) occurred later with onset ranging from 2.5
to 10 years with consistent electroencephalogram findings of
predominantly anterior bursts of low amplitude α-like activity
explanation: >-
Documents generalized seizures in 12/14 patients (supporting a
VERY_FREQUENT band) with characteristic anterior EEG findings.
genetic:
- name: ADGRG1
association: Loss of function
gene_term:
preferred_term: ADGRG1 (GPR56)
term:
id: hgnc:4512
label: ADGRG1
evidence:
- reference: PMID:15044805
reference_title: "G protein-coupled receptor-dependent development of human frontal cortex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
we show that mutations in GPR56, which encodes an orphan G
protein-coupled receptor (GPCR) with a large extracellular domain, cause a
human brain cortical malformation called bilateral frontoparietal
polymicrogyria (BFPP)
explanation: >-
Founding report identifying GPR56/ADGRG1 mutations as the cause of BFPP.
- reference: PMID:20929962
reference_title: "GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We identified homozygous GPR56 mutations in 14 patients from eight
consanguineous families with typical bilateral bifrontoparietal
polymicrogyria
explanation: >-
Confirms recessive (homozygous) GPR56 mutations in a multi-family BFPP
cohort, consistent with autosomal recessive loss of function.
treatments:
- name: Anti-Seizure Medication
description: >-
Symptomatic management of the drug-resistant epilepsy associated with BFPP
using standard anti-seizure medications selected by seizure type. No
disease-modifying therapy exists; management is supportive.
treatment_term:
preferred_term: pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
therapeutic_agent:
- preferred_term: levetiracetam
term:
id: CHEBI:6437
label: levetiracetam
- preferred_term: valproic acid
term:
id: CHEBI:39867
label: valproic acid
- name: Supportive and Rehabilitative Care
description: >-
Multidisciplinary supportive care including physical, occupational and
developmental therapies for the severe intellectual disability and motor
impairment.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
discussions:
- discussion_id: gap_adgrg1_human_regional_patterning_model_mismatch
prompt: >-
Does the Gpr56-null mouse, which faithfully reproduces pial basement-membrane
breach and neuronal overmigration but as a globally affected, naturally
lissencephalic cobblestone-like cortex, capture the human-specific,
regulatory-element-controlled regional patterning (frontoparietal-predominant
in classic BFPP, perisylvian when a non-coding regulatory element is
disrupted) that defines bilateral frontoparietal polymicrogyria, or does the
regional vulnerability gradient require human gyrencephalic models to resolve?
kind: HUMAN_MODEL_MISMATCH
status: OPEN
attaches_to:
- pathophysiology#Regional Cortical Patterning Dependence on GPR56 Expression
- pathophysiology#Neuronal Overmigration and Cortical Dyslamination
rationale: >-
The core overmigration mechanism of BFPP is well modeled in rodents: Gpr56-null
(and Col3a1-null) mice reproduce pial basement-membrane breach and overmigration
of neurons beyond the pial limit, so the model is valid for the pial-boundary
arm. What the mouse does not reproduce is the defining clinical feature of human
disease, its regional, anatomically patterned distribution. Human GPR56/ADGRG1
splice forms and the cis-regulatory architecture that restricts lateral cortical
expression differ markedly between mouse and human, and the gyrencephalic
regulatory element directs the restricted lateral expression whose disruption
produces perisylvian polymicrogyria. Because the rodent cortex is naturally
lissencephalic and lacks the human regulatory/splicing pattern, the
frontoparietal-versus-perisylvian regional gradient, the feature that
distinguishes BFPP from generic cobblestone malformation, cannot be assigned
from the knockout and remains an open human-model translational question.
evidence:
- reference: PMID:21768377
reference_title: "G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
Mutations in the GPR56 gene cause a malformed cerebral cortex in both humans
and mice that resembles cobblestone lissencephaly, which is characterized by
overmigration of neurons beyond the pial basement membrane.
explanation: >-
Establishes that the mouse knockout faithfully reproduces the human
overmigration / cobblestone arm, so the model is valid for the core
pial-boundary mechanism, which isolates the absent regional patterning as the
specific translational gap.
- reference: PMID:24531968
reference_title: Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning.
supports: SUPPORT
evidence_source: OTHER
snippet: >-
GPR56 splice forms are highly variable between mice and humans, and the
regulatory element of gyrencephalic mammals directs restricted lateral
cortical expression.
explanation: >-
Documents that the splicing and cis-regulatory architecture controlling
regional GPR56 expression differ between mouse and human, so the human
regional patterning that defines BFPP is not represented in the rodent model.
proposed_experiments:
- experiment_id: exp_adgrg1_humanized_regulatory_regional_patterning
name: Humanized GPR56 regulatory element and regionally patterned cortical-organoid experiment
description: >-
Test whether the human GPR56/ADGRG1 cis-regulatory element and splice
repertoire are required to generate region-specific cortical vulnerability by
comparing a knock-in mouse carrying the human regulatory element (and the
perisylvian regulatory deletion) against the conventional Gpr56-null, and by
patterning human iPSC-derived cortical organoids along anterior-posterior
identity to ask whether ADGRG1 loss or regulatory-element disruption produces
regionally graded overmigration.
experiment_type:
preferred_term: humanized-regulatory-element and patient-derived cortical organoid experiment
model_systems:
- name: Human iPSC-derived regionally patterned cortical organoid
description: >-
Human cortical organoids patterned toward frontal versus perisylvian/lateral
identity, carrying ADGRG1 loss of function or the perisylvian regulatory-element
deletion with isogenic corrected controls.
experimental_model_type: ORGANOID
namo_type: namo:Organoid
organism:
preferred_term: human
term:
id: NCBITaxon:9606
label: Homo sapiens
tissue_term:
preferred_term: cerebral cortex
term:
id: UBERON:0000956
label: cerebral cortex
cell_types:
- preferred_term: cortical radial glial cell
term:
id: CL:0013000
label: forebrain radial glial cell
- preferred_term: migrating cortical neuron
term:
id: CL:0000540
label: neuron
conditions:
- ADGRG1-related bilateral frontoparietal polymicrogyria
- cobblestone lissencephaly
cell_source: Patient-derived or CRISPR-engineered human induced pluripotent stem cells
culture_system: Regionally patterned three-dimensional cortical organoids with live-imaging overmigration assays
perturbations:
- name: ADGRG1 loss of function or perisylvian regulatory-element deletion
target: pathophysiology#Regional Cortical Patterning Dependence on GPR56 Expression
genes:
- preferred_term: ADGRG1
term:
id: hgnc:4512
label: ADGRG1
description: >-
Introduce ADGRG1 loss of function or the non-coding perisylvian
regulatory-element deletion into an isogenic human iPSC background, with
corrected controls.
readouts:
- name: Region-resolved neuronal overmigration across the pial boundary
target: pathophysiology#Neuronal Overmigration and Cortical Dyslamination
biological_processes:
- preferred_term: neuron migration
term:
id: GO:0001764
label: neuron migration
modifier: INCREASED
assays:
- preferred_term: live-cell imaging assay
direction: POSITIVE
- name: Regional GPR56 expression and cortical regionalization
target: pathophysiology#Regional Cortical Patterning Dependence on GPR56 Expression
biological_processes:
- preferred_term: cerebral cortex regionalization
term:
id: GO:0021796
label: cerebral cortex regionalization
modifier: ABNORMAL
assays:
- preferred_term: spatial transcriptomics assay
direction: NEGATIVE
notes: >-
Entry created from cortical-malformation epic 4098 (issue 4087), seeded from
Romero, Bahi-Buisson & Francis 2018 (Sem Cell Dev Biol 76:33-75). Modeled as a
coherent ADGRG1/GPR56 pial basement-membrane / radial glial endfoot
pathomechanism rather than lumped under generic polymicrogyria. The biallelic
COL3A1 (ligand-side) cobblestone-like malformation (PMID:28258187,
PMID:28742248) is a closely related but separate entry and is intentionally
not curated here. The GPR56-COL3A1, pial basement membrane breach, radial
glial endfoot detachment, and neuronal overmigration nodes now conform to the
pial basement membrane / radial glial endfoot failure module.
References & Deep Research
References
2Deep Research
1ADGRG1-related Bilateral Frontoparietal Polymicrogyria (BFPP): Comprehensive Disease Characteristics Report
Target disease
Disease name: ADGRG1-related Bilateral Frontoparietal Polymicrogyria (BFPP)
Category: Mendelian (autosomal recessive)
1. Disease information
1.1 Overview (what is the disease?)
ADGRG1-related bilateral frontoparietal polymicrogyria (BFPP) is a congenital malformation of cortical development characterized by bilateral frontoparietal polymicrogyria, typically showing an anterior-to-posterior gradient of severity, and commonly accompanied by white-matter signal abnormalities plus brainstem/cerebellar hypoplasia on MRI, with neurodevelopmental disability and high rates of epilepsy and oculomotor/cerebellar signs. (piao2005genotype–phenotypeanalysisof pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3)
A core definition of polymicrogyria used in BFPP literature is: “a cortical malformation characterized by supernumerary, small gyri with abnormal cortical lamination.” (piao2005genotype–phenotypeanalysisof pages 1-2)
1.2 Key identifiers and nomenclature
| Identifier type | ID | Preferred name | Synonyms/notes | Evidence/source |
|---|---|---|---|---|
| OMIM | 606854 | Bilateral frontoparietal polymicrogyria | Common abbreviation: BFPP; classic Mendelian cortical malformation linked to ADGRG1/GPR56 | Piao et al. 2005 (piao2005genotype–phenotypeanalysisof pages 1-2) |
| MONDO | MONDO_0000087 | polymicrogyria | Broader parent disease term used in OpenTargets disease-target association for ADGRG1 | OpenTargets association (OpenTargets Search: polymicrogyria,bilateral frontoparietal polymicrogyria-ADGRG1) |
| MONDO | MONDO_0017091 | bilateral polymicrogyria | Broader bilateral PMG term associated with ADGRG1 in OpenTargets | OpenTargets association (OpenTargets Search: polymicrogyria,bilateral frontoparietal polymicrogyria-ADGRG1) |
| OpenTargets target | ENSG00000205336 | ADGRG1 | Approved symbol ADGRG1; former symbol/name GPR56; disease-target evidence links ADGRG1 to polymicrogyria/bilateral polymicrogyria | OpenTargets association (OpenTargets Search: polymicrogyria,bilateral frontoparietal polymicrogyria-ADGRG1) |
| Other (gene nomenclature) | — | ADGRG1-related polymicrogyria syndrome | Also described as GPR56-related polymicrogyria; ADGRG1 formerly known as GPR56 | Khatib et al. 2024 (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
| Other (cytogenetic locus) | 16q12.2-21 | BFPP locus / ADGRG1-linked region | Historical linked interval for autosomal recessive BFPP before/alongside gene definition | Jansen & Andermann 2005 (jansen2005geneticsofthe pages 5-6); Piao et al. 2005 (piao2005genotype–phenotypeanalysisof pages 1-2) |
| Other (gene chromosomal location) | 16q21 | ADGRG1 | Gene location reported in recent family report; complements earlier BFPP linkage interval 16q12.2-21 | Khatib et al. 2024 (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3) |
Table: This table summarizes the key nomenclature and identifier anchors for ADGRG1-related bilateral frontoparietal polymicrogyria, including OMIM and MONDO mappings plus gene naming and chromosomal locus information. It is useful for harmonizing disease knowledge base entries across clinical and genomic resources.
Notes on missing identifiers: Orphanet, ICD-10/ICD-11, and MeSH identifiers specific to BFPP were not available in the retrieved full-text evidence; therefore, they are not reported here.
