46,XX Gonadal Dysgenesis
46,XX gonadal dysgenesis is a rare, genetically heterogeneous ovarian developmental disorder in phenotypically female individuals with a normal 46,XX karyotype. The shared disease mechanism is failure of ovarian development and folliculogenesis, producing streak or severely underdeveloped ovaries, estrogen deficiency, hypergonadotropic hypogonadism, absent spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and infertility. The entry includes isolated gene-defined forms and a Perrault-spectrum syndromic branch with sensorineural hearing impairment, but anchors ovarian developmental failure rather than broader 46,XX disorder-of-sex-development phenotype mentions caused by other mechanisms such as prenatal androgen excess.
Ask OpenScientist
Ask a research question about 46,XX Gonadal Dysgenesis. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Mappings
Definitions
1Show evidence (1 reference)
Inheritance
2Show evidence (2 references)
Show evidence (1 reference)
Subtypes
5Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Discussions and Knowledge Gaps
1Show evidence (1 reference)
Pathophysiology
8Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Pathograph
Phenotypes
7Ear 1
Show evidence (1 reference)
Endocrine 2
Show evidence (1 reference)
Show evidence (2 references)
Genitourinary 4
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
4Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Medical Actions
6Show evidence (2 references)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Clinical Trials
1Show evidence (1 reference)
Source YAML
click to showname: 46,XX Gonadal Dysgenesis
creation_date: "2026-04-14T05:40:11Z"
updated_date: "2026-05-31T10:35:00Z"
category: Mendelian
description: >-
46,XX gonadal dysgenesis is a rare, genetically heterogeneous ovarian
developmental disorder in phenotypically female individuals with a normal
46,XX karyotype. The shared disease mechanism is failure of ovarian
development and folliculogenesis, producing streak or severely
underdeveloped ovaries, estrogen deficiency, hypergonadotropic hypogonadism,
absent spontaneous pubertal development, primary amenorrhea, uterine
hypoplasia, and infertility. The entry includes isolated gene-defined forms
and a Perrault-spectrum syndromic branch with sensorineural hearing
impairment, but anchors ovarian developmental failure rather than broader
46,XX disorder-of-sex-development phenotype mentions caused by other
mechanisms such as prenatal androgen excess.
disease_term:
preferred_term: 46,XX gonadal dysgenesis
term:
id: MONDO:0009299
label: 46 XX gonadal dysgenesis
synonyms:
- 46 XX gonadal dysgenesis
- XX female gonadal dysgenesis
- 46,XX ovarian dysgenesis
- 46,XX pure gonadal dysgenesis
parents:
- Disorder of sex development
- Primary ovarian insufficiency
- Gonadal development disorder
definitions:
- name: Clinical disease framing for 46,XX gonadal dysgenesis
definition_type: CASE_DEFINITION
description: >-
46,XX gonadal dysgenesis is a disease-level ovarian developmental failure
state defined by a normal 46,XX karyotype, phenotypically female external
genitalia, underdeveloped or streak ovaries, absent spontaneous puberty,
primary amenorrhea, uterine hypoplasia, and hypergonadotropic
hypogonadism.
scope: >-
Disease anchor for isolated 46,XX gonadal dysgenesis rather than a generic
46,XX DSD phenotype bucket.
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically heterogeneous
disorder that is characterized by underdeveloped, dysfunctional ovaries,
with subsequent lack of spontaneous pubertal development, primary
amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.
explanation: >-
This abstract provides a compact disease-level definition for isolated
46,XX gonadal dysgenesis.
mappings:
mondo_mappings:
- term:
id: MONDO:0009299
label: 46 XX gonadal dysgenesis
mapping_predicate: skos:exactMatch
mapping_source: MONDO
mapping_justification: Primary MONDO disease identifier for 46,XX gonadal dysgenesis.
has_subtypes:
- name: ODG1-FSHR
display_name: Ovarian dysgenesis 1 (FSHR-related)
classification: gene
description: >-
Autosomal recessive FSHR-related ovarian dysgenesis caused by impaired
follicle-stimulating hormone receptor binding and signal transduction in
granulosa-cell pathways.
subtype_term:
preferred_term: ovarian dysgenesis 1
term:
id: MONDO:0024463
label: ovarian dysgenesis 1
genes:
- preferred_term: FSHR
term:
id: hgnc:3969
label: FSHR
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:7553856
reference_title: >-
Mutation in the follicle-stimulating hormone receptor gene causes
hereditary hypergonadotropic ovarian failure.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hypergonadotropic ovarian dysgenesis (ODG) with normal karyotype is a
heterogeneous condition that in some cases displays Mendelian recessive
inheritance.
explanation: >-
Establishes FSHR-related ovarian dysgenesis as a recessive subtype.
- name: ODG2-NUP107
display_name: Ovarian dysgenesis 2 (NUP107-related)
classification: gene
description: >-
Autosomal recessive NUP107-related subtype linking nuclear pore dysfunction
and defective oogenesis to isolated 46,XX gonadal dysgenesis.
subtype_term:
preferred_term: ovarian dysgenesis 2
term:
id: MONDO:0010349
label: ovarian dysgenesis 2
genes:
- preferred_term: NUP107
term:
id: hgnc:29914
label: NUP107
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Using homozygosity mapping and whole-exome sequencing, we identified a
recessive missense mutation in nucleoporin-107 (NUP107, c.1339G>A,
p.D447N).
explanation: >-
Defines the recessive NUP107-related subtype.
- name: ODG3-PSMC3IP
display_name: Ovarian dysgenesis 3 (PSMC3IP/HOP2-related)
classification: gene
description: >-
Autosomal recessive PSMC3IP-related subtype in which a homozygous HOP2
coactivator defect disrupts estrogen-driven transcription and follicular-pool
maintenance.
subtype_term:
preferred_term: ovarian dysgenesis 3
term:
id: MONDO:0013689
label: ovarian dysgenesis 3
genes:
- preferred_term: PSMC3IP
term:
id: hgnc:17928
label: PSMC3IP
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes
coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Affected females were homozygous for a 3 bp deletion (NM_016556.2,
c.600_602del) in the PSMC3IP gene, leading to deletion of a glutamic acid
residue (p.Glu201del) in the highly conserved C-terminal acidic domain.
explanation: >-
Defines the homozygous PSMC3IP-related subtype.
- name: ODG4-NR5A1
display_name: NR5A1-related 46,XX gonadal dysgenesis/ovarian insufficiency
classification: gene
description: >-
NR5A1-related ovarian developmental failure overlaps 46,XX gonadal
dysgenesis and primary ovarian insufficiency, most often through dominant
familial variants with variable penetrance and expressivity.
genes:
- preferred_term: NR5A1
term:
id: hgnc:7983
label: NR5A1
inheritance:
- name: Autosomal dominant with variable penetrance
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
evidence:
- reference: PMID:19246354
reference_title: Mutations in NR5A1 associated with ovarian insufficiency.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Families 1, 3, and 4 show a dominant mode of inheritance, whereas Family 2
shows a recessive mode.
explanation: >-
Supports dominant NR5A1 inheritance while preserving the reported
variability in penetrance and inheritance mode.
- name: Perrault-spectrum
display_name: Perrault-spectrum 46,XX gonadal dysgenesis
classification: syndromic
description: >-
Autosomal recessive syndromic branch with ovarian dysgenesis or primary
ovarian insufficiency in 46,XX individuals plus sensorineural hearing
impairment.
subtype_term:
preferred_term: Perrault syndrome
term:
id: MONDO:0017312
label: Perrault syndrome
genes:
- preferred_term: HARS2
term:
id: hgnc:4817
label: HARS2
- preferred_term: HSD17B4
term:
id: hgnc:5213
label: HSD17B4
- preferred_term: CLPP
term:
id: hgnc:2084
label: CLPP
- preferred_term: TWNK
term:
id: hgnc:1160
label: TWNK
- preferred_term: LARS2
term:
id: hgnc:17095
label: LARS2
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:32423379
reference_title: "LARS2-Perrault syndrome: a new case report and literature review."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Perrault syndrome (MIM: 233400) is a rare recessive genetically
heterogeneous disorder characterized by sensorineural hearing loss in males
and females and ovarian dysfunction in females [1].
explanation: >-
Supports the syndromic 46,XX gonadal-dysgenesis branch with hearing
impairment.
prevalence:
- population: Reported 46,XX gonadal dysgenesis cases
prevalence_class: RARE
percentage: Rare
evidence:
- reference: PMID:23087880
reference_title: "A rare cause for primary amenorrhea: Sporadic perrault syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Gonadal (ovarian) dysgenesis with normal chromosomes (46, XX), XX female
gonadal dysgenesis (XX-GD) is a rare genetically heterogeneous disorder.
explanation: >-
This report explicitly describes 46,XX gonadal dysgenesis as a rare
genetically heterogeneous disorder.
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
description: >-
Isolated FSHR-, NUP107-, and PSMC3IP-related forms are recessive, and
Perrault-spectrum 46,XX ovarian dysfunction with hearing impairment is also
recessive.
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Using homozygosity mapping and whole-exome sequencing, we identified a
recessive missense mutation in nucleoporin-107 (NUP107, c.1339G>A,
p.D447N).
explanation: >-
Establishes a recessive isolated subtype.
- reference: PMID:32423379
reference_title: "LARS2-Perrault syndrome: a new case report and literature review."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Perrault syndrome (MIM: 233400) is a rare recessive genetically
heterogeneous disorder characterized by sensorineural hearing loss in males
and females and ovarian dysfunction in females [1].
explanation: >-
Establishes recessive inheritance for the Perrault-spectrum branch.
- name: Autosomal dominant with variable penetrance
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
description: >-
NR5A1-related ovarian developmental failure can segregate dominantly with
incomplete penetrance and variable expressivity, while rare recessive NR5A1
families have also been described.
evidence:
- reference: PMID:19246354
reference_title: Mutations in NR5A1 associated with ovarian insufficiency.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Families 1, 3, and 4 show a dominant mode of inheritance, whereas Family 2
shows a recessive mode.
explanation: >-
Supports autosomal dominant inheritance for most NR5A1 families in this
series and flags the documented recessive exception.
progression:
- phase: Fetal ovarian developmental phase
age_range: fetal development
notes: >-
Causal lesions impair female gonad development, follicle-pool
establishment, or early oogenesis before the disorder is clinically
recognized.
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Impaired estrogenic signaling can lead to ovarian dysgenesis both by
affecting the size of the follicular pool created during fetal
development and by failing to counteract follicular atresia during
puberty.
explanation: >-
This supports an early developmental phase in which abnormal ovarian
signaling reduces the follicular pool before puberty.
- phase: Pubertal failure recognition phase
age_range: adolescence
notes: >-
The disorder commonly becomes clinically apparent when spontaneous puberty
fails and primary amenorrhea with hypergonadotropic ovarian failure is
recognized.