1.3 Synonyms / alternative names
Common disease names in the retrieved literature include: - “Bilateral frontoparietal polymicrogyria (BFPP)” (piao2005genotype–phenotypeanalysisof pages 1-2) - “ADGRG1-related polymicrogyria syndrome” (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3) - “GPR56-related polymicrogyria” / “GPR56 mutations” (ADGRG1 formerly known as GPR56) (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7)
1.4 Evidence provenance
Evidence in this report is derived from aggregated disease-level resources (e.g., OpenTargets) and aggregated literature evidence (case series + reviews), supplemented by individual patient/family case reports with molecular confirmation. (OpenTargets Search: polymicrogyria,bilateral frontoparietal polymicrogyria-ADGRG1, piao2005genotype–phenotypeanalysisof pages 3-5, carneiro2021casereportdiffuse pages 1-2)
2. Etiology
2.1 Disease causal factors
Primary cause: biallelic (typically homozygous) loss-of-function or deleterious variants in ADGRG1 (GPR56) causing abnormal cortical development with characteristic MRI findings and neurodevelopmental impairment. (piao2005genotype–phenotypeanalysisof pages 3-5, piao2005genotype–phenotypeanalysisof pages 1-2, chiang2011diseaseassociatedgpr56mutations pages 1-2)
Abstract quote (primary mechanistic genetics): Chiang et al. state, “Loss-of-function mutations in the gene encoding G protein-coupled receptor 56 (GPR56) lead to bilateral frontoparietal polymicrogyria (BFPP), an autosomal recessive disorder affecting brain development.” (chiang2011diseaseassociatedgpr56mutations pages 1-2)
2.2 Risk factors
- Consanguinity is a major familial/contextual risk factor for recessive BFPP and is common among reported pedigrees. (jansen2005geneticsofthe pages 5-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3)
- Family history of BFPP / known familial ADGRG1 variant increases recurrence risk consistent with autosomal recessive inheritance. (carneiro2021casereportdiffuse pages 1-2, carneiro2021casereportdiffuse pages 2-3)
2.3 Protective factors
No genetic or environmental protective factors were identified in the retrieved evidence.
2.4 Gene–environment interactions
No specific gene–environment interaction evidence was identified in the retrieved sources.
3. Phenotypes
3.1 Core clinical phenotype spectrum and frequencies
| Domain | Phenotype (plain) | Suggested HPO term(s) | Frequency/quantitative data | Typical onset/course | Key notes | Key sources (citation ids) |
|---|---|---|---|---|---|---|
| Neurodevelopment | Global developmental delay / psychomotor delay | HP:0001263 Developmental delay; HP:0001270 Motor delay | Reported as universal in classic BFPP series; mental retardation and motor developmental delay in 100% of 29 typical BFPP cases | Congenital/infantile onset; chronic, nonprogressive structural brain disorder with lifelong impairment | Core defining clinical feature across cohorts | (piao2005genotype–phenotypeanalysisof pages 3-5, piao2005genotype–phenotypeanalysisof pages 1-2, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3) |
| Cognition | Intellectual disability / severe cognitive impairment | HP:0001249 Intellectual disability; HP:0011342 Severe global developmental delay | Severe cognitive impairment in 79.3% of reviewed cases | Apparent in infancy/early childhood; persistent | Often accompanied by markedly limited language acquisition | (carneiro2021casereportdiffuse pages 3-5) |
| Epilepsy | Seizures / epilepsy | HP:0001250 Seizure; HP:0002373 Febrile seizures; HP:0002123 Generalized myoclonic seizure; HP:0002121 Absence seizure | 95% in 29-patient classic BFPP cohort; 88.2% (60/68) in review; refractory in 60.0% (36/60) of those with seizures; 90.4% (75/83) in 2024 review with refractory seizures in 54.7% (41/75) | Usually infancy to childhood onset; often chronic and can be drug-refractory | Lennox-Gastaut phenotype reported in some families; seizure types include atonic, atypical absence, myoclonic, tonic, spasms | (piao2005genotype–phenotypeanalysisof pages 3-5, carneiro2021casereportdiffuse pages 5-6, parrini2009bilateralfrontoparietalpolymicrogyria pages 6-8, parrini2009bilateralfrontoparietalpolymicrogyria pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
| Cerebellar / coordination | Cerebellar signs / ataxia | HP:0001251 Ataxia; HP:0002070 Cerebellar atrophy (imaging adjunct) | 94% in classic BFPP cohort; 92.6% in later literature review | Early childhood; generally persistent/nonprogressive relative to malformation | Reflects characteristic cerebellar involvement on MRI and exam | (piao2005genotype–phenotypeanalysisof pages 3-5, carneiro2021casereportdiffuse pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3) |
| Eye movement | Dysconjugate gaze / oculomotor abnormalities | HP:0000608 Abnormality of the eye movement; HP:0000486 Strabismus; HP:0000511 Nystagmus | Dysconjugate gaze in 88% of classic BFPP cohort; oculomotor abnormalities in 92.1% in later review; strabismus 59.5% among those with oculomotor findings | Usually recognized in infancy/childhood; persistent | Commonly includes strabismus and nystagmus | (piao2005genotype–phenotypeanalysisof pages 3-5, carneiro2021casereportdiffuse pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4) |
| Motor / pyramidal | Pyramidal signs / spasticity / brisk reflexes | HP:0002493 Upper motor neuron dysfunction; HP:0001257 Spasticity; HP:0001347 Hyperreflexia; HP:0002509 Limb hypertonia | Pyramidal signs present in 75.9% in review | Infantile/childhood onset; chronic | Can include spastic quadriparesis, ankle clonus, wide-based gait, hyperreflexia | (carneiro2021casereportdiffuse pages 3-5, parrini2009bilateralfrontoparietalpolymicrogyria pages 3-5) |
| Motor function | Ambulation outcome | HP:0002505 Impaired ambulation; HP:0002540 Delayed walking | Able to walk in 81.6% overall in 2021 review; median walking age 3.5 years; unable to walk 18.4%; 44/63 (~70%) able to walk in 2024 review | Delayed acquisition in childhood; variable ultimate attainment | Walking ability is variable and useful for severity stratification | (carneiro2021casereportdiffuse pages 3-5, carneiro2021casereportdiffuse pages 2-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6) |
| Tone / early presentation | Hypotonia, sometimes evolving to hypertonia | HP:0001252 Hypotonia; HP:0001276 Hypertonia | No pooled percentage available in cited excerpts | Often first year of life or noted at birth; chronic | Early pseudomyopathic presentation can mimic congenital muscular dystrophy | (carneiro2021casereportdiffuse pages 2-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4) |
| Growth / head size | Head circumference usually normal; occasional microcephaly or macrocephaly | HP:0000252 Microcephaly; HP:0000256 Macrocephaly | Normal head circumference 85.1% in 2021 review; 81% in 2024 review; microcephaly 12.7%, macrocephaly 6.3% in 2024 review | Congenital/childhood trait; generally stable descriptor | Head size is not usually markedly abnormal despite severe neurologic disease | (carneiro2021casereportdiffuse pages 5-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
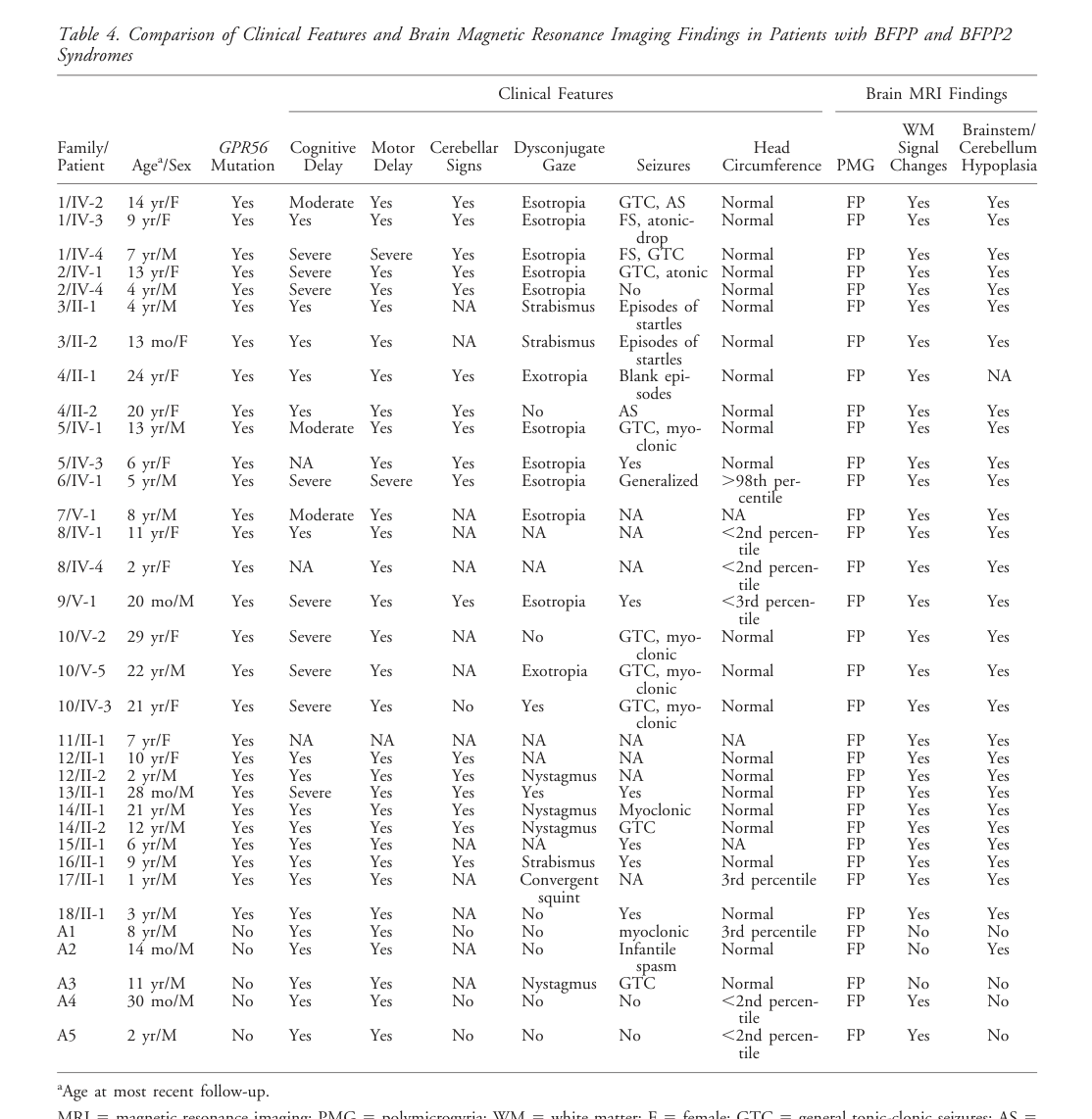
| Imaging / cortex | Bilateral frontoparietal polymicrogyria with anterior-posterior gradient | HP:0002126 Polymicrogyria; HP:0012650 Bilateral cerebral cortical dysgenesis | Hallmark MRI pattern in essentially all classic BFPP cases; all 29 classic cases had bilateral frontoparietal PMG | Congenital, static malformation | Symmetric bilateral PMG with decreasing anterior-to-posterior severity is the canonical radiologic signature | (piao2005genotype–phenotypeanalysisof pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, piao2005genotype–phenotypeanalysisof media 547d7d36) |
| Imaging / white matter | Patchy white-matter signal abnormalities / hypomyelination / reduced volume | HP:0002500 Abnormal cerebral white matter morphology; HP:0002188 Delayed CNS myelination | Present in all 29 classic cases as hallmark MRI finding; later reports describe diffuse hypomyelination in atypical severe cases | Congenital/static on serial imaging | May be patchy in classic BFPP or diffuse in atypical ADGRG1-related phenotypes | (piao2005genotype–phenotypeanalysisof pages 3-5, carneiro2021casereportdiffuse pages 2-3, piao2005genotype–phenotypeanalysisof media 547d7d36) |
| Imaging / posterior fossa | Brainstem and cerebellar hypoplasia | HP:0001321 Cerebellar hypoplasia; HP:0007366 Small pons | Present in all 29 classic cases as hallmark MRI finding | Congenital/static | Strong clue favoring ADGRG1-related BFPP over some other PMG subtypes | (piao2005genotype–phenotypeanalysisof pages 3-5, jansen2005geneticsofthe pages 5-6, piao2005genotype–phenotypeanalysisof media 547d7d36) |
| Severity / variability | Intrafamilial and interfamilial phenotypic variability | HP:0003812 Variable expressivity | Qualitative; no single pooled percentage | Lifelong, variable severity | Atypical diffuse polymicrogyria, pachygyria/lissencephaly-like changes, and severe non-ambulatory phenotypes have been reported | (carneiro2021casereportdiffuse pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4, lin2021atwinscase pages 7-10) |
Table: This table summarizes the core clinical and imaging phenotype spectrum reported for ADGRG1-related bilateral frontoparietal polymicrogyria, with frequencies drawn from classic and updated literature reviews. It is useful for knowledge-base curation because it aligns phenotype terms, quantitative prevalence, and suggested HPO mappings with supporting citations.