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically heterogeneous
disorder that is characterized by underdeveloped, dysfunctional ovaries,
with subsequent lack of spontaneous pubertal development, primary
amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.
explanation: >-
This abstract ties the clinical recognition phase to absent puberty and
primary amenorrhea in adolescence.
pathophysiology:
- name: Genetic disruption of female gonad development
description: >-
46,XX gonadal dysgenesis is genetically heterogeneous, but the shared
proximal lesion is failure of gene programs required for ovarian
development and maintenance.
biological_processes:
- preferred_term: female gonad development
term:
id: GO:0008585
label: female gonad development
modifier: DECREASED
- preferred_term: gonad development
term:
id: GO:0008406
label: gonad development
modifier: DECREASED
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
These results indicate a pivotal role for NUP107 in ovarian development
and suggest that nucleoporin defects may play a role in milder and more
common conditions such as premature ovarian failure.
explanation: >-
This directly supports ovarian developmental failure as a proximal
mechanism in 46,XX gonadal dysgenesis.
- reference: PMID:19246354
reference_title: Mutations in NR5A1 associated with ovarian insufficiency.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Mutations were associated with a range of ovarian anomalies, including
46,XX gonadal dysgenesis and 46,XX primary ovarian insufficiency.
explanation: >-
This shows that multiple ovarian-development genes can anchor the same
disease entity and its close overlap with ovarian insufficiency.
downstream:
- target: Impaired hormonal signaling and folliculogenesis
description: >-
Diverse causal lesions converge on disrupted ovarian signaling and
follicle maturation.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- defective ovarian transcriptional regulation
- impaired gonadotropin responsiveness
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
PSMC3IP joins previous genes known to be mutated in XX-GD, the FSH
receptor, and BMP15, highlighting the importance of hormonal signaling
in ovarian development and maintenance and suggesting a common pathway
perturbed in isolated XX-GD.
explanation: >-
This directly supports convergence on shared ovarian hormonal-signaling
mechanisms across multiple XX-GD genes.
- name: NUP107 nuclear-pore oogenesis defect
description: >-
NUP107-related ovarian dysgenesis links a nuclear pore component to
sex-specific defective oogenesis and ovarian development in 46,XX
individuals.
genes:
- preferred_term: NUP107
term:
id: hgnc:29914
label: NUP107
cell_types:
- preferred_term: oocyte
term:
id: CL:0000023
label: oocyte
cellular_components:
- preferred_term: nuclear pore
term:
id: GO:0005643
label: nuclear pore
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
NUP107 is a component of the nuclear pore complex, and the
NUP107-associated protein SEH1 is required for oogenesis in Drosophila.
explanation: >-
Supports the nuclear-pore and oogenesis mechanism for NUP107-related
disease.
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
In Drosophila, Nup107 knockdown in somatic gonadal cells resulted in female
sterility, whereas males were fully fertile.
explanation: >-
Supports a sex-specific ovarian requirement for NUP107 activity.
downstream:
- target: Streak gonads and ovarian failure
- name: PSMC3IP estrogen-receptor coactivation failure
description: >-
PSMC3IP/HOP2-related disease disrupts estrogen-driven nuclear-receptor
coactivation, reducing fetal follicular-pool establishment and increasing
vulnerability to follicular atresia during puberty.
genes:
- preferred_term: PSMC3IP
term:
id: hgnc:17928
label: PSMC3IP
cell_types:
- preferred_term: oocyte
term:
id: CL:0000023
label: oocyte
biological_processes:
- preferred_term: ovarian follicle development
term:
id: GO:0001541
label: ovarian follicle development
modifier: DECREASED
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes
coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
In cell lines, the p.Glu201del mutation abolished PSMC3IP activation of
estrogen-driven transcription.
explanation: >-
Directly supports loss of estrogen-driven transcriptional coactivation.
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes
coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Impaired estrogenic signaling can lead to ovarian dysgenesis both by
affecting the size of the follicular pool created during fetal development
and by failing to counteract follicular atresia during puberty.
explanation: >-
Links PSMC3IP-driven estrogenic-signaling failure to the developmental and
pubertal ovarian phenotype.
downstream:
- target: Impaired hormonal signaling and folliculogenesis
- name: FSHR receptor signaling resistance
description: >-
FSHR-related ovarian dysgenesis impairs follicle-stimulating hormone receptor
signaling, producing follicular arrest with hypergonadotropic ovarian failure.
genes:
- preferred_term: FSHR
term:
id: hgnc:3969
label: FSHR
cell_types:
- preferred_term: granulosa cell
term:
id: CL:0000501
label: granulosa cell
biological_processes:
- preferred_term: response to follicle-stimulating hormone
term:
id: GO:0032354
label: response to follicle-stimulating hormone
modifier: DECREASED
evidence:
- reference: PMID:7553856
reference_title: >-
Mutation in the follicle-stimulating hormone receptor gene causes
hereditary hypergonadotropic ovarian failure.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
Expression of the gene in transfected cells demonstrated a dramatic
reduction of binding capacity and signal transduction, but apparently
normal ligand-binding affinity of the mutated receptor.
explanation: >-
Directly supports receptor signaling resistance as the FSHR mechanism.
downstream:
- target: Impaired hormonal signaling and folliculogenesis
- name: NR5A1 steroidogenic transcription dosage defect
description: >-
NR5A1-related ovarian insufficiency reflects impaired transcriptional
activation of steroidogenic and follicle-growth genes in ovarian cells, with
variable penetrance and expressivity across families.
genes:
- preferred_term: NR5A1
term:
id: hgnc:7983
label: NR5A1
cell_types:
- preferred_term: granulosa cell
term:
id: CL:0000501
label: granulosa cell
biological_processes:
- preferred_term: estrogen biosynthetic process
term:
id: GO:0006703
label: estrogen biosynthetic process
modifier: DECREASED
evidence:
- reference: PMID:19246354
reference_title: Mutations in NR5A1 associated with ovarian insufficiency.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
Our data show that mutated forms of NR5A1, detected in subjects with
anomalies of ovarian development and function, show quantitative
impairment in the transactivation of two of these factors (CYP11A1 and
CYP19A1).
explanation: >-
Directly supports impaired NR5A1 transcriptional output as the molecular
mechanism.
downstream:
- target: Streak gonads and ovarian failure
- name: Impaired hormonal signaling and folliculogenesis
description: >-
A major shared mechanistic branch is reduced ovarian hormone-response and
follicular maintenance, including impaired FSH signaling and defective
estrogen-responsive transcription.
cell_types:
- preferred_term: granulosa cell
term:
id: CL:0000501
label: granulosa cell
- preferred_term: oocyte
term:
id: CL:0000023
label: oocyte
biological_processes:
- preferred_term: response to follicle-stimulating hormone
term:
id: GO:0032354
label: response to follicle-stimulating hormone
modifier: DECREASED
- preferred_term: ovarian follicle development
term:
id: GO:0001541
label: ovarian follicle development
modifier: DECREASED
evidence:
- reference: PMID:7553856
reference_title: >-
Mutation in the follicle-stimulating hormone receptor gene causes
hereditary hypergonadotropic ovarian failure.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
Expression of the gene in transfected cells demonstrated a dramatic
reduction of binding capacity and signal transduction, but apparently
normal ligand-binding affinity of the mutated receptor.
explanation: >-
This directly supports impaired FSH signaling as one disease-causing
mechanistic branch in 46,XX gonadal dysgenesis.
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Impaired estrogenic signaling can lead to ovarian dysgenesis both by
affecting the size of the follicular pool created during fetal
development and by failing to counteract follicular atresia during
puberty.
explanation: >-
This supports defective estrogen-responsive transcription and accelerated
follicular loss as another shared mechanistic branch.
downstream:
- target: Streak gonads and ovarian failure
description: >-
Chronic failure of follicle development and ovarian signaling produces
streak or severely underdeveloped ovaries with loss of reproductive
function.
causal_link_type: DIRECT
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically
heterogeneous disorder characterized by lack of spontaneous pubertal
development, primary amenorrhea, uterine hypoplasia, and
hypergonadotropic hypogonadism as a result of streak gonads.
explanation: >-
This directly links impaired ovarian signaling to streak gonads and
ovarian failure.
- name: Streak gonads and ovarian failure
description: >-
The convergent anatomical outcome is a streak or severely underdeveloped
ovary with little or no effective steroidogenic or follicular function.
biological_processes:
- preferred_term: estrogen biosynthetic process
term:
id: GO:0006703
label: estrogen biosynthetic process
modifier: DECREASED
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically heterogeneous
disorder that is characterized by underdeveloped, dysfunctional ovaries,
with subsequent lack of spontaneous pubertal development, primary
amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.
explanation: >-
This supports underdeveloped dysfunctional ovaries as the shared anatomic
lesion of the disease.
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically
heterogeneous disorder characterized by lack of spontaneous pubertal
development, primary amenorrhea, uterine hypoplasia, and
hypergonadotropic hypogonadism as a result of streak gonads.
explanation: >-
This explicitly identifies streak gonads as the proximate cause of the
endocrine and reproductive phenotype.
downstream:
- target: Gonadal Dysgenesis
description: >-
Streak or severely underdeveloped dysfunctional ovaries are the defining
gonadal dysgenesis phenotype.
causal_link_type: DIRECT
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
underdeveloped, dysfunctional ovaries
explanation: >-
The NUP107 family report identifies underdeveloped dysfunctional ovaries
as the defining structural lesion of XX gonadal dysgenesis.
- target: Female Infertility
description: >-
Streak gonads and ovarian failure prevent normal ovulation and
reproductive capacity.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Loss of functional ovarian tissue prevents follicular maturation and ovulation.
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
hypergonadotropic hypogonadism as a result of streak gonads
explanation: >-
The PSMC3IP report identifies streak gonads as the cause of the
endocrine reproductive failure; infertility follows from loss of
functional ovarian tissue.
- target: Hypoestrogenic hypergonadotropic state
description: >-
Ovarian failure lowers estrogen output and disinhibits pituitary
gonadotropin secretion.
causal_link_type: DIRECT
evidence:
- reference: PMID:8855829
reference_title: >-
Clinical features of primary ovarian failure caused by a point mutation
in the follicle-stimulating hormone receptor gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Clinically, both groups of patients were characterized by primary or
early secondary amenorrhea, variable development of secondary sex
characteristics, and high serum levels of FSH and LH.
explanation: >-
This directly supports the endocrine consequence of ovarian failure as
amenorrhea with elevated gonadotropins.
- name: Hypoestrogenic hypergonadotropic state
description: >-
Loss of ovarian steroidogenic function results in estrogen deficiency and
compensatory elevation of pituitary gonadotropins, driving pubertal failure
and amenorrhea.
biological_processes:
- preferred_term: gonadotropin secretion
term:
id: GO:0032274
label: gonadotropin secretion
modifier: INCREASED
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically heterogeneous
disorder that is characterized by underdeveloped, dysfunctional ovaries,
with subsequent lack of spontaneous pubertal development, primary
amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.
explanation: >-
This supports the downstream hypoestrogenic hypergonadotropic endocrine
state as a core mechanistic consequence.
- reference: PMID:8855829
reference_title: >-
Clinical features of primary ovarian failure caused by a point mutation
in the follicle-stimulating hormone receptor gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Clinically, both groups of patients were characterized by primary or
early secondary amenorrhea, variable development of secondary sex
characteristics, and high serum levels of FSH and LH.
explanation: >-
This links the endocrine state to amenorrhea and impaired pubertal
development in a mechanistically defined XX-GD subset.
downstream:
- target: Hypergonadotropic Hypogonadism
description: >-
Ovarian steroidogenic failure drives compensatory pituitary gonadotropin
elevation.
causal_link_type: DIRECT
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
uterine hypoplasia, and hypergonadotropic hypogonadism
explanation: >-
The NUP107 family report includes hypergonadotropic hypogonadism as a
core consequence of underdeveloped dysfunctional ovaries.
- target: Delayed Puberty
description: >-
Estrogen deficiency from ovarian failure prevents spontaneous pubertal
development.
causal_link_type: DIRECT
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
lack of spontaneous pubertal development
explanation: >-
The NUP107 family report directly states lack of spontaneous pubertal
development in XX gonadal dysgenesis.
- target: Primary Amenorrhea
description: >-
Pubertal ovarian failure prevents menarche, producing primary amenorrhea.
causal_link_type: DIRECT
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
spontaneous pubertal development, primary amenorrhea, uterine hypoplasia
explanation: >-
The PSMC3IP family report directly lists primary amenorrhea in the
reproductive phenotype resulting from streak gonads.
- target: Uterine Hypoplasia
description: >-
Sustained hypoestrogenism limits uterine growth, producing uterine
hypoplasia.
causal_link_type: DIRECT
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism
explanation: >-
The NUP107 family report includes uterine hypoplasia in the downstream
phenotype of underdeveloped dysfunctional ovaries.
genetic:
- name: NUP107
gene_term:
preferred_term: NUP107
term:
id: hgnc:29914
label: NUP107
association: CAUSATIVE
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Using homozygosity mapping and whole-exome sequencing, we identified a
recessive missense mutation in nucleoporin-107 (NUP107, c.1339G>A,
p.D447N).
explanation: >-
NUP107-related 46,XX gonadal dysgenesis segregates recessively in the
reported family.
features: >-
Recessive NUP107 missense variants cause isolated 46,XX gonadal
dysgenesis with defective oogenesis and ovarian developmental failure.
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Using homozygosity mapping and whole-exome sequencing, we identified a
recessive missense mutation in nucleoporin-107 (NUP107, c.1339G>A,
p.D447N).
explanation: >-
This identifies NUP107 as a causal recessive gene for familial 46,XX
gonadal dysgenesis.
- name: PSMC3IP
gene_term:
preferred_term: PSMC3IP
term:
id: hgnc:17928
label: PSMC3IP
association: CAUSATIVE
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Most cases are unexplained but thought to be autosomal recessive.
explanation: >-
The PSMC3IP family is part of the recessive isolated XX-GD pattern and
had homozygous affected females.
features: >-
Homozygous PSMC3IP/HOP2 variants abolish coactivation of
estrogen-driven transcription and cause isolated XX gonadal dysgenesis.
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Affected females were homozygous for a 3 bp deletion (NM_016556.2,
c.600_602del) in the PSMC3IP gene, leading to deletion of a glutamic acid
residue (p.Glu201del) in the highly conserved C-terminal acidic domain.
explanation: >-
This establishes PSMC3IP as a causal gene in a familial 46,XX gonadal
dysgenesis pedigree.
- name: FSHR
gene_term:
preferred_term: FSHR
term:
id: hgnc:3969
label: FSHR
association: CAUSATIVE
inheritance:
- name: Autosomal recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: PMID:7553856
reference_title: >-
Mutation in the follicle-stimulating hormone receptor gene causes
hereditary hypergonadotropic ovarian failure.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hypergonadotropic ovarian dysgenesis (ODG) with normal karyotype is a
heterogeneous condition that in some cases displays Mendelian recessive
inheritance.
explanation: >-
Supports recessive inheritance for FSHR-related ovarian dysgenesis.
features: >-
Recessive FSHR variants define an FSH-resistant ovarian dysgenesis subset
with impaired receptor signaling and elevated gonadotropins.
evidence:
- reference: PMID:7553856
reference_title: >-
Mutation in the follicle-stimulating hormone receptor gene causes
hereditary hypergonadotropic ovarian failure.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We conclude that the mutation causes ODG in these families.
explanation: >-
This provides direct causal evidence for FSHR-mediated ovarian
dysgenesis.
- name: NR5A1
gene_term:
preferred_term: NR5A1
term:
id: hgnc:7983
label: NR5A1
association: CAUSATIVE
inheritance:
- name: Autosomal dominant with variable penetrance
inheritance_term:
preferred_term: Autosomal dominant inheritance
term:
id: HP:0000006
label: Autosomal dominant inheritance
evidence:
- reference: PMID:19246354
reference_title: Mutations in NR5A1 associated with ovarian insufficiency.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Families 1, 3, and 4 show a dominant mode of inheritance, whereas Family
2 shows a recessive mode.
explanation: >-
Supports dominant NR5A1 inheritance with documented variability.
features: >-
NR5A1 variants span 46,XX gonadal dysgenesis to primary ovarian
insufficiency, illustrating disease overlap within ovarian developmental
failure.
evidence:
- reference: PMID:19246354
reference_title: Mutations in NR5A1 associated with ovarian insufficiency.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Mutations were associated with a range of ovarian anomalies, including
46,XX gonadal dysgenesis and 46,XX primary ovarian insufficiency.
explanation: >-
This shows that NR5A1 can anchor both 46,XX gonadal dysgenesis and the
adjacent ovarian-insufficiency phenotype space.
phenotypes:
- category: Reproductive
name: Gonadal Dysgenesis
description: >-
Streak or severely underdeveloped ovaries are the defining structural
lesion of the disease.
diagnostic: true
phenotype_term:
preferred_term: gonadal dysgenesis
term:
id: HP:0000133
label: Gonadal dysgenesis
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically heterogeneous
disorder that is characterized by underdeveloped, dysfunctional ovaries,
with subsequent lack of spontaneous pubertal development, primary
amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.
explanation: >-
This directly supports gonadal dysgenesis as the structural hallmark
phenotype.
- category: Reproductive
name: Delayed Puberty
description: >-
Spontaneous pubertal development fails because ovarian estrogen production
is inadequate.
diagnostic: true
phenotype_term:
preferred_term: delayed puberty
term:
id: HP:0000823
label: Delayed puberty
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically heterogeneous
disorder that is characterized by underdeveloped, dysfunctional ovaries,
with subsequent lack of spontaneous pubertal development, primary
amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.
explanation: >-
The abstract explicitly supports absent spontaneous pubertal development,
which maps to delayed puberty at the phenotype level.
- category: Reproductive
name: Primary Amenorrhea
description: >-
Menarche does not occur because ovarian failure prevents normal pubertal
maturation.
diagnostic: true
phenotype_term:
preferred_term: primary amenorrhea
term:
id: HP:0000786
label: Primary amenorrhea
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically
heterogeneous disorder characterized by lack of spontaneous pubertal
development, primary amenorrhea, uterine hypoplasia, and
hypergonadotropic hypogonadism as a result of streak gonads.
explanation: >-
This directly supports primary amenorrhea as a core diagnostic phenotype.
- category: Endocrine
name: Hypergonadotropic Hypogonadism
description: >-
Ovarian failure lowers sex-steroid output and drives compensatory
gonadotropin elevation.
diagnostic: true
phenotype_term:
preferred_term: hypergonadotropic hypogonadism
term:
id: HP:0000815
label: Hypergonadotropic hypogonadism
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically heterogeneous
disorder that is characterized by underdeveloped, dysfunctional ovaries,
with subsequent lack of spontaneous pubertal development, primary
amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.
explanation: >-
This explicitly identifies hypergonadotropic hypogonadism as part of the
core XX-GD phenotype.
- reference: PMID:8855829
reference_title: >-
Clinical features of primary ovarian failure caused by a point mutation
in the follicle-stimulating hormone receptor gene.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Clinically, both groups of patients were characterized by primary or
early secondary amenorrhea, variable development of secondary sex
characteristics, and high serum levels of FSH and LH.
explanation: >-
This supports elevated gonadotropins as the endocrine signature of a
mechanistically defined XX-GD subset.
- category: Reproductive
name: Uterine Hypoplasia
description: >-
Uterine growth remains poor when ovarian estrogen production is absent or
severely reduced.
diagnostic: true
phenotype_term:
preferred_term: uterine hypoplasia
term:
id: HP:0008684
label: Aplasia/hypoplasia of the uterus
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically heterogeneous
disorder that is characterized by underdeveloped, dysfunctional ovaries,
with subsequent lack of spontaneous pubertal development, primary
amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.
explanation: >-
This directly supports uterine hypoplasia as part of the core disease
phenotype.
- category: Reproductive
name: Female Infertility
description: >-
Streak gonads and ovarian failure prevent normal ovulation and reproductive
capacity.
phenotype_term:
preferred_term: female infertility
term:
id: HP:0008222
label: Female infertility
evidence:
- reference: PMID:21963259
reference_title: >-
XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that
abolishes coactivation of estrogen-driven transcription.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
XX female gonadal dysgenesis (XX-GD) is a rare, genetically
heterogeneous disorder characterized by lack of spontaneous pubertal
development, primary amenorrhea, uterine hypoplasia, and
hypergonadotropic hypogonadism as a result of streak gonads.
explanation: >-
Streak gonads imply loss of functional ovarian tissue and support female
infertility as a downstream reproductive consequence.
- category: Hearing
name: Sensorineural hearing impairment
subtype: Perrault-spectrum
description: >-
Sensorineural hearing impairment distinguishes Perrault-spectrum disease from
isolated 46,XX gonadal dysgenesis and should prompt audiology and syndromic
genetic evaluation.
phenotype_term:
preferred_term: Sensorineural hearing impairment
term:
id: HP:0000407
label: Sensorineural hearing impairment
evidence:
- reference: PMID:32423379
reference_title: "LARS2-Perrault syndrome: a new case report and literature review."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Perrault syndrome (MIM: 233400) is a rare recessive genetically
heterogeneous disorder characterized by sensorineural hearing loss in males
and females and ovarian dysfunction in females [1].
explanation: >-
Supports hearing impairment as the key syndromic feature of the
Perrault-spectrum subtype.
diagnosis:
- name: 46,XX karyotype confirmation
presence: Normal 46,XX complement without Turner mosaicism or Y-chromosome material.
description: >-
Karyotyping confirms the 46,XX disease boundary, excludes Turner-spectrum
causes of gonadal dysgenesis, and helps prevent applying 46,XY tumor-risk
assumptions to isolated 46,XX disease.
diagnosis_term:
preferred_term: karyotyping
term:
id: MAXO:0001611
label: karyotyping
evidence:
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: The chromosome study confirmed normal 46, XX karyotype.
explanation: >-
Supports early DSD diagnostic workup, including karyotype confirmation.
- reference: PMID:28216916
reference_title: "A rare case of 46,XX gonadal dysgenesis and Mayer-Rokitansky-Kuster-Hauser syndrome."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: 46,XX karyotype complement
explanation: >-
Confirms that the case definition relies on a 46,XX karyotype.
- name: Gonadotropin and ovarian hormone profile
presence: Elevated FSH/LH with low estradiol and low ovarian reserve markers where measured.
description: >-
Hormone evaluation distinguishes hypoestrogenic hypergonadotropic gonadal
dysgenesis from MRKH and other causes of primary amenorrhea with normal
ovarian function.
diagnosis_term:
preferred_term: circulating hormone measurement
term:
id: MAXO:0035005
label: circulating hormone measurement
evidence:
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hormone profiles and karyotype should be investigated in all cases of the
presumptive diagnosis of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome or
Mullerian agenesis.
explanation: >-
Directly supports hormone-profile testing in the primary-amenorrhea
differential.