Key quantitative phenotype statistics from landmark and updated reviews include: - In a classic cohort of 29 typical BFPP cases, cerebellar signs (94%), dysconjugate gaze (88%), and seizures (95%) were highly prevalent; cognitive and motor developmental delay were universal. (piao2005genotype–phenotypeanalysisof pages 3-5) - In an updated literature synthesis (as captured in the Carneiro 2021 excerpts), seizures were reported in 88.2%, with 60.0% of those being drug-refractory; severe cognitive impairment was reported in 79.3%; oculomotor findings in 92.1%. (carneiro2021casereportdiffuse pages 5-6, carneiro2021casereportdiffuse pages 3-5) - In the Khatib 2024 review excerpt, seizures were present in 75/83 (90.4%), refractory in 41/75 (54.7%). (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7)
3.2 Phenotype characteristics (onset, progression, QoL impact)
- Onset: Congenital/infantile, with developmental delay/hypotonia often evident within the first year; seizures typically begin in infancy or childhood. (carneiro2021casereportdiffuse pages 2-3, parrini2009bilateralfrontoparietalpolymicrogyria pages 3-5)
- Course: The malformation is structural and nonprogressive, but functional outcomes vary; epilepsy can be refractory and can contribute to developmental decompensation (epileptic encephalopathy-like course). (carneiro2021casereportdiffuse pages 3-5, parrini2009bilateralfrontoparietalpolymicrogyria pages 6-8)
- Quality-of-life impact: Severe intellectual disability, refractory epilepsy, impaired ambulation, and oculomotor dysfunction can markedly limit independence and communication; quantitative QoL metrics (e.g., EQ-5D/SF-36) were not identified in the retrieved literature.
3.3 Suggested HPO terms
HPO suggestions are included per-phenotype in Artifact-01.
4. Genetic / molecular information
4.1 Causal gene(s)
ADGRG1 (former name GPR56) is the established causal gene for classic BFPP, with autosomal recessive inheritance. (piao2005genotype–phenotypeanalysisof pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3)
4.2 Pathogenic variant spectrum
| Gene (HGNC symbol) | Inheritance | Variant class / region | Example variants (HGVS / legacy protein) | Notable genotype-phenotype notes | Population / consanguinity / founder info | Key counts / classification | Evidence type | Key sources |
|---|---|---|---|---|---|---|---|---|
| ADGRG1 (formerly GPR56) | Autosomal recessive | Disease-causing variants in classic BFPP are typically biallelic, often homozygous | Historical protein-level examples span extracellular and 7TM regions | In the landmark genotype-phenotype study, homozygous GPR56 mutations were identified in all 29 patients with typical BFPP; BFPP is therefore a canonical recessive ADGRG1-related cortical malformation (piao2005genotype–phenotypeanalysisof pages 3-5, piao2005genotype–phenotypeanalysisof pages 1-2) | Consanguinity is common in reported families, but homozygous variants were also seen in some apparently nonconsanguineous pedigrees (jansen2005geneticsofthe pages 5-6, piao2005genotype–phenotypeanalysisof pages 3-5) | OMIM BFPP 606854; ADGRG1 linked to BFPP/polymicrogyria across human cohorts (piao2005genotype–phenotypeanalysisof pages 1-2, OpenTargets Search: polymicrogyria,bilateral frontoparietal polymicrogyria-ADGRG1) | Human clinical / human genetics | (piao2005genotype–phenotypeanalysisof pages 3-5, piao2005genotype–phenotypeanalysisof pages 1-2, jansen2005geneticsofthe pages 5-6) |
| ADGRG1 | Autosomal recessive | Extracellular-region missense cluster: N-terminal ECD, GPS motif, extracellular loops of 7TM | p.Arg38Gln (R38Q), p.Arg38Trp (R38W), p.Tyr88Cys (Y88C), p.Cys91Ser (C91S), p.Cys346Ser (C346S), p.Trp349Ser (W349S), p.Arg565Trp (R565W), p.Leu640Arg (L640R) | BFPP-associated missense variants cluster in extracellular regions and act through multiple loss-of-function mechanisms including reduced surface expression, ER retention, defective glycosylation, impaired GPS proteolysis, altered receptor shedding, loss of ligand interaction, and altered lipid-raft distribution (chiang2011diseaseassociatedgpr56mutations pages 8-10, chiang2011diseaseassociatedgpr56mutations pages 3-5, chiang2011diseaseassociatedgpr56mutations pages 10-11, ke2008biochemicalcharacterizationof pages 1-2, chiang2011diseaseassociatedgpr56mutations pages 1-2) | Families reported across Arabic-speaking Middle Eastern, Pakistani, Indian, Afghani, Canadian, Turkish, Italian, Israeli, and Hispanic American backgrounds; several alleles appear recurrent/founder-like in specific pedigrees (piao2005genotype–phenotypeanalysisof pages 3-5) | Missense variants were absent from 260 control chromosomes in the 2005 cohort (piao2005genotype–phenotypeanalysisof pages 3-5) | Human clinical + in vitro functional | (chiang2011diseaseassociatedgpr56mutations pages 8-10, chiang2011diseaseassociatedgpr56mutations pages 3-5, chiang2011diseaseassociatedgpr56mutations pages 10-11, ke2008biochemicalcharacterizationof pages 1-2, chiang2011diseaseassociatedgpr56mutations pages 1-2, piao2005genotype–phenotypeanalysisof pages 3-5) |
| ADGRG1 | Autosomal recessive | Truncating variants (nonsense / frameshift / deletion) | Historical 7-bp deletion; nonsense and frameshift alleles noted across BFPP pedigrees | Truncating alleles are associated with severe disease and are common among the most motor-impaired/non-ambulatory cases; loss of function is an established disease mechanism (carneiro2021casereportdiffuse pages 2-3, carneiro2021casereportdiffuse pages 5-6, carneiro2021casereportdiffuse pages 3-5) | Many truncating-variant families arise in consanguineous settings, though compound heterozygosity is also reported in the wider ADGRG1 literature (carneiro2021casereportdiffuse pages 5-6, carneiro2021casereportdiffuse pages 3-5) | By 2021, 77 patients from 47 pedigrees with 34 distinct ADGRG1 variants had been reported (carneiro2021casereportdiffuse pages 3-5) | Human clinical / literature review | (carneiro2021casereportdiffuse pages 3-5, carneiro2021casereportdiffuse pages 2-3, carneiro2021casereportdiffuse pages 5-6) |
| ADGRG1 | Autosomal recessive | Nonsense variant in 7TM domain | NM_001145771.2:c.1504C>T; NP_005673.3:p.Arg502Ter / p.Arg502*; dbSNP rs746634404 | Reported in a child with diffuse polymicrogyria without the classic anterior-posterior gradient, diffuse hypomyelination, pontine/cerebellar hypoplasia, profound developmental impairment, and refractory epilepsy, expanding the ADGRG1 phenotypic spectrum beyond classic BFPP (carneiro2021casereportdiffuse pages 2-3, carneiro2021casereportdiffuse pages 1-2, carneiro2021casereportdiffuse pages 3-5) | Found homozygously in a consanguineous family; both parents were heterozygous carriers (carneiro2021casereportdiffuse pages 1-2, carneiro2021casereportdiffuse pages 2-3) | Very rare in gnomAD (f = 0.0000119 in cited report); classified as pathogenic with very strong ACMG evidence in the case report (carneiro2021casereportdiffuse pages 2-3) | Human clinical + diagnostic genetics | (carneiro2021casereportdiffuse pages 2-3, carneiro2021casereportdiffuse pages 1-2, carneiro2021casereportdiffuse pages 3-5) |
| ADGRG1 | Autosomal recessive | Novel missense variant | NM_201525.4:c.308T>C; p.Leu103Pro | Identified in a large Syrian consanguineous family with ADGRG1-related polymicrogyria/BFPP; affected individuals showed early developmental delay, severe cognitive/motor impairment, oculomotor findings, and often refractory seizures with intrafamilial variability (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6) | Reported in a consanguineous Syrian family with five affected individuals (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4) | Absent from gnomAD v2.1.1 and predicted damaging; classified as pathogenic in study workflow using ACMG-based interpretation (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3) | Human clinical / exome sequencing | (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6) |
| ADGRG1 | Autosomal recessive | Recurrent pathogenic missense variant | c.1693C>T; p.Arg565Trp (legacy R565W) | Seen in BFPP and in severe overlapping phenotypes; associated reports include Lennox-Gastaut syndrome / drug-refractory epilepsy in some families and extensive polymicrogyria with hindbrain abnormalities in others (parrini2009bilateralfrontoparietalpolymicrogyria pages 6-8, parrini2009bilateralfrontoparietalpolymicrogyria pages 3-5, shaath2024integratinggenomesequencing pages 2-4) | Previously reported in a consanguineous Bedouin family; also detected homozygously in monozygotic twins from a consanguineous family (parrini2009bilateralfrontoparietalpolymicrogyria pages 6-8, shaath2024integratinggenomesequencing pages 4-5, shaath2024integratinggenomesequencing pages 2-4) | In the 2024 twin report, ClinVar pathogenic (VCV000005831.23) with CADD 29.5 (shaath2024integratinggenomesequencing pages 4-5, shaath2024integratinggenomesequencing pages 2-4) | Human clinical + curated clinical variant evidence | (parrini2009bilateralfrontoparietalpolymicrogyria pages 6-8, parrini2009bilateralfrontoparietalpolymicrogyria pages 3-5, shaath2024integratinggenomesequencing pages 4-5, shaath2024integratinggenomesequencing pages 2-4) |
| ADGRG1 | Autosomal recessive | Cohort-level variant spectrum | Missense, nonsense, frameshift, deletion; recurrent alleles plus private family-specific variants | The recognized phenotype is broad: classic bilateral frontoparietal PMG with white-matter and hindbrain abnormalities, but also atypical diffuse PMG, pachygyria/lissencephaly-like presentations, and variable ambulation/cognitive outcomes (carneiro2021casereportdiffuse pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4) | Early literature: 8 independent mutations in 22 radiologically and clinically confirmed patients from 12 families, with 9 families showing close parental consanguinity and families of Middle Eastern and French Canadian origin; broader 2005 sampling was geographically diverse (jansen2005geneticsofthe pages 5-6, piao2005genotype–phenotypeanalysisof pages 3-5) | 2005 landmark: all 29 typical BFPP cases mutation-positive; 2021 review: 77 patients / 47 pedigrees / 34 variants (piao2005genotype–phenotypeanalysisof pages 3-5, carneiro2021casereportdiffuse pages 3-5) | Human clinical / literature review | (piao2005genotype–phenotypeanalysisof pages 3-5, carneiro2021casereportdiffuse pages 3-5, jansen2005geneticsofthe pages 5-6) |
Table: This table summarizes the core human genetic evidence linking ADGRG1 (GPR56) to bilateral frontoparietal polymicrogyria, including inheritance, variant classes, representative alleles, and cohort-level statistics. It is useful for quickly mapping variant-level findings to phenotypic interpretation, population context, and evidence type.