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The initial laboratory investigation in our hospital revealed
hypergonadotrophic hypogonadism (FSH 130 IU/L, LH 2 IU/L, serum estradiol
<5 pg/mL) with confirmed 46, XX karyotype.
explanation: >-
Provides the expected endocrine pattern in a 46,XX gonadal dysgenesis
diagnostic workup.
- name: Pubertal staging and estrogenization assessment
presence: Absent or incomplete breast development and other delayed secondary sex characteristics.
description: >-
Tanner staging and focused physical examination document pubertal failure
and provide a quick clinical clue to severe estrogen deficiency.
diagnosis_term:
preferred_term: physical examination
term:
id: MAXO:0000527
label: physical examination
evidence:
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Determining the presence or absence of secondary sexual characteristics
especially breast development is a simple first step to ensure the presence
of circulating estrogen level (3).
explanation: >-
Supports pubertal and secondary-sex-characteristic assessment as part of
diagnosis.
- name: Pelvic ultrasound and MRI
presence: Streak or nonvisualized ovaries, uterine hypoplasia, or apparent uterine nonvisualization under estrogen deficiency.
description: >-
Pelvic imaging evaluates uterus, ovaries, streak gonads, and possible
Mullerian-anomaly overlap. MRI can clarify ultrasound findings, and repeat
imaging after estrogen replacement may prevent misclassification as MRKH.
diagnosis_term:
preferred_term: pelvis MRI
term:
id: MAXO:0035034
label: pelvis MRI
evidence:
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Internal genitalia (ovaries, uterus, and upper two third of vagina) and
streak gonad could not be identified on the pelvic magnetic resonance
imaging (MRI).
explanation: >-
Supports pelvic MRI for internal reproductive-structure evaluation.
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Subsequent identification of the uterus needs to be re-evaluated after at
least 6–12 months of estrogen replacement.
explanation: >-
Supports repeat imaging after estrogen replacement when severe
hypoestrogenism obscures uterine structures.
- name: Baseline bone-density assessment
presence: Osteopenia, osteoporosis, or prolonged untreated estrogen deficiency.
description: >-
Baseline and follow-up DXA are relevant because delayed diagnosis and
untreated hypoestrogenism can cause early bone loss.
diagnosis_term:
preferred_term: Dual-energy X-ray absorptiometry procedure
term:
id: MAXO:0020004
label: Dual-energy X-ray absorptiometry procedure
evidence:
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Bone density scan (DEXA scan) revealed osteoporosis at the lumbar spine (T
score −2.7) and osteopenia at the hip (T score −1.5).
explanation: >-
Documents clinically relevant bone-density loss after delayed diagnosis.
- name: Molecular testing for hereditary forms
presence: Pathogenic variants in FSHR, NUP107, PSMC3IP, NR5A1, or Perrault-spectrum genes.
description: >-
Gene panel or exome testing should cover isolated 46,XX ovarian dysgenesis
genes and syndromic Perrault genes when hearing loss or family history
suggests that branch.
diagnosis_term:
preferred_term: gene panel testing
term:
id: MAXO:0001615
label: gene panel testing
evidence:
- reference: PMID:26485283
reference_title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Using homozygosity mapping and whole-exome sequencing, we identified a
recessive missense mutation in nucleoporin-107 (NUP107, c.1339G>A,
p.D447N).
explanation: >-
Demonstrates molecular diagnosis for a hereditary isolated subtype.
- reference: PMID:32423379
reference_title: "LARS2-Perrault syndrome: a new case report and literature review."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Systematic genetic screening of children with hearing loss allows the
early identification of a Perrault syndrome in order to ensure specific
endocrinological surveillance and management to prevent secondary
complications.
explanation: >-
Supports molecular testing when the syndromic hearing-loss branch is
suspected.
- name: Audiology assessment for Perrault-spectrum disease
presence: Sensorineural hearing loss or pathogenic variants in Perrault-spectrum genes.
description: >-
Audiology evaluation distinguishes Perrault-spectrum disease from isolated
46,XX gonadal dysgenesis and guides hearing support.
diagnosis_term:
preferred_term: audiologist evaluation
term:
id: MAXO:0000734
label: audiologist evaluation
evidence:
- reference: PMID:32423379
reference_title: "LARS2-Perrault syndrome: a new case report and literature review."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Hearing loss should be assessed and treated by a multidisciplinary team
including an audiologist and otolaryngologist.
explanation: >-
Supports audiology assessment and hearing-care referral for the
Perrault-spectrum subtype.
treatments:
- name: Pubertal induction and long-term hormone replacement
description: >-
Estrogen-based pubertal induction followed by estrogen-progestogen
maintenance is used to initiate and sustain puberty, support secondary
sexual characteristics, and reduce long-term consequences of
hypoestrogenism.
treatment_term:
preferred_term: sex hormone modifying agent therapy
term:
id: MAXO:0000282
label: sex hormone modifying agent therapy
target_phenotypes:
- preferred_term: Delayed puberty
term:
id: HP:0000823
label: Delayed puberty
- preferred_term: Hypergonadotropic hypogonadism
term:
id: HP:0000815
label: Hypergonadotropic hypogonadism
evidence:
- reference: PMID:36300209
reference_title: "Hypogonadism in adolescent girls: treatment and long-term effects."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Girls with hypogonadism require hormone replacement therapy to initiate and
sustain puberty.
explanation: >-
Supports pubertal induction and maintenance hormone replacement for
hypogonadal adolescents.
- reference: PMID:35142292
reference_title: >-
A rare case of 46,XX gonadal dysgenesis, Mayer-Rokitansky-Kuster-Hauser
syndrome, pituitary and thyroid hypoplasia.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hormone replacement therapy remains the only therapeutic option for the
development of secondary sexual characteristics and the prevention of
osteoporosis.
explanation: >-
This directly supports hormone replacement therapy as core conservative
management for 46,XX gonadal dysgenesis.
- name: Cyclic estrogen-progestogen maintenance when uterus is present
description: >-
Once estrogenized uterine tissue is present, cyclic progestogen is used with
estrogen to oppose endometrial effects during long-term replacement.
treatment_term:
preferred_term: sex hormone modifying agent therapy
term:
id: MAXO:0000282
label: sex hormone modifying agent therapy
target_phenotypes:
- preferred_term: Hypergonadotropic hypogonadism
term:
id: HP:0000815
label: Hypergonadotropic hypogonadism
evidence:
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Treatment with estrogen and progesterone are required for reducing the risk
of endometrial hyperplasia and carcinoma, which will increase due to
long-term application of estrogen without opposition.
explanation: >-
Supports cyclic progestogen addition to estrogen replacement when uterine
tissue is present.
- name: Bone-health monitoring and calcium-vitamin D support
description: >-
Delayed diagnosis and prolonged hypoestrogenism can cause osteopenia or
osteoporosis, so bone-density monitoring plus calcium and vitamin D support
should accompany hormone replacement.
treatment_term:
preferred_term: Dual-energy X-ray absorptiometry procedure
term:
id: MAXO:0020004
label: Dual-energy X-ray absorptiometry procedure
evidence:
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Early initiation of estrogen therapy is essential to minimize bone loss.
Moreover, lifelong oral calcium and vitamin D supplement should be given to
maintain bone health.
explanation: >-
Supports bone-focused management in hypoestrogenic 46,XX gonadal
dysgenesis.
- reference: PMID:24945456
reference_title: "Committee opinion no. 605: primary ovarian insufficiency in adolescents and young women."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
The goals of hormonal therapy extend beyond simply symptom relief to levels
that support bone, cardiovascular, and sexual health.
explanation: >-
General adolescent POI guidance supports bone-health goals for hormone
therapy.
- name: Fertility and reproductive counseling
description: >-
Counseling should cover infertility expectations, reproductive endocrinology
referral, donor-oocyte or gestational-surrogacy pathways when relevant, and
recurrence-risk implications for inherited subtypes.
treatment_term:
preferred_term: behavioral counseling
term:
id: MAXO:0000077
label: behavioral counseling
target_phenotypes:
- preferred_term: Female infertility
term:
id: HP:0008222
label: Female infertility
evidence:
- reference: PMID:24945456
reference_title: "Committee opinion no. 605: primary ovarian insufficiency in adolescents and young women."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Patients and their families should be counseled on the effect of the
patient's condition on future fertility, on the risk of comorbidities
associated with primary ovarian insufficiency, and on the condition's
potential for genetic inheritance.
explanation: >-
Supports fertility and inheritance counseling in adolescent ovarian
insufficiency.
- reference: PMID:31809259
reference_title: >-
Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal
dysgenesis: a missed opportunity for prevention of osteoporosis.
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
She has been informed about future options for having children by adoption
or gestational surrogacy.
explanation: >-
Case-level evidence supports reproductive-option counseling, though it is
not a comparative intervention study.
- name: Psychologic support and longitudinal follow-up
description: >-
Diagnosis can affect self-esteem, emotional health, fertility expectations,
and identity; longitudinal care should include psychologic counseling and at
least annual follow-up for ovarian-insufficiency consequences.
treatment_term:
preferred_term: behavioral counseling
term:
id: MAXO:0000077
label: behavioral counseling
evidence:
- reference: PMID:24945456
reference_title: "Committee opinion no. 605: primary ovarian insufficiency in adolescents and young women."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Psychologic counseling also should be offered because impaired self-esteem
and emotional distress have been reported after diagnosis of primary
ovarian insufficiency.
explanation: >-
Supports psychologic counseling after diagnosis.
- reference: PMID:24945456
reference_title: "Committee opinion no. 605: primary ovarian insufficiency in adolescents and young women."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Once primary ovarian insufficiency is diagnosed, patients should be
evaluated at least annually.
explanation: >-
Supports longitudinal follow-up.
- name: Audiology and hearing-support management for Perrault-spectrum cases
description: >-
Perrault-spectrum patients need multidisciplinary hearing assessment and
intervention planning in parallel with endocrine surveillance.
treatment_term:
preferred_term: audiologist evaluation
term:
id: MAXO:0000734
label: audiologist evaluation
target_phenotypes:
- preferred_term: Sensorineural hearing impairment
term:
id: HP:0000407
label: Sensorineural hearing impairment
evidence:
- reference: PMID:32423379
reference_title: "LARS2-Perrault syndrome: a new case report and literature review."
supports: SUPPORT
evidence_source: OTHER
snippet: >-
Hearing loss should be assessed and treated by a multidisciplinary team
including an audiologist and otolaryngologist.
explanation: >-
Supports audiology-led hearing management for the Perrault-spectrum
subtype.
clinical_trials:
- name: NCT06518746
phase: NOT_APPLICABLE
status: RECRUITING
description: >-
Pilot interventional study evaluating gonadal tissue cryopreservation for
fertility preservation or restoration of hormonal function in patients with
gonadal dysgenesis/DSD who are at risk of decreased fertility potential or
malignancy.
target_phenotypes:
- preferred_term: female infertility
term:
id: HP:0008222
label: Female infertility
evidence:
- reference: clinicaltrials:NCT06518746
reference_title: Gonadal Dysgenesis Tissue Cryopreservation for Fertility Preservation
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The overall objective of this study is to determine the safety and
efficacy of gonadal tissue cryopreservation as a method of preserving
fertility and/or restoring hormonal function in patients with gonadal
dysgenesis who are at risk of decreased fertility potential or malignancy.
explanation: >-
ClinicalTrials.gov describes an active gonadal tissue cryopreservation
study directly relevant to infertility risk in gonadal dysgenesis.
discussions:
- discussion_id: gap_46xx_gd_oligogenic_modifier_burden
prompt: >-
In genetically unsolved 46,XX gonadal dysgenesis, how often do combined
heterozygous variants in ovarian maturation genes act as modifiers or
oligogenic contributors rather than independent monogenic causes?
kind: KNOWLEDGE_GAP
status: OPEN
attaches_to:
- pathophysiology#Genetic disruption of female gonad development
rationale: >-
Case-level data support a possible synergic FIGLA/NOBOX/NR5A1 model, but
the evidence is not yet strong enough to reclassify each variant as a
general causal mechanism for this disease entity.
evidence:
- reference: PMID:33101191
reference_title: >-
The Potential Synergic Effect of a Complex Pattern of Multiple Inherited
Genetic Variants as a Pathogenic Factor for Ovarian Dysgenesis: A Case
Report.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Our results support the hypothesis that the severity of the clinical
picture of the proband, resulting in complete ovarian dysgenesis, may be
due to a synergic detrimental effect of inherited genetic variants.
explanation: >-
This supports preserving the oligogenic modifier model as an open
disease-mechanism question rather than a settled single-gene assertion.
references:
- reference: DOI:10.1016/j.ajhg.2011.09.006
title: XX Ovarian Dysgenesis Is Caused by a PSMC3IP/HOP2 Mutation that Abolishes Coactivation of Estrogen-Driven Transcription
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings: []
- reference: DOI:10.1016/j.ecl.2024.01.009
title: Primary Amenorrhea and Premature Ovarian Insufficiency
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings:
- statement: Primary Amenorrhea and Premature Ovarian Insufficiency
supporting_text: Primary Amenorrhea and Premature Ovarian Insufficiency
- reference: DOI:10.1086/422103
title: Hypergonadotropic Ovarian Failure Associated with an Inherited Mutation of Human Bone Morphogenetic Protein-15 (BMP15) Gene
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings:
- statement: Hypergonadotropic Ovarian Failure Associated with an Inherited Mutation of Human Bone Morphogenetic Protein-15 (BMP15) Gene
supporting_text: Hypergonadotropic Ovarian Failure Associated with an Inherited Mutation of Human Bone Morphogenetic Protein-15 (BMP15) Gene
- reference: DOI:10.1093/humrep/dew025
title: 'A novel follicle-stimulating hormone receptor mutation causing primary ovarian failure: a fertility application of whole exome sequencing'
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings:
- statement: 'A novel follicle-stimulating hormone receptor mutation causing primary ovarian failure: a fertility application of whole exome sequencing'
supporting_text: 'A novel follicle-stimulating hormone receptor mutation causing primary ovarian failure: a fertility application of whole exome sequencing'
- reference: DOI:10.1172/JCI83553
title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings: []
- reference: DOI:10.2350/14-04-1471-pb.1
title: Perspectives in Pediatric Pathology, Chapter 5. Gonadal Dysgenesis
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings:
- statement: One of the most challenging areas in pediatric testicular pathology is the appropriate understanding and pathological diagnosis of disorders of sexual development (DSD), and in particular, the issue of gonadal dysgenesis.
supporting_text: One of the most challenging areas in pediatric testicular pathology is the appropriate understanding and pathological diagnosis of disorders of sexual development (DSD), and in particular, the issue of gonadal dysgenesis.
evidence:
- reference: DOI:10.2350/14-04-1471-pb.1
reference_title: Perspectives in Pediatric Pathology, Chapter 5. Gonadal Dysgenesis
supports: SUPPORT
evidence_source: OTHER
snippet: One of the most challenging areas in pediatric testicular pathology is the appropriate understanding and pathological diagnosis of disorders of sexual development (DSD), and in particular, the issue of gonadal dysgenesis.
explanation: Deep research cited this publication as relevant literature for 46 XX Gonadal Dysgenesis.
- reference: DOI:10.3389/fendo.2020.540683
title: 'The Potential Synergic Effect of a Complex Pattern of Multiple Inherited Genetic Variants as a Pathogenic Factor for Ovarian Dysgenesis: A Case Report'
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings:
- statement: 'The Potential Synergic Effect of a Complex Pattern of Multiple Inherited Genetic Variants as a Pathogenic Factor for Ovarian Dysgenesis: A Case Report'
supporting_text: 'The Potential Synergic Effect of a Complex Pattern of Multiple Inherited Genetic Variants as a Pathogenic Factor for Ovarian Dysgenesis: A Case Report'
- reference: DOI:10.3389/fendo.2024.1354759
title: Diagnosis and management of non-CAH 46,XX disorders/differences in sex development
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings:
- statement: Prenatal-onset androgen excess leads to abnormal sexual development in 46,XX individuals.
supporting_text: Prenatal-onset androgen excess leads to abnormal sexual development in 46,XX individuals.
evidence:
- reference: DOI:10.3389/fendo.2024.1354759
reference_title: Diagnosis and management of non-CAH 46,XX disorders/differences in sex development
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: Prenatal-onset androgen excess leads to abnormal sexual development in 46,XX individuals.
explanation: Deep research cited this publication as relevant literature for 46 XX Gonadal Dysgenesis.
- reference: DOI:10.3389/fendo.2024.1372887
title: Long-term outcomes in non-CAH 46,XX DSD
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings:
- statement: Differences/disorders of sex development (DSD) comprise a large group of rare congenital conditions.
supporting_text: Differences/disorders of sex development (DSD) comprise a large group of rare congenital conditions.
evidence:
- reference: DOI:10.3389/fendo.2024.1372887
reference_title: Long-term outcomes in non-CAH 46,XX DSD
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: Differences/disorders of sex development (DSD) comprise a large group of rare congenital conditions.
explanation: Deep research cited this publication as relevant literature for 46 XX Gonadal Dysgenesis.
- reference: DOI:10.3389/fendo.2024.1402579
title: '46,XX Differences of Sex Development outside congenital adrenal hyperplasia: pathogenesis, clinical aspects, puberty, sex hormone replacement therapy and fertility outcomes'
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings:
- statement: The term ‘differences of sex development’ (DSD) refers to a group of congenital conditions that are associated with atypical development of chromosomal, gonadal, and/or anatomical sex.
supporting_text: The term ‘differences of sex development’ (DSD) refers to a group of congenital conditions that are associated with atypical development of chromosomal, gonadal, and/or anatomical sex.
evidence:
- reference: DOI:10.3389/fendo.2024.1402579
reference_title: '46,XX Differences of Sex Development outside congenital adrenal hyperplasia: pathogenesis, clinical aspects, puberty, sex hormone replacement therapy and fertility outcomes'
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: The term ‘differences of sex development’ (DSD) refers to a group of congenital conditions that are associated with atypical development of chromosomal, gonadal, and/or anatomical sex.
explanation: Deep research cited this publication as relevant literature for 46 XX Gonadal Dysgenesis.
- reference: DOI:10.3390/cimb46050274
title: 'Role of NR5A1 Gene Mutations in Disorders of Sex Development: Molecular and Clinical Features'
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-falcon.md
findings:
- statement: 'Role of NR5A1 Gene Mutations in Disorders of Sex Development: Molecular and Clinical Features'
supporting_text: Disorders/differences of sex development (DSDs) are defined as broad, heterogenous groups of congenital conditions characterized by atypical development of genetic, gonadal, or phenotypic sex accompanied by abnormal development of internal and/or external genitalia.
evidence:
- reference: DOI:10.3390/cimb46050274
reference_title: 'Role of NR5A1 Gene Mutations in Disorders of Sex Development: Molecular and Clinical Features'
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: Disorders/differences of sex development (DSDs) are defined as broad, heterogenous groups of congenital conditions characterized by atypical development of genetic, gonadal, or phenotypic sex accompanied by abnormal development of internal and/or external genitalia.
explanation: Deep research cited this publication as relevant literature for 46 XX Gonadal Dysgenesis.
- reference: PMID:19246354
title: Mutations in NR5A1 associated with ovarian insufficiency.
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-openai.md
findings: []
- reference: PMID:21963259
title: XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription.
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-openai.md
findings: []
- reference: PMID:23087880
title: "A rare cause for primary amenorrhea: Sporadic perrault syndrome."
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-openai.md
findings: []
- reference: PMID:26485283
title: A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis.
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-openai.md
findings: []
- reference: PMID:31809259
title: 'Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal dysgenesis: a missed opportunity for prevention of osteoporosis.'
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-openai.md
findings:
- statement: >-
Hormone profiles and karyotype evaluation help distinguish 46,XX gonadal
dysgenesis from MRKH syndrome in primary amenorrhea.
supporting_text: >-
Hormone profiles and karyotype should be investigated in all cases of the
presumptive diagnosis of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome or
Mullerian agenesis.
evidence:
- reference: PMID:31809259
reference_title: 'Misdiagnosis of Mullerian agenesis in a patient with 46, XX gonadal dysgenesis: a missed opportunity for prevention of osteoporosis.'
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Hormone profiles and karyotype should be investigated in all cases of
the presumptive diagnosis of Mayer-Rokitansky-Kuster-Hauser (MRKH)
syndrome or Mullerian agenesis.
explanation: Deep research cited this publication as relevant literature for 46 XX Gonadal Dysgenesis.
- reference: PMID:35142292
title: "A rare case of 46,XX gonadal dysgenesis, Mayer-Rokitansky-Kuster-Hauser syndrome, pituitary and thyroid hypoplasia."
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-openai.md
findings: []
- reference: PMID:7553856
title: Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure.
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-openai.md
findings: []
- reference: PMID:8855829
title: Clinical features of primary ovarian failure caused by a point mutation in the follicle-stimulating hormone receptor gene.
found_in:
- 46_XX_Gonadal_Dysgenesis-deep-research-openai.md
findings: []
References & Deep Research
References
19Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Deep Research
2Question: You are an expert researcher providing comprehensive, well-cited information.
Provide detailed information focusing on: 1. Key concepts and definitions with current understanding 2. Recent developments and latest research (prioritize 2023-2024 sources) 3. Current applications and real-world implementations 4. Expert opinions and analysis from authoritative sources 5. Relevant statistics and data from recent studies
Format as a comprehensive research report with proper citations. Include URLs and publication dates where available. Always prioritize recent, authoritative sources and provide specific citations for all major claims.
Disease Characteristics Research Template
Target Disease
- Disease Name: 46,XX Gonadal Dysgenesis
- MONDO ID: (if available)
- Category: Mendelian
Research Objectives
Please provide a comprehensive research report on 46,XX Gonadal Dysgenesis covering all of the disease characteristics listed below. This report will be used to populate a disease knowledge base entry. Be thorough and cite primary literature (PMID preferred) for all claims.
For each section, suggested databases/resources are listed. These are the first places you should search for information on each topic.
1. Disease Information
Search first: OMIM, Orphanet, ICD-10/ICD-11, MeSH, PubMed
- What is the disease? Provide a concise overview.
- What are the key identifiers? (OMIM, Orphanet, ICD-10/ICD-11, MeSH, Mondo)
- What are the common synonyms and alternative names?