Key genetic findings: - Landmark genotype–phenotype analysis identified homozygous GPR56 mutations in all 29 patients with typical BFPP (defining ADGRG1 as a major Mendelian cause of this imaging phenotype). (piao2005genotype–phenotypeanalysisof pages 1-2) - BFPP-associated missense variants cluster in extracellular regions (ECD/GPS/extracellular loops), with additional truncating variants and occasional regulatory variants (e.g., promoter deletion affecting expression in developing neurons). (ke2008biochemicalcharacterizationof pages 1-2, murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2) - By the time of the 2021 review excerpt, ADGRG1 BFPP-spectrum variants had been reported in 77 patients from 47 pedigrees with 34 distinct variants. (carneiro2021casereportdiffuse pages 3-5)
4.3 Functional consequences
Many BFPP-associated variants cause loss-of-function via impaired receptor maturation/processing/trafficking and impaired ligand interactions (see Mechanism/Pathophysiology). (chiang2011diseaseassociatedgpr56mutations pages 3-5, chiang2011diseaseassociatedgpr56mutations pages 10-11)
4.4 Modifier genes / multilocus disease
The retrieved excerpts note that broader sequencing (exome/genome) can reveal additional pathogenic variants that may modify severity in malformation syndromes; specific validated modifier genes for ADGRG1-BFPP were not established in the retrieved excerpts. (carneiro2021casereportdiffuse pages 3-5)
4.5 Epigenetics / chromosomal abnormalities
No disease-specific epigenetic signatures or recurrent chromosomal abnormalities were identified in the retrieved evidence.
5. Environmental information
No convincing disease-specific environmental, lifestyle, or infectious etiologic triggers were identified in the retrieved evidence for ADGRG1-related BFPP as a Mendelian disorder. However, congenital infections (e.g., CMV/HSV) can mimic the MRI pattern and must be considered in differential diagnosis. (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6)
6. Mechanism / pathophysiology
6.1 Molecular mechanism: ADGRG1 receptor biology and BFPP loss-of-function
ADGRG1/GPR56 is an adhesion GPCR with an extracellular domain (ECD), a GPCR proteolysis site (GPS/GAIN) and a 7TM signaling domain; BFPP-associated missense variants occur in extracellular regions and can cause disease through multiple convergent loss-of-function mechanisms. (chiang2011diseaseassociatedgpr56mutations pages 1-2)
Primary functional mechanisms in BFPP variants (cellular/biochemical): - Defective processing and trafficking: BFPP mutants frequently show ER retention, EndoH-sensitive glycosylation, reduced surface expression, and likely increased degradation. (chiang2011diseaseassociatedgpr56mutations pages 3-5, chiang2011diseaseassociatedgpr56mutations pages 5-6) - Loss of GPS proteolysis: GPS-site mutants (e.g., C346S, W349S) can become uncleaved single-chain forms and fail to reach the cell surface. (chiang2011diseaseassociatedgpr56mutations pages 3-5, ke2008biochemicalcharacterizationof pages 1-2) - Impaired ligand interactions / adhesion: N-terminal variants can abolish or reduce binding to a protein ligand and impair ligand-dependent adhesion. (chiang2011diseaseassociatedgpr56mutations pages 8-10, chiang2011diseaseassociatedgpr56mutations pages 7-8) - Membrane microdomain mislocalization: Some extracellular-loop variants (e.g., R565W, L640R) alter β-subunit behavior and lipid-raft distribution, supporting pathogenic mechanisms beyond simple surface expression loss. (chiang2011diseaseassociatedgpr56mutations pages 8-10)
6.2 Signaling pathways and ligand modalities (current understanding; 2023–2024 emphasis)
ADGRG1 signaling is strongly linked to Gα12/Gα13 → RhoA pathways: - Abstract quote (2024): “GPR56 constitutively activates both G12 and G13.” (jallouli2024gproteinselectivity pages 1-2) - Abstract quote (2024): “[10C7 antibody] led to an activation that favors G13 over G12.” (jallouli2024gproteinselectivity pages 1-2)
Endogenous ligand/binding-partner biology relevant to development and myelination includes: - Collagen III (basement membrane ligand): ADGRG1 binding to collagen III in the basement membrane is linked to Gα12/13–RhoA-dependent inhibition of neuronal migration, and loss of this regulation contributes to BFPP cortical malformation. (einspahr2022pathophysiologicalimpactof pages 5-9) - Transglutaminase-2 (TG2): TG2 is an ADGRG1 ligand/binding partner in multiple contexts, including oligodendrocyte lineage signaling (microglia→OPC) and epithelial migration systems. (giera2018microglialtransglutaminase2drives pages 1-2, giera2018microglialtransglutaminase2drives pages 3-5, bauer2024mesenchymaltransglutaminase2 pages 1-2)
Recent mechanistic development (2024, epithelial context): Bauer et al. describe TG2–GPR56 as a ligand–receptor pair that activates RhoA/ROCK and ADAM17, leading to EGFR transactivation and rapid keratinocyte migration, illustrating mechanistic routes by which extracellular ligands can drive ADGRG1 signaling. (bauer2024mesenchymaltransglutaminase2 pages 1-2, bauer2024mesenchymaltransglutaminase2 pages 2-5, bauer2024mesenchymaltransglutaminase2 pages 6-9)
6.3 Causal chain from gene defect to clinical phenotype (integrated)
1) Biallelic ADGRG1 variants → impaired receptor processing/trafficking and/or impaired extracellular ligand interactions (collagen III; protein ligands), reducing effective receptor function at the cell surface. (chiang2011diseaseassociatedgpr56mutations pages 3-5, chiang2011diseaseassociatedgpr56mutations pages 10-11, chiang2011diseaseassociatedgpr56mutations pages 1-2)
2) Reduced ADGRG1 function perturbs Gα12/13–RhoA signaling that normally regulates neuronal migration, cortical lamination, and pial basement membrane integrity. (einspahr2022pathophysiologicalimpactof pages 5-9, murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2)
3) Developmental disruption yields frontoparietal-predominant polymicrogyria, abnormal cortical lamination, associated white-matter abnormalities, and posterior fossa involvement (pons/vermian/cerebellar hypoplasia), producing a characteristic radiologic pattern. (piao2005genotype–phenotypeanalysisof pages 3-5, piao2005genotype–phenotypeanalysisof media 547d7d36)
4) The resulting circuit malformation and developmental perturbations manifest as global developmental delay/intellectual disability, seizures (often refractory), oculomotor abnormalities, pyramidal signs/spasticity, and cerebellar signs. (piao2005genotype–phenotypeanalysisof pages 3-5, carneiro2021casereportdiffuse pages 5-6)
6.4 Suggested ontology terms (mechanism, anatomy, cells)
GO Biological Process (suggested): - Neuronal migration (GO:0001764) (supported conceptually by Gα12/13–RhoA migration regulation and cortical malformation) (einspahr2022pathophysiologicalimpactof pages 5-9) - Cell adhesion (GO:0007155) (ligand-dependent adhesion and receptor–ligand interactions) (chiang2011diseaseassociatedgpr56mutations pages 10-11) - Myelination / glial development (GO:0042552; oligodendrocyte precursor proliferation related processes) (giera2018microglialtransglutaminase2drives pages 1-2, ackerman2018gpr56adgrg1regulatesdevelopment pages 1-2)
Cell Ontology (CL; suggested): - Radial glial cell (CL:0000243) (pial basement membrane/endfeet abnormalities in Gpr56 loss model) (murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2) - Neural progenitor cell (CL:0011020) (reduced proliferation in mouse loss models; BFPP developmental mechanism) (murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2) - GABAergic interneuron (CL:0000099) (e1m promoter activity in developing GABAergic neurons; epilepsy link) (murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2) - Oligodendrocyte precursor cell (CL:0002453) (TG2→ADGRG1 promotes OPC proliferation and remyelination) (giera2018microglialtransglutaminase2drives pages 1-2) - Microglial cell (CL:0000129) (microglial TG2 source) (giera2018microglialtransglutaminase2drives pages 3-5) - Schwann cell (CL:0000688) (peripheral myelin roles) (ackerman2018gpr56adgrg1regulatesdevelopment pages 1-2)
UBERON (suggested): - Cerebral cortex (UBERON:0000956), frontal lobe (UBERON:0001870), parietal lobe (UBERON:0001872) (piao2005genotype–phenotypeanalysisof pages 3-5) - Pons (UBERON:0000988), cerebellar vermis (UBERON:0002124), cerebellum (UBERON:0002037) (piao2005genotype–phenotypeanalysisof pages 3-5, piao2005genotype–phenotypeanalysisof media 547d7d36) - Cerebral white matter (UBERON:0002314) (piao2005genotype–phenotypeanalysisof pages 3-5)
7. Anatomical structures affected
7.1 Organ/system level
Primary affected system is the central nervous system, especially: - Bilateral frontoparietal cerebral cortex (polymicrogyria) (piao2005genotype–phenotypeanalysisof pages 3-5) - White matter (patchy signal change/hypomyelination) (piao2005genotype–phenotypeanalysisof pages 3-5, carneiro2021casereportdiffuse pages 2-3) - Brainstem and cerebellum (pons/vermian/cerebellar hypoplasia) (piao2005genotype–phenotypeanalysisof pages 3-5, piao2005genotype–phenotypeanalysisof media 547d7d36)
7.2 Tissue/cell level
Evidence from mammalian models indicates involvement of neurodevelopmental and glial lineages (radial glia, neuronal progenitors, migrating neurons), and later roles in myelination involving Schwann cells and oligodendrocyte lineage cells. (murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2, ackerman2018gpr56adgrg1regulatesdevelopment pages 1-2, giera2018microglialtransglutaminase2drives pages 1-2)
7.3 Subcellular level
Mutant receptor biology implicates ER retention/misprocessing (EndoH sensitivity) and altered membrane microdomain distribution (lipid rafts). (chiang2011diseaseassociatedgpr56mutations pages 3-5, chiang2011diseaseassociatedgpr56mutations pages 8-10)
8. Temporal development
- Typical onset: congenital/infancy with early psychomotor delay; seizures often begin in infancy or early childhood. (carneiro2021casereportdiffuse pages 2-3, parrini2009bilateralfrontoparietalpolymicrogyria pages 3-5)
- Progression: structural malformation is static; clinical trajectory is variable and can be complicated by refractory epilepsy and developmental plateau/regression. (carneiro2021casereportdiffuse pages 3-5, carneiro2021casereportdiffuse pages 1-2)
9. Inheritance and population
9.1 Inheritance
BFPP is autosomal recessive, frequently observed in consanguineous pedigrees; classic cohorts show biallelic/homozygous ADGRG1 variants in typical BFPP. (jansen2005geneticsofthe pages 5-6, piao2005genotype–phenotypeanalysisof pages 1-2)
9.2 Epidemiology
True prevalence/incidence estimates specific to ADGRG1-related BFPP were not identified in the retrieved evidence.
9.3 Population and geographic distribution
Reported mutation-positive BFPP families in the landmark cohort included Arabic-speaking Middle Eastern, Pakistani, Indian, Afghani, Canadian, Turkish, Italian, Israeli, and Hispanic American backgrounds. (piao2005genotype–phenotypeanalysisof pages 3-5)
9.4 Statistics supporting clinical relevance in PMG cohorts (recent)
In a large polymicrogyria genetics study (275 families), a genetic etiology was found in 32.7% (90/275); ADGRG1/GPR56 was listed among known PMG genes detected in the cohort, though per-gene counts were not extractable from the retrieved excerpts. (akula2023exomesequencingand pages 2-3)
10. Diagnostics
| Category | Item | Details/real-world implementation | Suggested ontology term (LOINC/MAXO where applicable) | Evidence/notes | Key sources (citation ids) |
|---|---|---|---|---|---|
| Diagnostics | Brain MRI | Core radiologic pattern in classic ADGRG1-related BFPP is symmetric bilateral frontoparietal polymicrogyria with a decreasing anterior-to-posterior gradient, patchy/random white-matter abnormalities, and cerebellar/brainstem hypoplasia; atypical cases may show diffuse polymicrogyria, hypomyelination, thin corpus callosum, ventriculomegaly, or pachygyria/lissencephaly-like changes. MRI is the frontline real-world diagnostic modality. | MAXO: brain MRI; LOINC-style concept: Magnetic resonance imaging of brain | Hallmark imaging clue; brainstem/cerebellar involvement helps distinguish ADGRG1-related disease from some broader PMG categories. Early MRI can occasionally be normal, so repeat imaging may be needed. | (piao2005genotype–phenotypeanalysisof pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, piao2005genotype–phenotypeanalysisof media 547d7d36, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6, carneiro2021casereportdiffuse pages 2-3) |
| Diagnostics | EEG | EEG is used for seizure characterization and longitudinal management; reported findings include predominantly frontal spikes/spike-waves, multifocal spikes, generalized slow spike-and-wave complexes, and tonic seizures during sleep in severe epileptic encephalopathy/Lennox-Gastaut presentations. | LOINC-style concept: Electroencephalogram study; MAXO: electroencephalographic monitoring | Supports phenotyping of high-burden epilepsy; abnormalities are variable and reflect seizure syndrome rather than disease specificity. | (carneiro2021casereportdiffuse pages 2-3, parrini2009bilateralfrontoparietalpolymicrogyria pages 3-5, carneiro2021casereportdiffuse pages 1-2) |
| Diagnostics | Whole-exome sequencing (WES) | Recommended high-yield molecular test in suspected ADGRG1-related BFPP, especially with PMG plus cerebellar/brainstem findings or consanguinity. In the 2024 family report, WES used Illumina NovaSeq paired-end sequencing, alignment with BWA-MEM2, SNV/indel calling with GATK, and annotation/classification with ACMG-based pipelines. | MAXO: exome sequencing | Useful early when neuroradiologic interpretation is uncertain or when imaging overlaps infection/cobblestone phenotypes. | (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
| Diagnostics | Targeted NGS panel | Real-world alternative/adjunct to WES: targeted next-generation sequencing panels for brain morphogenesis/malformations of cortical development can detect ADGRG1 variants, as shown in the 2021 case. | MAXO: targeted gene panel sequencing | Practical in clinical neurogenetics workflows where phenotype strongly suggests a malformation-of-cortical-development disorder. | (carneiro2021casereportdiffuse pages 1-2, carneiro2021casereportdiffuse pages 2-3) |
| Diagnostics | Sanger segregation testing | After candidate variant detection, familial segregation by Sanger sequencing is used to confirm biallelic inheritance and carrier status in parents/siblings. | MAXO: Sanger sequencing; MAXO: genetic testing of family members | Important for confirming autosomal recessive inheritance and informing recurrence risk/cascade testing. | (carneiro2021casereportdiffuse pages 1-2, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, carneiro2021casereportdiffuse pages 2-3) |
| Diagnostics | Variant interpretation | Variant interpretation should follow ACMG/AMP principles, incorporating rarity in population databases, predicted loss-of-function mechanism, in silico tools, and segregation. Examples include p.Arg502Ter classified with very strong pathogenic evidence and p.Leu103Pro interpreted through an ACMG-based workflow. | MAXO: genetic variant interpretation | ADGRG1 loss of function is a recognized disease mechanism, supporting pathogenic classification of truncating alleles. | (carneiro2021casereportdiffuse pages 2-3, shaath2024integratinggenomesequencing pages 4-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3) |
| Diagnostics | CNV/SV analysis | Exome-era workflows may include exome-based CNV/SV calling (e.g., CoNIFER, 3bCNV in the 2024 report) to avoid missing structural contributors when single-nucleotide testing is unrevealing. | MAXO: copy number variation analysis | Not a classic major mechanism for ADGRG1-BFPP based on retrieved evidence, but included in modern rare-disease diagnostic pipelines. | (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
| Diagnostics | Ancillary exclusion testing | In complex cases, clinicians have used chromosomal testing, fragile X/imprinting studies, mtDNA testing, metabolic screening, and muscle biopsy to exclude alternative diagnoses. | MAXO: metabolic testing; MAXO: muscle biopsy | Particularly useful when presentation mimics congenital muscular dystrophy/cobblestone complex or metabolic disease. | (carneiro2021casereportdiffuse pages 2-3) |
| Differential diagnosis | Congenital CMV/HSV and other congenital infections | BFPP MRI may mimic congenital CMV or HSV because of polymicrogyria, ventriculomegaly, and white-matter abnormalities; infectious workup and molecular testing help avoid misdiagnosis. | MAXO: infectious disease differential diagnosis; MAXO: brain MRI | Important diagnostic pitfall emphasized in recent literature, especially where expert neuroradiology access is limited. | (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
| Differential diagnosis | Dystroglycanopathies / cobblestone muscular dystrophy | ADGRG1-related disease can present with early hypotonia and MRI overlap resembling cobblestone complex; normal muscle biopsy and the characteristic posterior fossa/brainstem pattern favor ADGRG1-related BFPP. | MAXO: muscle biopsy; MAXO: differential diagnosis | Early “pseudomyopathic” presentation is a recurring clinical trap. | (carneiro2021casereportdiffuse pages 2-3) |
| Differential diagnosis | Other PMG subtypes / BGP / pachygyria-lissencephaly spectrum | Atypical ADGRG1 cases may lack the canonical gradient and instead show diffuse PMG or pachygyria/lissencephaly-like changes, requiring broad malformation-of-cortical-development differential diagnosis. | MAXO: exome sequencing; MAXO: neuroradiologic review | Broad genomic testing helps resolve overlap syndromes and possible multilocus disease. | (carneiro2021casereportdiffuse pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4) |
| Differential diagnosis | False reassurance from normal early MRI | One reported patient had a normal MRI in infancy despite later-confirmed ADGRG1-related disease; repeat imaging should be considered if clinical suspicion remains high. | MAXO: follow-up brain MRI | Normal early imaging does not exclude the diagnosis. | (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6) |
| Management/Treatment | Antiseizure therapy | Seizure treatment is symptomatic and individualized. Reported medications include valproate (initially effective in one case), vigabatrin, levetiracetam, topiramate, clonazepam, and perampanel; many patients have refractory epilepsy. | MAXO: antiseizure medication therapy | No disease-modifying therapy identified in retrieved literature; seizure burden is often high and drug resistance common. | (carneiro2021casereportdiffuse pages 2-3, carneiro2021casereportdiffuse pages 1-2, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
| Management/Treatment | Developmental and supportive care | Real-world care typically includes multidisciplinary neurodevelopmental support for motor, cognitive, speech/language, feeding, and visual/oculomotor impairments, although disease-specific protocols are not detailed in the retrieved ADGRG1 papers. | MAXO: supportive care; MAXO: physical therapy; MAXO: speech therapy; MAXO: occupational therapy | Supportive management is inferred from the severe, lifelong neurodevelopmental phenotype and standard PMG care principles. | (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, carneiro2021casereportdiffuse pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
| Management/Treatment | Longitudinal neurologic follow-up | Ongoing follow-up is needed for epilepsy evolution, ambulation, tone/pyramidal signs, cerebellar features, and developmental progress; some patients show regression with epileptic decompensation. | MAXO: neurologic follow-up | Clinical course is static structurally but functionally variable, especially with refractory epilepsy. | (carneiro2021casereportdiffuse pages 1-2, carneiro2021casereportdiffuse pages 3-5) |
| Management/Treatment | Clinical research implementation | Observational study NCT01488461 (“Phenotypic and Genotypic Studies in Congenital and Early Onset Ataxias”) posted 2011-12-08 and completed 2014-10 included sequencing of GPR56 in patients with suggestive congenital ataxia features. | MAXO: enrollment in observational study; MAXO: genetic testing | Illustrates real-world incorporation of GPR56/ADGRG1 testing into neurogenetics research/diagnostic pathways for cerebellar ataxia syndromes. | (NCT01488461 chunk 1) |
| Prevention | Genetic counseling | Genetic counseling is strongly recommended for affected families, especially in consanguineous settings, to explain autosomal recessive inheritance, recurrence risk, testing options, and family planning. | MAXO: genetic counseling | Explicitly emphasized in the 2024 family report and supported by segregation-based diagnosis. | (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
| Prevention | Carrier testing / cascade testing | Once the familial ADGRG1 variant is known, targeted testing of parents, siblings, and extended relatives can identify carriers and clarify reproductive risk. | MAXO: carrier screening; MAXO: cascade genetic testing | Practical prevention strategy for rare recessive disease in extended families. | (carneiro2021casereportdiffuse pages 1-2, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, carneiro2021casereportdiffuse pages 2-3) |
| Prevention | Prenatal diagnosis / reproductive planning | Although not elaborated as formal protocols in all retrieved papers, family reports show direct reproductive utility of molecular diagnosis; one cited family used fetal testing in a subsequent pregnancy, and counseling papers explicitly frame genetic diagnosis as aiding future reproductive choices. | MAXO: prenatal genetic testing; MAXO: reproductive counseling; MAXO: preimplantation genetic testing | Appropriate once a familial pathogenic ADGRG1 variant is established. | (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) |
| Prevention | Consanguinity-informed risk assessment | In populations/families with consanguinity, earlier consideration of autosomal recessive BFPP and earlier exome/panel testing may shorten diagnostic delay and reduce recurrence through informed planning. | MAXO: risk assessment; MAXO: exome sequencing | Consanguinity is a prominent feature in many reported pedigrees. | (jansen2005geneticsofthe pages 5-6, piao2005genotype–phenotypeanalysisof pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3) |
Table: This table summarizes practical diagnostic workflows, common differential-diagnosis pitfalls, symptomatic management, and prevention/counseling strategies for ADGRG1-related bilateral frontoparietal polymicrogyria. It is useful for translating case-report and cohort evidence into a knowledge-base-ready clinical implementation view.