- Is the information derived from individual patients (e.g., EHR) or aggregated disease-level resources?
2. Etiology
- Disease Causal Factors: What are the primary causes? (genetic, environmental, infectious, mechanistic)
- Risk Factors:
Search first: PubMed, Cochrane Library, UpToDate, clinical guidelines, ClinVar, ClinGen, GWAS Catalog, PheGenI, CTD, CDC, WHO, epidemiological databases
- Genetic risk factors (causal variants, susceptibility loci, modifier genes)
- Environmental risk factors (toxins, lifestyle, occupational exposures, age, sex, family history)
- Protective Factors:
Search first: PubMed, Cochrane Library, clinical trial databases, GWAS Catalog, gnomAD, WHO, CDC, nutrition databases
- Genetic protective factors (protective variants, modifier alleles)
- Environmental protective factors (diet, lifestyle, exposures that reduce risk)
- Gene-Environment Interactions: How do genetic and environmental factors interact to influence disease?
Search first: CTD, PubMed, PheGenI, GxE databases
3. Phenotypes
Search first: HPO (Human Phenotype Ontology), OMIM, Orphanet, PubMed, clinicaltrials.gov, MedDRA, SNOMED CT, DECIPHER, LOINC
For each phenotype, provide: - Phenotype type: symptoms, clinical signs, physical manifestations, behavioral changes, or laboratory abnormalities
For symptoms/signs: HPO, OMIM, Orphanet, PubMed For behavioral changes: HPO, DSM, RDoC (Research Domain Criteria), PubMed For laboratory abnormalities: LOINC, SNOMED CT, LabTests Online, PubMed - Phenotype characteristics: Search first: OMIM, Orphanet, HPO, PubMed - Age of symptom onset (neonatal, childhood, adult-onset, late-onset) - Symptom severity (mild, moderate, severe, variable) - Symptom progression (stable, progressive, episodic, fluctuating) - Frequency among affected individuals (percentage or qualitative) - Quality of life impact: Effects on daily functioning and well-being (per-phenotype when possible) Search first: EQ-5D database, SF-36, WHO QOL databases, PubMed - Suggest HPO (Human Phenotype Ontology) terms for each phenotype
4. Genetic/Molecular Information
- Causal Genes: Gene mutations or chromosomal abnormalities responsible for disease (gene symbols, OMIM IDs)
Search first: OMIM, ClinVar, HGMD, Ensembl, NCBI Gene
- Pathogenic Variants:
- Affected genes (gene symbols, HGNC IDs) > Search first: OMIM, NCBI Gene, Ensembl, HGNC, UniProt, GeneCards
- Variant classification (pathogenic, likely pathogenic, VUS per ACMG/AMP guidelines) > Search first: ClinVar, ClinGen, ACMG/AMP guidelines, VarSome
- Variant type/class (missense, frameshift, nonsense, splice-site, structural)
- Allele frequency in population databases > Search first: gnomAD, 1000 Genomes, ExAC, TOPMed, dbSNP
- Somatic vs germline origin > Search first: COSMIC (somatic), ClinVar, ICGC, TCGA
- Functional consequences (loss of function, gain of function, dominant negative)
- Modifier Genes: Genes that modify disease severity or expression
- Epigenetic Information: DNA methylation, histone modifications, chromatin changes affecting disease
Search first: ENCODE, Roadmap Epigenomics, MethBase, DiseaseMeth
- Chromosomal Abnormalities: Large-scale genetic changes (aneuploidy, translocations, inversions)
Search first: DECIPHER, ClinVar, ECARUCA, UCSC Genome Browser
5. Environmental Information
- Environmental Factors: Non-genetic contributing factors (toxins, radiation, pollution, occupational exposure)
Search first: CTD (Comparative Toxicogenomics Database), TOXNET, PubMed, EPA databases
- Lifestyle Factors: Behavioral factors (smoking, diet, exercise, alcohol consumption)
Search first: CDC databases, WHO, PubMed, NHANES
- Infectious Agents: If applicable, pathogens causing or triggering disease (bacteria, viruses, fungi, parasites)
Search first: NCBI Taxonomy, ViPR, BV-BRC, MicrobeDB, GIDEON
6. Mechanism / Pathophysiology
- Molecular Pathways: Specific signaling cascades or biochemical pathways involved (Wnt, MAPK, mTOR, PI3K-AKT, etc.)
Search first: KEGG, Reactome, WikiPathways, PathBank, BioCyc
- Cellular Processes: Cell-level mechanisms (apoptosis, autophagy, cell cycle dysregulation, inflammation, etc.)
Search first: Gene Ontology (GO), Reactome, KEGG, PubMed
- Protein Dysfunction: How protein structure or function is altered (misfolding, aggregation, loss of function, gain of function)
Search first: UniProt, PDB (Protein Data Bank), InterPro, Pfam, AlphaFold
- Metabolic Changes: Alterations in metabolic processes (energy metabolism, lipid metabolism, amino acid metabolism)
Search first: KEGG, BioCyc, HMDB (Human Metabolome Database), BRENDA
- Immune System Involvement: Role of immune response (autoimmunity, immunodeficiency, chronic inflammation)
Search first: ImmPort, Immunome Database, IEDB, Gene Ontology
- Tissue Damage Mechanisms: How tissues/ are injured (oxidative stress, ischemia, fibrosis, necrosis)
Search first: PubMed, Gene Ontology, Reactome
- Biochemical Abnormalities: Specific molecular defects (enzyme deficiencies, receptor dysfunction, ion channel defects)
Search first: BRENDA, UniProt, KEGG, OMIM, PubMed
- Epigenetic Changes: DNA methylation, histone modifications affecting gene expression in disease
Search first: ENCODE, Roadmap Epigenomics, MethBase, DiseaseMeth
- Molecular Profiling (if available):
- Transcriptomics/gene expression changes > Search first: GEO (Gene Expression Omnibus), ArrayExpress, GTEx, Human Cell Atlas, SRA
- Proteomics findings > Search first: PRIDE, ProteomeXchange, Human Protein Atlas, STRING, BioGRID
- Metabolomics signatures > Search first: MetaboLights, Metabolomics Workbench, HMDB, METLIN
- Lipidomics alterations > Search first: LIPID MAPS, SwissLipids, LipidHome, Metabolomics Workbench
- Genomic structural features > Search first: UCSC Genome Browser, Ensembl, NCBI, dbVar, DGV
- Advanced Technologies (if applicable):
- Single-cell analysis findings (cell-type specific mechanisms, cellular heterogeneity) > Search first: Human Cell Atlas, Single Cell Portal, GEO, CELLxGENE
- Spatial transcriptomics findings > Search first: GEO, Spatial Research, Vizgen, 10x Genomics data
- Multi-omics integration results > Search first: TCGA, ICGC, cBioPortal, LinkedOmics, PubMed
- Functional genomics screens (CRISPR, RNAi) > Search first: DepMap, GenomeRNAi, PubMed, BioGRID ORCS
For each mechanism, describe: - The causal chain from initial trigger to clinical manifestation - Which mechanisms are upstream vs downstream - What cell types and biological processes are involved - Suggest GO terms for biological processes and CL terms for cell types
7. Anatomical Structures Affected
- Organ Level:
- Primary organs directly affected
- Secondary organ involvement (complications, secondary effects)
- Body systems involved (cardiovascular, nervous, digestive, respiratory, endocrine, etc.)
Search first: Uberon, FMA (Foundational Model of Anatomy), OMIM, HPO, ICD-11, MeSH, SNOMED CT
- Tissue and Cell Level:
- Specific tissue types affected (epithelial, connective, muscle, nervous)
- Specific cell populations targeted (with Cell Ontology terms)
Search first: Uberon, Human Protein Atlas, Cell Ontology, Human Cell Atlas, CellMarker, PanglaoDB
- Subcellular Level:
- Cellular compartments involved (mitochondria, nucleus, ER, lysosomes) (with GO Cellular Component terms)
Search first: Gene Ontology (Cellular Component), UniProt, Human Protein Atlas
- Localization:
- Specific anatomical sites (with UBERON terms) > Search first: FMA, Uberon, NeuroNames (for brain), SNOMED CT
- Lateralization (unilateral, bilateral, asymmetric) > Search first: HPO, clinical literature, imaging databases
8. Temporal Development
- Onset:
- Typical age of onset (congenital, pediatric, adult, geriatric)
- Onset pattern (acute, subacute, chronic, insidious)
Search first: OMIM, Orphanet, HPO, PubMed
- Progression:
- Disease stages (early, intermediate, advanced, end-stage) > Search first: Cancer Staging Manual (AJCC), WHO classifications, PubMed
- Progression rate (rapid, slow, variable)
- Disease course pattern (episodic, relapsing-remitting, progressive, stable)
- Disease duration (self-limited, chronic lifelong)
Search first: Disease registries, longitudinal cohort databases, natural history studies, PubMed, Orphanet, OMIM
- Patterns:
- Remission patterns (spontaneous, treatment-induced) > Search first: Clinical trial databases, disease registries, PubMed
- Critical periods (time windows of vulnerability or opportunity for intervention) > Search first: PubMed, developmental biology databases, clinical guidelines
9. Inheritance and Population
- Epidemiology:
- Prevalence (cases per 100,000 at given time)
- Incidence (new cases per 100,000 per year)
Search first: Orphanet, CDC, WHO, GBD (Global Burden of Disease), national registries, SEER, disease registries
- For Genetic Etiology:
- Inheritance pattern (AD, AR, X-linked, mitochondrial, multifactorial, polygenic) > Search first: OMIM, Orphanet, ClinVar, GTR (Genetic Testing Registry)
- Penetrance (complete, incomplete, age-dependent) > Search first: ClinVar, OMIM, PubMed, ClinGen
- Expressivity (variable, consistent) > Search first: OMIM, ClinVar, PubMed
- Genetic anticipation (increasing severity in successive generations) > Search first: OMIM, PubMed (especially for repeat expansion disorders)
- Germline mosaicism > Search first: ClinVar, OMIM, genetic counseling literature, PubMed
- Founder effects (population-specific mutations) > Search first: gnomAD, population genetics databases, PubMed
- Consanguinity role > Search first: OMIM, population studies, genetic counseling resources
- Carrier frequency > Search first: gnomAD, carrier screening databases, GeneReviews, GTR
- Population Demographics:
- Affected populations (ethnic or demographic groups with higher prevalence) > Search first: gnomAD, 1000 Genomes, PAGE Study, PubMed, population registries
- Geographic distribution (endemic areas, regional variation) > Search first: WHO, CDC, GBD, Orphanet, geographic epidemiology databases
- Geographic distribution of specific variants
- Sex ratio (male:female) > Search first: Disease registries, OMIM, PubMed, epidemiological databases
- Age distribution of affected individuals > Search first: CDC, disease registries, SEER, Orphanet
10. Diagnostics
- Clinical Tests:
- Laboratory tests (blood, urine, tissue chemistry, specific enzyme assays) > Search first: LOINC, LabTests Online, PubMed
- Biomarkers (proteins, metabolites, genetic markers, circulating biomarkers) > Search first: FDA Biomarker List, BEST (Biomarkers, EndpointS, and other Tools), PubMed
- Imaging studies (X-ray, CT, MRI, PET, ultrasound) > Search first: RadLex, DICOM, Radiopaedia, imaging databases
- Functional tests (pulmonary function, cardiac stress tests) > Search first: LOINC, clinical guidelines, PubMed
- Electrophysiology (EEG, EMG, ECG, nerve conduction studies) > Search first: LOINC, clinical neurophysiology databases, PubMed
- Biopsy findings (histopathology, immunohistochemistry) > Search first: SNOMED CT, College of American Pathologists resources, PubMed
- Pathology findings (microscopic examination) > Search first: SNOMED CT, Digital Pathology databases, PubMed
- Genetic Testing:
Search first: GTR (Genetic Testing Registry), GeneReviews, ClinGen
- Overview of recommended genetic testing approach
- Whole genome sequencing (WGS) utility > Search first: GTR, ClinVar, GEL (Genomics England), gnomAD