Key diagnostic principles: - Diagnosis is typically suspected from MRI pattern and confirmed by molecular testing (panel or WES) with segregation and ACMG-based interpretation. (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, carneiro2021casereportdiffuse pages 2-3) - Important diagnostic pitfalls include mimicry of congenital CMV/HSV and cobblestone/dystroglycanopathy-like presentations. (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6, carneiro2021casereportdiffuse pages 2-3)
11. Outcome / prognosis
- Many individuals have severe intellectual disability and motor impairment; however, ambulation is achieved in a majority in aggregated series (e.g., 81.6% in one literature synthesis; 44/63 ~70% in another), indicating variable functional outcomes. (carneiro2021casereportdiffuse pages 3-5, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6)
- Epilepsy is common and frequently refractory (e.g., 54.7–60% refractory among seizure-affected individuals in aggregated datasets), which is a major driver of morbidity. (carneiro2021casereportdiffuse pages 5-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7)
- Disease-specific mortality rates for BFPP were not identified in the retrieved evidence; one severe multi-system twin presentation with ADGRG1 p.Arg565Trp (plus an additional variant) died before age 2, illustrating potential severity in complex/overlapping cases rather than typical BFPP prognosis. (shaath2024integratinggenomesequencing pages 2-4, shaath2024integratinggenomesequencing pages 5-7)
12. Treatment
12.1 Pharmacotherapy (symptomatic)
No disease-modifying pharmacotherapy specific to ADGRG1-BFPP was identified in the retrieved evidence. Current treatment is symptomatic, primarily targeting seizures.
Antiseizure medications (ASMs): - Valproate was “initially responsive” in one ADGRG1 case report; other reported ASMs used across severe cases include vigabatrin, levetiracetam, topiramate, clonazepam, and perampanel, with many patients remaining refractory. (carneiro2021casereportdiffuse pages 2-3, carneiro2021casereportdiffuse pages 1-2) - A separate ADGRG1 regulatory-variant–associated polymicrogyria phenotype (perisylvian-restricted) is reported to have vigabatrin efficacy in GPR56-mutated patients (reported in model-focused review context). (murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2)
Suggested MAXO terms (treatment): - MAXO: antiseizure medication therapy (carneiro2021casereportdiffuse pages 1-2) - MAXO: supportive care / multidisciplinary rehabilitation (inferred standard of care; disease-specific trials absent) (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3)
12.2 Surgical/interventional
No ADGRG1-specific epilepsy surgery outcomes were identified in the retrieved evidence excerpts.
12.3 Experimental therapeutics
No interventional clinical trials targeting ADGRG1-BFPP were identified. An observational study in congenital/early-onset ataxias included sequencing GPR56 in participants with suggestive features (NCT01488461; first posted 2011-12-08, completion 2014-10). (NCT01488461 chunk 1)
13. Prevention
Primary prevention of the genetic disorder is not possible without reproductive interventions; prevention focuses on recurrence reduction through genetic counseling and family testing. - Genetic counseling and carrier/cascade testing are recommended and emphasized, especially in consanguineous families. (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7) - Prenatal testing / reproductive planning is enabled once a familial pathogenic ADGRG1 variant is established; family reports highlight reproductive utility of molecular diagnosis. (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7)
Suggested MAXO terms (prevention): - MAXO: genetic counseling; MAXO: carrier screening; MAXO: prenatal genetic testing (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3)
14. Other species / natural disease
No naturally occurring veterinary disease analogs explicitly tied to ADGRG1 variants were identified in the retrieved evidence.
15. Model organisms
15.1 Model systems and key phenotypes
- Mouse (Gpr56 loss): reported to cause reduced progenitor proliferation, radial glial endfeet/pial basement membrane abnormalities, neuron overmigration, disorganized lamination, and a “cobblestone-like malformation.” (murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2)
- Zebrafish/rodent glial models: ADGRG1/GPR56 is implicated as a conserved regulator of myelination and glial development; in PNS Schwann cells, GPR56-dependent RhoA signaling supports radial sorting and myelin thickness/organization, and mutants show progressive neuropathy-like phenotypes. (ackerman2018gpr56adgrg1regulatesdevelopment pages 1-2, ackerman2018gpr56adgrg1regulatesdevelopment pages 10-11)
- Microglia–OPC axis (mouse): microglia-derived TG2 signals to ADGRG1 on OPCs (with laminin) promoting OPC proliferation and improving remyelination in murine demyelination models. (giera2018microglialtransglutaminase2drives pages 1-2, giera2018microglialtransglutaminase2drives pages 3-5)
- Common marmoset (transgenic promoter model): a polymicrogyria-associated GPR56 promoter element preferentially drives expression in developing GABAergic neurons, supporting interneuron involvement in epilepsy linked to GPR56 regulatory variants. (murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2)
15.2 Model applications and limitations
These models support mechanistic interrogation of (i) cortical lamination and migration, (ii) ECM ligand interactions and Gα12/13–RhoA signaling, and (iii) myelination/glia–glia signaling (microglia→OPC; Schwann cells). However, no model fully recapitulates the complete human BFPP MRI signature (frontoparietal gradient plus hindbrain hypoplasia) in the retrieved excerpts, and species differences in ligand interactions (e.g., TG2 binding specificity in some systems) have been reported in mechanistic studies. (chiang2011diseaseassociatedgpr56mutations pages 7-8, ackerman2018gpr56adgrg1regulatesdevelopment pages 1-2)
Recent developments (2023–2024 prioritized highlights)
- 2024 (family + review): identification of a novel homozygous ADGRG1 variant (p.Leu103Pro) in a large consanguineous family; detailed modern WES pipeline and emphasis on genetic counseling and diagnostic pitfalls (MRI mimicry of infections; occasional normal infancy MRI). (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3, khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6)
- 2024 (multi-omics rare disease): integrated WGS + untargeted metabolomics in twins with homozygous ADGRG1 p.Arg565Trp and complex overlapping phenotype; metabolomics flagged 180 clinically relevant biochemicals with substantial lipid (48%) and amino-acid pathway (29%) perturbations, and evidence for oxidative stress/glutathione depletion markers and urea-cycle/arginine–proline metabolism involvement. (shaath2024integratinggenomesequencing pages 5-7, shaath2024integratinggenomesequencing pages 7-8, shaath2024integratinggenomesequencing pages 4-5)
- 2024 (signaling pharmacology): GPR56 ligand modalities show biased G-protein selectivity (G13 vs G12) while converging on Rho pathway activation, providing tools and conceptual frameworks potentially relevant to future targeted modulation strategies (not yet applied clinically to BFPP). (jallouli2024gproteinselectivity pages 1-2, jallouli2024gproteinselectivity pages 11-13)
- 2023 (cohort genetics): large polymicrogyria cohort study demonstrated 32.7% genetic diagnostic yield and highlighted convergent mechanistic categories including ion-conducting proteins; ADGRG1 remains a canonical known PMG gene within such cohorts. (akula2023exomesequencingand pages 2-3)
Expert synthesis / interpretation (authoritative-source-driven)
Taken together, the human genetics literature establishes ADGRG1 as a high-confidence Mendelian cause of BFPP with a recognizable radiogenomic signature and high burden of epilepsy and developmental disability (piao2005genotype–phenotypeanalysisof pages 3-5, piao2005genotype–phenotypeanalysisof pages 1-2). Functional biochemical studies support that many disease alleles are “processing/trafficking” loss-of-function variants in an adhesion GPCR requiring precise proteolytic maturation and extracellular interactions; this molecular fragility plausibly explains why both extracellular missense and truncating variants can converge on a similar neurodevelopmental outcome (chiang2011diseaseassociatedgpr56mutations pages 3-5, ke2008biochemicalcharacterizationof pages 1-2, chiang2011diseaseassociatedgpr56mutations pages 1-2). Recent signaling studies (2024) deepen mechanistic resolution of how distinct ADGRG1 activation modalities (antibody vs small molecule vs constitutive) can yield biased G-protein engagement while preserving Rho-pathway output, suggesting that future therapeutic exploration would likely need to consider ligand modality, receptor cleavage state, and cell-type-specific signaling context (jallouli2024gproteinselectivity pages 1-2, jallouli2024gproteinselectivity pages 7-9). At present, however, clinical management remains supportive and seizure-focused, and prevention is primarily via genetic counseling and reproductive planning in affected families. (khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7, carneiro2021casereportdiffuse pages 1-2)
References
-
(piao2005genotype–phenotypeanalysisof pages 3-5): Xianhua Piao, Bernard S. Chang, Adria Bodell, Katelyn Woods, Bruria BenZeev, Meral Topcu, Renzo Guerrini, Hadassa Goldberg‐Stern, Laszlo Sztriha, William B. Dobyns, A. James Barkovich, and Christopher A. Walsh. Genotype–phenotype analysis of human frontoparietal polymicrogyria syndromes. Annals of Neurology, 58:680-687, Nov 2005. URL: https://doi.org/10.1002/ana.20616, doi:10.1002/ana.20616. This article has 174 citations and is from a highest quality peer-reviewed journal.
-
(khatib2024adgrg1relatedpolymicrogyriasyndrome pages 1-3): Dalida El Khatib, Moussa Hojeij, Sandra Sabbagh, Cybel Mehawej, Eliane Chouery, Seung Woo Ryu, JiHye Kim, and Andre Mégarbané. Adgrg1-related polymicrogyria syndrome: report on a large consanguineous family with a novel variant and review. Egyptian Journal of Medical Human Genetics, Apr 2024. URL: https://doi.org/10.1186/s43042-024-00499-1, doi:10.1186/s43042-024-00499-1. This article has 1 citations and is from a peer-reviewed journal.
-
(piao2005genotype–phenotypeanalysisof pages 1-2): Xianhua Piao, Bernard S. Chang, Adria Bodell, Katelyn Woods, Bruria BenZeev, Meral Topcu, Renzo Guerrini, Hadassa Goldberg‐Stern, Laszlo Sztriha, William B. Dobyns, A. James Barkovich, and Christopher A. Walsh. Genotype–phenotype analysis of human frontoparietal polymicrogyria syndromes. Annals of Neurology, 58:680-687, Nov 2005. URL: https://doi.org/10.1002/ana.20616, doi:10.1002/ana.20616. This article has 174 citations and is from a highest quality peer-reviewed journal.
-
(OpenTargets Search: polymicrogyria,bilateral frontoparietal polymicrogyria-ADGRG1): Open Targets Query (polymicrogyria,bilateral frontoparietal polymicrogyria-ADGRG1, 2 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(khatib2024adgrg1relatedpolymicrogyriasyndrome pages 6-7): Dalida El Khatib, Moussa Hojeij, Sandra Sabbagh, Cybel Mehawej, Eliane Chouery, Seung Woo Ryu, JiHye Kim, and Andre Mégarbané. Adgrg1-related polymicrogyria syndrome: report on a large consanguineous family with a novel variant and review. Egyptian Journal of Medical Human Genetics, Apr 2024. URL: https://doi.org/10.1186/s43042-024-00499-1, doi:10.1186/s43042-024-00499-1. This article has 1 citations and is from a peer-reviewed journal.
-
(jansen2005geneticsofthe pages 5-6): An C. Jansen and E. Andermann. Genetics of the polymicrogyria syndromes. Journal of Medical Genetics, 42:369-378, May 2005. URL: https://doi.org/10.1136/jmg.2004.023952, doi:10.1136/jmg.2004.023952. This article has 205 citations and is from a domain leading peer-reviewed journal.