- Whole exome sequencing (WES) utility > Search first: GTR, ClinVar, OMIM, GeneMatcher
- Gene panels (which panels, which genes) > Search first: GTR, ClinVar, laboratory-specific databases
- Single gene testing > Search first: GTR, ClinVar, OMIM, GeneReviews
- Chromosomal microarray (CMA) > Search first: DECIPHER, ClinVar, dbVar, ECARUCA
- Karyotyping > Search first: Chromosome Abnormality Database, ClinVar, cytogenetics resources
- FISH > Search first: ClinVar, cytogenetics databases, PubMed
- Mitochondrial DNA testing > Search first: MITOMAP, MSeqDR, ClinVar, GTR
- Repeat expansion testing > Search first: GTR, ClinVar, repeat expansion databases, PubMed
- Omics-Based Diagnostics (if applicable):
- RNA sequencing / transcriptomics > Search first: GEO, ArrayExpress, GTEx, RNA-seq databases
- Proteomics > Search first: PRIDE, ProteomeXchange, FDA Biomarker database
- Metabolomics > Search first: MetaboLights, Metabolomics Workbench, HMDB
- Epigenomics > Search first: GEO, ENCODE, Roadmap Epigenomics, MethBase
- Liquid biopsy > Search first: COSMIC, ClinVar, liquid biopsy databases, PubMed
- Clinical Criteria:
- Standardized diagnostic criteria (DSM, ICD, society guidelines) > Search first: DSM-5, ICD-11, clinical society guidelines, UpToDate
- Differential diagnosis (other conditions to rule out, with distinguishing features) > Search first: DynaMed, UpToDate, clinical decision support systems
- Screening:
- Screening methods for asymptomatic individuals (newborn screening, carrier screening, cascade screening) > Search first: ACMG recommendations, CDC newborn screening, GTR
11. Outcome/Prognosis
- Survival and Mortality:
- Survival rate (5-year, 10-year, overall) > Search first: SEER, cancer registries, disease-specific registries, PubMed
- Life expectancy (with and without treatment if applicable) > Search first: Orphanet, disease registries, actuarial databases, PubMed
- Mortality rate > Search first: CDC, WHO, GBD, national mortality databases
- Disease-specific mortality (deaths directly attributable to disease) > Search first: Disease registries, CDC Wonder, GBD, PubMed
- Morbidity and Function:
- Morbidity (disease-related disability and health impacts) > Search first: GBD, WHO, disability databases, PubMed
- Disability outcomes (long-term functional impairments) > Search first: ICF (International Classification of Functioning), disability registries
- Quality of life measures (EQ-5D, SF-36, PROMIS, disease-specific tools) > Search first: EQ-5D database, SF-36, PROMIS, PubMed
- Disease Course:
- Complications (secondary problems: infections, organ failure, etc.) > Search first: ICD codes, disease registries, clinical databases, PubMed
- Recovery potential (likelihood and extent of recovery, with vs without treatment) > Search first: Natural history studies, rehabilitation databases, PubMed
- Prediction:
- Prognostic factors (age, disease severity, biomarkers, treatment response) > Search first: Prognostic models databases, clinical calculators, PubMed
- Prognostic biomarkers (molecular markers predicting disease course) > Search first: FDA Biomarker database, PubMed, cancer prognostic databases
12. Treatment
- Pharmacotherapy:
- Pharmacological treatments (drug names, drug classes, mechanisms of action) > Search first: DrugBank, RxNorm, ATC classification, DailyMed, FDA databases
- Pharmacogenomics (how genetic variants affect drug metabolism, efficacy, toxicity) > Search first: PharmGKB, CPIC (Clinical Pharmacogenetics), FDA Table of PGx Biomarkers
- Advanced Therapeutics:
- Gene therapy (viral vectors, CRISPR, gene replacement, gene editing) > Search first: ClinicalTrials.gov, FDA gene therapy database, ASGCT resources
- Cell therapy (stem cell transplant, CAR-T, cellular therapeutics) > Search first: ClinicalTrials.gov, FDA cell therapy database, FACT standards
- RNA-based therapies (ASOs, siRNA, mRNA therapies) > Search first: ClinicalTrials.gov, FDA approvals, PubMed
- Targeted therapies (treatments directed at specific molecular targets) > Search first: My Cancer Genome, OncoKB, ClinicalTrials.gov, FDA approvals
- Immunotherapies (checkpoint inhibitors, monoclonal antibodies) > Search first: Cancer Immunotherapy Database, FDA approvals, ClinicalTrials.gov
- Surgical and Interventional:
- Surgical interventions (types of surgery, timing, outcomes) > Search first: CPT codes, surgical registries, clinical guidelines, PubMed
- Supportive and Rehabilitative:
- Supportive care (symptom management, pain control, nutrition) > Search first: Clinical guidelines, Cochrane Library, PubMed
- Rehabilitation (physical therapy, occupational therapy, speech therapy) > Search first: Rehabilitation medicine databases, clinical guidelines, PubMed
- Experimental:
- Experimental treatments in clinical trials (with NCT identifiers if available) > Search first: ClinicalTrials.gov, EU Clinical Trials Register, WHO ICTRP
- Treatment Outcomes:
- Treatment response rates > Search first: Clinical trial databases, FDA reviews, systematic reviews, PubMed
- Side effects and adverse events > Search first: FDA Adverse Event Reporting System (FAERS), MedWatch, PubMed
- Treatment Strategy:
- Treatment algorithms (clinical pathways, decision trees) > Search first: Clinical practice guidelines, NCCN Guidelines, UpToDate
- Combination therapies > Search first: ClinicalTrials.gov, treatment guidelines, PubMed
- Personalized medicine approaches (genotype-guided treatment) > Search first: My Cancer Genome, CIViC, PharmGKB, precision medicine databases
For each treatment, suggest MAXO (Medical Action Ontology) terms where applicable.
13. Prevention
- Prevention Levels:
- Primary prevention (preventing disease occurrence: vaccination, risk factor modification) > Search first: CDC, WHO, USPSTF recommendations, Cochrane Library
- Secondary prevention (early detection and treatment: screening programs, early intervention) > Search first: USPSTF, CDC screening guidelines, WHO
- Tertiary prevention (preventing complications in those with disease) > Search first: Clinical guidelines, disease management protocols, PubMed
- Immunization: Vaccine strategies (if applicable)
Search first: CDC vaccine schedules, WHO immunization, FDA vaccine database
- Screening and Early Detection:
- Screening programs (population-based: newborn screening, cancer screening) > Search first: CDC screening programs, USPSTF, cancer screening databases
- Genetic screening (carrier screening, preimplantation genetic diagnosis, prenatal testing) > Search first: ACMG recommendations, ACOG guidelines, GTR
- Risk stratification (identifying high-risk individuals for targeted prevention) > Search first: Risk prediction models, clinical calculators, PubMed
- Behavioral Interventions: Lifestyle modifications to reduce risk
Search first: CDC, WHO, behavioral intervention databases, Cochrane Library
- Counseling: Genetic counseling (risk assessment, family planning guidance)
Search first: NSGC resources, ACMG guidelines, GeneReviews
- Public Health:
- Public health interventions (sanitation, vector control, health education) > Search first: CDC, WHO, public health databases, PubMed
- Environmental interventions (reducing environmental risk factors) > Search first: EPA databases, WHO environmental health, PubMed
- Prophylaxis: Preventive medications or procedures
Search first: Clinical guidelines, FDA approvals, PubMed
14. Other Species / Natural Disease
- Taxonomy: Species affected (with NCBI Taxon identifiers)
Search first: NCBI Taxonomy
- Breed: Specific breeds affected (with VBO identifiers if applicable)
Search first: VBO (Vertebrate Breed Ontology)
- Gene: Orthologous genes in other species (with NCBI Gene IDs)
Search first: NCBI Gene
- Natural Disease:
- Naturally occurring disease in other species (companion animals, wildlife) > Search first: OMIA (Online Mendelian Inheritance in Animals), VetCompass, PubMed
- Veterinary relevance and importance in animal health > Search first: OMIA, veterinary databases, PubMed
- Comparative Biology:
- Comparative pathology (similarities and differences across species) > Search first: OMIA, comparative pathology databases, PubMed
- Evolutionary conservation of disease mechanisms > Search first: HomoloGene, OrthoMCL, Alliance of Genome Resources
- Transmission (if applicable):
- Zoonotic potential > Search first: CDC zoonotic diseases, WHO zoonoses, GIDEON
- Cross-species susceptibility > Search first: NCBI Taxonomy, veterinary databases, PubMed
15. Model Organisms
- Model Types:
- Model organism type (mammalian, invertebrate, cellular, in vitro) > Search first: Alliance of Genome Resources, model organism databases
- Specific model systems (mouse, rat, zebrafish, Drosophila, C. elegans, yeast, cell lines, organoids, iPSCs) > Search first: MGI, RGD, ZFIN, FlyBase, WormBase, SGD, ATCC, Cellosaurus
- Induced models (drug treatment, surgical intervention, environmental manipulation) > Search first: MGI, model organism databases, PubMed
- Genetic Models:
- Types available (knockout, knock-in, transgenic, conditional, humanized) > Search first: MGI, IMPC, KOMP, EuMMCR, IMSR
- Model Characteristics:
- Phenotype recapitulation (how well model reproduces human disease features) > Search first: Model organism databases, comparative studies, PubMed
- Model limitations (aspects of human disease not captured) > Search first: Model organism databases, PubMed, review articles
- Applications:
- Research applications (what aspects of disease can be studied) > Search first: Model organism databases, PubMed
- Resources:
- Model databases > Search first: MGI, RGD, ZFIN, FlyBase, WormBase, IMSR, EMMA, MMRRC
Citation Requirements
- Cite primary literature (PMID preferred) for all mechanistic and clinical claims
- Prioritize recent reviews and landmark papers
- Include direct quotes from abstracts where possible to support key statements
- Distinguish evidence source types: human clinical, model organism, in vitro, computational
Output Format
Structure your response as a comprehensive narrative organized by the sections above. For each section, provide: - Factual content with specific details (numbers, percentages, gene names, variant nomenclature) - Ontology term suggestions (HPO, GO, CL, UBERON, CHEBI, MAXO, MONDO) where applicable - Evidence citations with PMIDs - Direct quotes from abstracts to support key claims - Clear indication when information is not available or not applicable for this disease
This report will be used to populate a disease knowledge base entry with: - Pathophysiology descriptions with causal chains - Gene/protein annotations (HGNC, GO terms) - Phenotype associations (HP terms) with frequencies - Cell type involvement (CL terms) - Anatomical locations (UBERON terms) - Chemical entities (CHEBI terms) - Treatment annotations (MAXO terms) - Evidence items with PMIDs and exact abstract quotes - Epidemiology, prognosis, diagnostic, and prevention information - Animal model descriptions with phenotype recapitulation details
Comprehensive Disease Characteristics Research Report: 46,XX Gonadal Dysgenesis
Target disease