-
(carneiro2021casereportdiffuse pages 1-2): Fábio Carneiro, Júlia Duarte, Francisco Laranjeira, Sofia Barbosa-Gouveia, María-Luz Couce, and Maria José Fonseca. Case report: diffuse polymicrogyria associated with a novel adgrg1 variant. Frontiers in Pediatrics, Aug 2021. URL: https://doi.org/10.3389/fped.2021.728077, doi:10.3389/fped.2021.728077. This article has 7 citations.
-
(chiang2011diseaseassociatedgpr56mutations pages 1-2): Nien-Yi Chiang, Cheng-Chih Hsiao, Yi-Shu Huang, Hsin-Yi Chen, I-Ju Hsieh, Gin-Wen Chang, and Hsi-Hsien Lin. Disease-associated gpr56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. Journal of Biological Chemistry, 286:14215-14225, Apr 2011. URL: https://doi.org/10.1074/jbc.m110.183830, doi:10.1074/jbc.m110.183830. This article has 103 citations and is from a domain leading peer-reviewed journal.
-
(carneiro2021casereportdiffuse pages 2-3): Fábio Carneiro, Júlia Duarte, Francisco Laranjeira, Sofia Barbosa-Gouveia, María-Luz Couce, and Maria José Fonseca. Case report: diffuse polymicrogyria associated with a novel adgrg1 variant. Frontiers in Pediatrics, Aug 2021. URL: https://doi.org/10.3389/fped.2021.728077, doi:10.3389/fped.2021.728077. This article has 7 citations.
-
(carneiro2021casereportdiffuse pages 3-5): Fábio Carneiro, Júlia Duarte, Francisco Laranjeira, Sofia Barbosa-Gouveia, María-Luz Couce, and Maria José Fonseca. Case report: diffuse polymicrogyria associated with a novel adgrg1 variant. Frontiers in Pediatrics, Aug 2021. URL: https://doi.org/10.3389/fped.2021.728077, doi:10.3389/fped.2021.728077. This article has 7 citations.
-
(carneiro2021casereportdiffuse pages 5-6): Fábio Carneiro, Júlia Duarte, Francisco Laranjeira, Sofia Barbosa-Gouveia, María-Luz Couce, and Maria José Fonseca. Case report: diffuse polymicrogyria associated with a novel adgrg1 variant. Frontiers in Pediatrics, Aug 2021. URL: https://doi.org/10.3389/fped.2021.728077, doi:10.3389/fped.2021.728077. This article has 7 citations.
-
(parrini2009bilateralfrontoparietalpolymicrogyria pages 6-8): Elena Parrini, Anna Rita Ferrari, Thomas Dorn, Christopher A. Walsh, and Renzo Guerrini. Bilateral frontoparietal polymicrogyria, lennox‐gastaut syndrome, and gpr56 gene mutations. Epilepsia, 50:1344-1353, Jun 2009. URL: https://doi.org/10.1111/j.1528-1167.2008.01787.x, doi:10.1111/j.1528-1167.2008.01787.x. This article has 75 citations and is from a domain leading peer-reviewed journal.
-
(parrini2009bilateralfrontoparietalpolymicrogyria pages 3-5): Elena Parrini, Anna Rita Ferrari, Thomas Dorn, Christopher A. Walsh, and Renzo Guerrini. Bilateral frontoparietal polymicrogyria, lennox‐gastaut syndrome, and gpr56 gene mutations. Epilepsia, 50:1344-1353, Jun 2009. URL: https://doi.org/10.1111/j.1528-1167.2008.01787.x, doi:10.1111/j.1528-1167.2008.01787.x. This article has 75 citations and is from a domain leading peer-reviewed journal.
-
(khatib2024adgrg1relatedpolymicrogyriasyndrome pages 3-4): Dalida El Khatib, Moussa Hojeij, Sandra Sabbagh, Cybel Mehawej, Eliane Chouery, Seung Woo Ryu, JiHye Kim, and Andre Mégarbané. Adgrg1-related polymicrogyria syndrome: report on a large consanguineous family with a novel variant and review. Egyptian Journal of Medical Human Genetics, Apr 2024. URL: https://doi.org/10.1186/s43042-024-00499-1, doi:10.1186/s43042-024-00499-1. This article has 1 citations and is from a peer-reviewed journal.
-
(khatib2024adgrg1relatedpolymicrogyriasyndrome pages 4-6): Dalida El Khatib, Moussa Hojeij, Sandra Sabbagh, Cybel Mehawej, Eliane Chouery, Seung Woo Ryu, JiHye Kim, and Andre Mégarbané. Adgrg1-related polymicrogyria syndrome: report on a large consanguineous family with a novel variant and review. Egyptian Journal of Medical Human Genetics, Apr 2024. URL: https://doi.org/10.1186/s43042-024-00499-1, doi:10.1186/s43042-024-00499-1. This article has 1 citations and is from a peer-reviewed journal.
-
(piao2005genotype–phenotypeanalysisof media 547d7d36): Xianhua Piao, Bernard S. Chang, Adria Bodell, Katelyn Woods, Bruria BenZeev, Meral Topcu, Renzo Guerrini, Hadassa Goldberg‐Stern, Laszlo Sztriha, William B. Dobyns, A. James Barkovich, and Christopher A. Walsh. Genotype–phenotype analysis of human frontoparietal polymicrogyria syndromes. Annals of Neurology, 58:680-687, Nov 2005. URL: https://doi.org/10.1002/ana.20616, doi:10.1002/ana.20616. This article has 174 citations and is from a highest quality peer-reviewed journal.
-
(lin2021atwinscase pages 7-10): Wenxin Lin, Yingying Chai, Xia Zhang, Tingting Huang, Guo Zheng, Gang Zhang, Fang Peng, and Yanjun Huang. A twins case of lissencephaly with gpr56 compound heterozygous mutations and literatures review. ArXiv, Jan 2021. URL: https://doi.org/10.21203/rs.3.rs-144069/v1, doi:10.21203/rs.3.rs-144069/v1. This article has 0 citations.
-
(chiang2011diseaseassociatedgpr56mutations pages 8-10): Nien-Yi Chiang, Cheng-Chih Hsiao, Yi-Shu Huang, Hsin-Yi Chen, I-Ju Hsieh, Gin-Wen Chang, and Hsi-Hsien Lin. Disease-associated gpr56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. Journal of Biological Chemistry, 286:14215-14225, Apr 2011. URL: https://doi.org/10.1074/jbc.m110.183830, doi:10.1074/jbc.m110.183830. This article has 103 citations and is from a domain leading peer-reviewed journal.
-
(chiang2011diseaseassociatedgpr56mutations pages 3-5): Nien-Yi Chiang, Cheng-Chih Hsiao, Yi-Shu Huang, Hsin-Yi Chen, I-Ju Hsieh, Gin-Wen Chang, and Hsi-Hsien Lin. Disease-associated gpr56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. Journal of Biological Chemistry, 286:14215-14225, Apr 2011. URL: https://doi.org/10.1074/jbc.m110.183830, doi:10.1074/jbc.m110.183830. This article has 103 citations and is from a domain leading peer-reviewed journal.
-
(chiang2011diseaseassociatedgpr56mutations pages 10-11): Nien-Yi Chiang, Cheng-Chih Hsiao, Yi-Shu Huang, Hsin-Yi Chen, I-Ju Hsieh, Gin-Wen Chang, and Hsi-Hsien Lin. Disease-associated gpr56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. Journal of Biological Chemistry, 286:14215-14225, Apr 2011. URL: https://doi.org/10.1074/jbc.m110.183830, doi:10.1074/jbc.m110.183830. This article has 103 citations and is from a domain leading peer-reviewed journal.
-
(ke2008biochemicalcharacterizationof pages 1-2): Ning Ke, Hongwen Ma, Gundo Diedrich, John Chionis, Guohong Liu, De-Hua Yu, Flossie Wong-Staal, and Qi-Xiang Li. Biochemical characterization of genetic mutations of gpr56 in patients with bilateral frontoparietal polymicrogyria (bfpp). Biochemical and biophysical research communications, 366 2:314-20, Feb 2008. URL: https://doi.org/10.1016/j.bbrc.2007.11.071, doi:10.1016/j.bbrc.2007.11.071. This article has 35 citations and is from a peer-reviewed journal.
-
(shaath2024integratinggenomesequencing pages 2-4): Rulan Shaath, Aljazi Al-Maraghi, Haytham Ali, Jehan AlRayahi, Adam D. Kennedy, Karen L. DeBalsi, Sura Hussein, Najwa Elbashir, Sujitha S. Padmajeya, Sasirekha Palaniswamy, Sarah H. Elsea, Ammira A. Akil, Noha A. Yousri, and Khalid A. Fakhro. Integrating genome sequencing and untargeted metabolomics in monozygotic twins with a rare complex neurological disorder. Metabolites, 14:152, Mar 2024. URL: https://doi.org/10.3390/metabo14030152, doi:10.3390/metabo14030152. This article has 5 citations.
-
(shaath2024integratinggenomesequencing pages 4-5): Rulan Shaath, Aljazi Al-Maraghi, Haytham Ali, Jehan AlRayahi, Adam D. Kennedy, Karen L. DeBalsi, Sura Hussein, Najwa Elbashir, Sujitha S. Padmajeya, Sasirekha Palaniswamy, Sarah H. Elsea, Ammira A. Akil, Noha A. Yousri, and Khalid A. Fakhro. Integrating genome sequencing and untargeted metabolomics in monozygotic twins with a rare complex neurological disorder. Metabolites, 14:152, Mar 2024. URL: https://doi.org/10.3390/metabo14030152, doi:10.3390/metabo14030152. This article has 5 citations.
-
(murayama2020thepolymicrogyriaassociatedgpr56 pages 1-2): Ayako Y. Murayama, Ken-ichiro Kuwako, Junko Okahara, Byoung-Il Bae, Misako Okuno, Hiromi Mashiko, Tomomi Shimogori, Christopher A. Walsh, Erika Sasaki, and Hideyuki Okano. The polymicrogyria-associated gpr56 promoter preferentially drives gene expression in developing gabaergic neurons in common marmosets. Scientific Reports, Dec 2020. URL: https://doi.org/10.1038/s41598-020-78608-4, doi:10.1038/s41598-020-78608-4. This article has 17 citations and is from a peer-reviewed journal.
-
(chiang2011diseaseassociatedgpr56mutations pages 5-6): Nien-Yi Chiang, Cheng-Chih Hsiao, Yi-Shu Huang, Hsin-Yi Chen, I-Ju Hsieh, Gin-Wen Chang, and Hsi-Hsien Lin. Disease-associated gpr56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. Journal of Biological Chemistry, 286:14215-14225, Apr 2011. URL: https://doi.org/10.1074/jbc.m110.183830, doi:10.1074/jbc.m110.183830. This article has 103 citations and is from a domain leading peer-reviewed journal.
-
(chiang2011diseaseassociatedgpr56mutations pages 7-8): Nien-Yi Chiang, Cheng-Chih Hsiao, Yi-Shu Huang, Hsin-Yi Chen, I-Ju Hsieh, Gin-Wen Chang, and Hsi-Hsien Lin. Disease-associated gpr56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. Journal of Biological Chemistry, 286:14215-14225, Apr 2011. URL: https://doi.org/10.1074/jbc.m110.183830, doi:10.1074/jbc.m110.183830. This article has 103 citations and is from a domain leading peer-reviewed journal.
-
(jallouli2024gproteinselectivity pages 1-2): Raida Jallouli, Ana L. Moreno-Salinas, Andréanne Laniel, Brian Holleran, Charlotte Avet, Joan Jacob, Trang Hoang, Christine Lavoie, Kendra S. Carmon, Michel Bouvier, and Richard Leduc. G protein selectivity profile of gpr56/adgrg1 and its effect on downstream effectors. Cellular and Molecular Life Sciences: CMLS, Sep 2024. URL: https://doi.org/10.1007/s00018-024-05416-8, doi:10.1007/s00018-024-05416-8. This article has 10 citations.
-
(einspahr2022pathophysiologicalimpactof pages 5-9): Jeanette M. Einspahr and Douglas G. Tilley. Pathophysiological impact of the adhesion g protein-coupled receptor family. Aug 2022. URL: https://doi.org/10.1152/ajpcell.00445.2021, doi:10.1152/ajpcell.00445.2021. This article has 31 citations.
-
(giera2018microglialtransglutaminase2drives pages 1-2): Stefanie Giera, Rong Luo, Yanqin Ying, Sarah D Ackerman, Sung-Jin Jeong, Hannah M Stoveken, Christopher J Folts, Christina A Welsh, Gregory G Tall, Beth Stevens, Kelly R Monk, and Xianhua Piao. Microglial transglutaminase-2 drives myelination and myelin repair via gpr56/adgrg1 in oligodendrocyte precursor cells. eLife, May 2018. URL: https://doi.org/10.7554/elife.33385, doi:10.7554/elife.33385. This article has 164 citations and is from a domain leading peer-reviewed journal.
-
(giera2018microglialtransglutaminase2drives pages 3-5): Stefanie Giera, Rong Luo, Yanqin Ying, Sarah D Ackerman, Sung-Jin Jeong, Hannah M Stoveken, Christopher J Folts, Christina A Welsh, Gregory G Tall, Beth Stevens, Kelly R Monk, and Xianhua Piao. Microglial transglutaminase-2 drives myelination and myelin repair via gpr56/adgrg1 in oligodendrocyte precursor cells. eLife, May 2018. URL: https://doi.org/10.7554/elife.33385, doi:10.7554/elife.33385. This article has 164 citations and is from a domain leading peer-reviewed journal.
-
(bauer2024mesenchymaltransglutaminase2 pages 1-2): Lea Bauer, Jessica Edwards, Andreas Heil, Sharon Dewitt, Heike Biebermann, Daniel Aeschlimann, and Vera Knäuper. Mesenchymal transglutaminase 2 activates epithelial adam17: link to g-protein-coupled receptor 56 (adgrg1) signalling. International Journal of Molecular Sciences, 25:2329, Feb 2024. URL: https://doi.org/10.3390/ijms25042329, doi:10.3390/ijms25042329. This article has 5 citations.
-
(bauer2024mesenchymaltransglutaminase2 pages 2-5): Lea Bauer, Jessica Edwards, Andreas Heil, Sharon Dewitt, Heike Biebermann, Daniel Aeschlimann, and Vera Knäuper. Mesenchymal transglutaminase 2 activates epithelial adam17: link to g-protein-coupled receptor 56 (adgrg1) signalling. International Journal of Molecular Sciences, 25:2329, Feb 2024. URL: https://doi.org/10.3390/ijms25042329, doi:10.3390/ijms25042329. This article has 5 citations.
-
(bauer2024mesenchymaltransglutaminase2 pages 6-9): Lea Bauer, Jessica Edwards, Andreas Heil, Sharon Dewitt, Heike Biebermann, Daniel Aeschlimann, and Vera Knäuper. Mesenchymal transglutaminase 2 activates epithelial adam17: link to g-protein-coupled receptor 56 (adgrg1) signalling. International Journal of Molecular Sciences, 25:2329, Feb 2024. URL: https://doi.org/10.3390/ijms25042329, doi:10.3390/ijms25042329. This article has 5 citations.
-
(ackerman2018gpr56adgrg1regulatesdevelopment pages 1-2): Sarah D. Ackerman, Rong Luo, Yannick Poitelon, Amit Mogha, Breanne L. Harty, Mitchell D’Rozario, Nicholas E. Sanchez, Asvin K.K. Lakkaraju, Paul Gamble, Jun Li, Jun Qu, Matthew R. MacEwan, Wilson Zachary Ray, Adriano Aguzzi, M. Laura Feltri, Xianhua Piao, and Kelly R. Monk. Gpr56/adgrg1 regulates development and maintenance of peripheral myelin. The Journal of Experimental Medicine, 215:941-961, Mar 2018. URL: https://doi.org/10.1084/jem.20161714, doi:10.1084/jem.20161714. This article has 73 citations.
-
(akula2023exomesequencingand pages 2-3): Shyam K. Akula, Allen Y. Chen, Jennifer E. Neil, Diane D. Shao, Alisa Mo, Norma K. Hylton, Stephanie DiTroia, Vijay S. Ganesh, Richard S. Smith, Katherine O’Kane, Rebecca C. Yeh, Jack H. Marciano, Samantha Kirkham, Connor J. Kenny, Janet H. T. Song, Muna Al Saffar, Francisca Millan, David J. Harris, Andrea V. Murphy, Kara C. Klemp, Stephen R. Braddock, Harrison Brand, Isaac Wong, Michael E. Talkowski, Anne O’Donnell-Luria, Abbe Lai, Robert Sean Hill, Ganeshwaran H. Mochida, Ryan N. Doan, A. James Barkovich, Edward Yang, Dina Amrom, Eva Andermann, Annapurna Poduri, Christopher A. Walsh, Bassam Abu-Libdeh, Lihadh Al-Gazali, Muna Al Saffar, Edith Alva Moncayo, Dina Amrom, Eva Anderman, Anna-Kaisa Anttonen, Saunder Barnes, Sara Barnett, Todd Barron, Brenda J. Barry, Lina Basel-Vanagaite, Laila Bastaki, Luis Bello-Espinosa, Tawfeg Ben-Omran, Matthew Bernard, Carsten G. Bonneman, Blaise Bourgeois, Stephen Brown, Roberto H. Caraballo, Gergory Cascino, Michael Clarke, Monika Cohen, Yanick Crow, Bernard Dan, Kira A. Dies, William B. Dobyns, François Dubeau, Christelle El Achkar, Gregory M. Enns, Laurence Faivre, Laura Flores-Sarnat, John Gaitanis, Kuchukhidze Giorgi, Andrew Green, Alan Guberman, Renzo Guerrini, Micheil Innes, Richard Jacobsen, Sebastian Jacquemont, Samir Khalil, Joerg Klepper, Dimitri Kranic, Kalpathy Krishnamoorthy, Anna-Elina Lehesjoki, Dorit Lev, Richard J. Leventer, Emily Lisi, Valerie Loik Ramey, Sally Ann Lynch, Laila Mahmoud, David Manchester, David Mandelbaum, Daphna Marom, Deborah Marsden, Mayra Martinez Ojeda, Amira Masri, Livija Medne, Denis Melanson, David T. Miller, Anna Minster, Edward Neilan, Dang Khoa Nguyen, Heather E. Olson, Ignacio Pascual-Castroviejo, Philip L. Pearl, Daniela Pilz, Nada Quercia, Salmo Raskin, Miriam Regev, Lance Rodan, Cynthia Rooney, Michael Rutlin, Mustafa Sahin, Mustafa A. Salih, Pierre Sarda, Harvey B. Sarnat, Ingrid Scheffer, Joseph Shieh, Sharon E. Smith, Janet S. Soul, Siddharth Srivastava, Laszlo Sztriha, Donatella Tampieri, John Tolmie, Meral Topçu, Eugen Trinka, John Tsai, Jack Tsao, Sheila Unger, Iris Unterberger, Goekhan Uyanik, Kette Valente, Thomas Voit, Louise Wilson, and Grace Yoon. Exome sequencing and the identification of new genes and shared mechanisms in polymicrogyria. JAMA Neurology, 80:980, Sep 2023. URL: https://doi.org/10.1001/jamaneurol.2023.2363, doi:10.1001/jamaneurol.2023.2363. This article has 39 citations and is from a highest quality peer-reviewed journal.
-
(NCT01488461 chunk 1): Phenotypic and Genotypic Studies in Congenital and Early Onset Ataxias. Assistance Publique - Hôpitaux de Paris. 2012. ClinicalTrials.gov Identifier: NCT01488461
-
(shaath2024integratinggenomesequencing pages 5-7): Rulan Shaath, Aljazi Al-Maraghi, Haytham Ali, Jehan AlRayahi, Adam D. Kennedy, Karen L. DeBalsi, Sura Hussein, Najwa Elbashir, Sujitha S. Padmajeya, Sasirekha Palaniswamy, Sarah H. Elsea, Ammira A. Akil, Noha A. Yousri, and Khalid A. Fakhro. Integrating genome sequencing and untargeted metabolomics in monozygotic twins with a rare complex neurological disorder. Metabolites, 14:152, Mar 2024. URL: https://doi.org/10.3390/metabo14030152, doi:10.3390/metabo14030152. This article has 5 citations.
-
(ackerman2018gpr56adgrg1regulatesdevelopment pages 10-11): Sarah D. Ackerman, Rong Luo, Yannick Poitelon, Amit Mogha, Breanne L. Harty, Mitchell D’Rozario, Nicholas E. Sanchez, Asvin K.K. Lakkaraju, Paul Gamble, Jun Li, Jun Qu, Matthew R. MacEwan, Wilson Zachary Ray, Adriano Aguzzi, M. Laura Feltri, Xianhua Piao, and Kelly R. Monk. Gpr56/adgrg1 regulates development and maintenance of peripheral myelin. The Journal of Experimental Medicine, 215:941-961, Mar 2018. URL: https://doi.org/10.1084/jem.20161714, doi:10.1084/jem.20161714. This article has 73 citations.
-
(shaath2024integratinggenomesequencing pages 7-8): Rulan Shaath, Aljazi Al-Maraghi, Haytham Ali, Jehan AlRayahi, Adam D. Kennedy, Karen L. DeBalsi, Sura Hussein, Najwa Elbashir, Sujitha S. Padmajeya, Sasirekha Palaniswamy, Sarah H. Elsea, Ammira A. Akil, Noha A. Yousri, and Khalid A. Fakhro. Integrating genome sequencing and untargeted metabolomics in monozygotic twins with a rare complex neurological disorder. Metabolites, 14:152, Mar 2024. URL: https://doi.org/10.3390/metabo14030152, doi:10.3390/metabo14030152. This article has 5 citations.
-
(jallouli2024gproteinselectivity pages 11-13): Raida Jallouli, Ana L. Moreno-Salinas, Andréanne Laniel, Brian Holleran, Charlotte Avet, Joan Jacob, Trang Hoang, Christine Lavoie, Kendra S. Carmon, Michel Bouvier, and Richard Leduc. G protein selectivity profile of gpr56/adgrg1 and its effect on downstream effectors. Cellular and Molecular Life Sciences: CMLS, Sep 2024. URL: https://doi.org/10.1007/s00018-024-05416-8, doi:10.1007/s00018-024-05416-8. This article has 10 citations.
-
(jallouli2024gproteinselectivity pages 7-9): Raida Jallouli, Ana L. Moreno-Salinas, Andréanne Laniel, Brian Holleran, Charlotte Avet, Joan Jacob, Trang Hoang, Christine Lavoie, Kendra S. Carmon, Michel Bouvier, and Richard Leduc. G protein selectivity profile of gpr56/adgrg1 and its effect on downstream effectors. Cellular and Molecular Life Sciences: CMLS, Sep 2024. URL: https://doi.org/10.1007/s00018-024-05416-8, doi:10.1007/s00018-024-05416-8. This article has 10 citations.