Disease name: 46,XX gonadal dysgenesis (also referred to in the literature as 46,XX gonadal dysgenesis, 46,XX ovarian dysgenesis, XX gonadal dysgenesis (XX-GD), XX ovarian dysgenesis, and sometimes discussed on a spectrum with primary ovarian insufficiency (POI) when presenting as primary amenorrhea with hypergonadotropic hypogonadism). (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2, martin2020clinicalandmolecular pages 63-66)
Category: Mendelian (genetically heterogeneous; both recessive and dominant mechanisms reported; some X-linked). (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2, luppino2024roleofnr5a1 pages 7-8, yatsenko2024primaryamenorrheaand pages 16-17)
MONDO / OMIM / Orphanet / ICD / MeSH identifiers: Not reliably extractable from the currently retrieved full-text evidence set using the available tools; the report below is grounded in primary literature and recent reviews that explicitly define the condition and its genetics. (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2)
Evidence provenance note: The content below is primarily derived from aggregated literature sources (peer‑reviewed reviews and primary research), not from EHRs. (abalı2024diagnosisandmanagement pages 1-2, grouthier2024longtermoutcomesin pages 2-3)
1. Disease information
1.1 Concise overview (current understanding)
46,XX gonadal dysgenesis is a disorder of ovarian development and/or function in individuals with a 46,XX karyotype, typically characterized by lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism (elevated gonadotropins with gonadal failure). (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2)
A widely cited clinical framing is that affected individuals present in adolescence with failure of pubertal progression (e.g., minimal breast development), primary amenorrhea, low estrogen, and markedly elevated FSH/LH due to loss of ovarian negative feedback. (weinbergshukron2015amutationin pages 1-2, martin2020clinicalandmolecular pages 63-66)
1.2 Key synonyms / alternative names
- XX gonadal dysgenesis (XX-GD) (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2)
- XX ovarian dysgenesis (zangen2011xxovariandysgenesis pages 1-2)
- 46,XX ovarian dysgenesis (zangen2011xxovariandysgenesis pages 1-2)
- 46,XX pure gonadal dysgenesis (used in some clinical discussions/case‑based literature on the POI/ovarian dysgenesis spectrum). (cattoni2020thepotentialsynergic pages 1-2)
2. Etiology
2.1 Primary causal factors
Primary cause is genetic, involving defects in pathways of ovarian determination, follicle formation/maintenance, gonadotropin signaling, and/or meiosis/DNA repair. (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2, yatsenko2024primaryamenorrheaand pages 16-17)
Key mechanistic gene categories emphasized by primary studies and recent reviews: - Gonadotropin signaling / receptor resistance: e.g., FSHR loss-of-function leading to FSH resistance and hypergonadotropic ovarian failure. (martin2020clinicalandmolecular pages 63-66) - Meiosis and recombination / follicle pool establishment: e.g., PSMC3IP (HOP2) and NUP107. (zangen2011xxovariandysgenesis pages 1-2, weinbergshukron2015amutationin pages 1-2) - Ovarian developmental transcriptional regulators and maintenance factors: e.g., NR5A1, FIGLA, NOBOX, FOXL2, and pro‑ovarian pathway genes such as WNT4/RSPO1 (reported in the XX‑GD genetic landscape). (luppino2024roleofnr5a1 pages 7-8, cattoni2020thepotentialsynergic pages 1-2, weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2, abalı2024diagnosisandmanagement pages 6-7)
2.2 Risk factors
Because 46,XX gonadal dysgenesis is primarily genetic, “risk” is largely determined by family history and carrier status (depending on inheritance). (zangen2011xxovariandysgenesis pages 1-2, weinbergshukron2015amutationin pages 1-2)
The broader POI literature provides population-level context: non‑iatrogenic POI has estimated prevalence increasing with age (approx. 1:10,000 before age 20; 1:1,000 before age 30; 1:100 before age 40), with chromosomal abnormalities accounting for about ~9% in one synthesis. (cattoni2020thepotentialsynergic pages 1-2)
2.3 Protective factors / gene–environment interactions
No specific protective factors or gene–environment interactions were identified in the retrieved evidence set for 46,XX gonadal dysgenesis specifically; broader POI literature emphasizes multifactorial contributions in many cases, but XX‑GD itself is often described as Mendelian and rare. (zangen2011xxovariandysgenesis pages 1-2, cattoni2020thepotentialsynergic pages 1-2)
3. Phenotypes
3.1 Core phenotype (human clinical)
Hallmark phenotype constellation: - Absent or incomplete puberty / lack of spontaneous pubertal development (symptom/sign). (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) - Primary amenorrhea (symptom). (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) - Hypergonadotropic hypogonadism (laboratory abnormality): very high gonadotropins (FSH/LH) with ovarian failure. (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2, martin2020clinicalandmolecular pages 63-66) - Uterine hypoplasia (anatomical finding), and ovaries may be not visualized on ultrasound/MRI in some cases. (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2)
Quantitative example (from NUP107-associated XX-GD): LH reported in the range 38–60 IU/L and FSH 50–92 IU/L in affected individuals; an example proband had LH 52 IU/L and FSH 87 IU/L; uterus ~4 cm and ovaries not visualized on imaging. (weinbergshukron2015amutationin pages 1-2)
3.2 Phenotype characteristics
- Age of onset: typically adolescence, presenting as absent/delayed puberty and primary amenorrhea. (weinbergshukron2015amutationin pages 1-2, martin2020clinicalandmolecular pages 63-66)
- Severity: ranges from complete ovarian dysgenesis (no pubertal development) to milder POI spectrum with partial residual function; XX‑GD is described as a severe end of ovarian insufficiency spectrum. (zangen2011xxovariandysgenesis pages 1-2, cattoni2020thepotentialsynergic pages 1-2)
- Progression/course: usually chronic/lifelong hypogonadism unless treated hormonally; fertility is typically severely impaired. (grouthier2024longtermoutcomesin pages 2-3, cattoni2020thepotentialsynergic pages 1-2)
3.3 Suggested HPO terms (non-exhaustive)
Based on the phenotype descriptions in primary papers and reviews: - Primary amenorrhea (HPO: HP:0000786) (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) - Delayed puberty (HP:0000821) / Absent puberty (HP:0000875) (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) - Hypergonadotropic hypogonadism (HP:0000044) (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) - Uterine hypoplasia (HP:0000130) (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) - Streak gonads (often described in XX‑GD clinical definitions; map to gonadal dysgenesis concept; HPO frequently used: Gonadal dysgenesis HP:0000130?—note: exact HPO term IDs should be verified in an ontology browser; the concept is directly described in-source). (zangen2011xxovariandysgenesis pages 1-2)
3.4 Quality of life impact
For the broader non‑CAH 46,XX DSD group (which includes XX gonadal dysgenesis and monogenic POI), long‑term quality of life data are emphasized as sparse: “data … remain scarce” and adult QoL assessment is noted as lacking accurate data in this rare group. (grouthier2024longtermoutcomesin pages 2-3)
4. Genetic / molecular information
4.1 Causal genes (high-confidence examples from primary literature)
The genetic architecture is heterogeneous. Primary studies provide strong evidence for Mendelian forms including: - NUP107 (AR): recessive missense mutation segregating with XX‑GD; functional model (Drosophila) supports ovarian developmental requirement. (weinbergshukron2015amutationin pages 1-2) - PSMC3IP/HOP2 (AR): homozygous deletion; functional assays show loss of estrogen-driven transcriptional coactivation. (zangen2011xxovariandysgenesis pages 1-2) - FSHR (AR): rare; WES-identified homozygous missense variant with demonstrated membrane trafficking/signaling impairment in vitro and hypergonadotropic amenorrhea in affected sisters. (martin2020clinicalandmolecular pages 63-66)
Recent reviews further emphasize additional implicated genes and pathways (often overlapping with POI genetics) including NR5A1, FIGLA, NOBOX, FOXL2, BMP15, and pro‑ovarian pathway regulators (e.g., WNT4/RSPO1) in the ovarian dysgenesis/POI spectrum. (luppino2024roleofnr5a1 pages 7-8, yatsenko2024primaryamenorrheaand pages 16-17, zangen2011xxovariandysgenesis pages 1-2)
4.2 Pathogenic variants and functional consequences (examples)
- FSHR p.Asp408Tyr (c.1222G>T) in two affected sisters (homozygous): flow cytometry showed ~48% reduction in cell-surface receptor signal and ~50% reduction in FSH-stimulated cAMP signal in mutant‑transfected cells, consistent with impaired signaling/trafficking. (martin2020clinicalandmolecular pages 63-66)
- PSMC3IP p.Glu201del (homozygous 3-bp deletion): functional assays showed the mutation “abolished PSMC3IP activation of estrogen-driven transcription”, supporting a loss‑of‑function mechanism. (zangen2011xxovariandysgenesis pages 1-2)
- NUP107 p.D447N (homozygous missense): functional studies in Drosophila showed female sterility when Nup107 was knocked down in somatic gonadal cells, and the human-corresponding mutant allele led to near‑complete sterility and ovarian/egg chamber defects. (weinbergshukron2015amutationin pages 1-2)
4.3 Inheritance patterns (summary)
- Autosomal recessive inheritance is emphasized for several severe XX‑GD genes (e.g., NUP107, PSMC3IP, FSHR) and is suggested to account for a substantial portion of unexplained severe cases in some families/consanguinity settings. (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2, martin2020clinicalandmolecular pages 63-66)
- Autosomal dominant / heterozygous contributions are relevant particularly for POI-spectrum genes such as NR5A1, with reported pathogenic variants in 0.26%–8% of sporadic POI in different studies and 2.8% in a cohort of 142 women with ovarian deficiency/diminished ovarian reserve/unexplained infertility. (luppino2024roleofnr5a1 pages 7-8)
- X-linked inheritance is noted for some genes implicated in ovarian failure/POI such as BMP15 (summarized as X‑linked recessive in XX‑GD landscape discussions). (weinbergshukron2015amutationin pages 1-2)
4.4 Oligogenic / modifier models
A 2020 case report proposed that the severe phenotype (complete ovarian dysgenesis) could reflect a synergic detrimental effect of inherited variants across FIGLA, NOBOX, and NR5A1, with relatives carrying subsets showing variable residual ovarian function. (cattoni2020thepotentialsynergic pages 1-2)
A broader 2023 review discusses oligogenic inheritance in DSD and highlights the challenges of interpreting combined variants; although this is not limited to XX‑GD, it supports the plausibility of multi‑hit models in sex development disorders. (stancampiano202446xxdifferencesof pages 4-5)
5. Environmental information
No specific environmental/lifestyle/infectious causal factors were identified for 46,XX gonadal dysgenesis in the retrieved evidence set; the condition is presented as primarily genetic. (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2)
6. Mechanism / pathophysiology
6.1 Causal chain (from gene defect to phenotype)
A simplified mechanistic chain consistent with primary genetic examples: 1. Primary genetic defect (e.g., meiotic recombination factor PSMC3IP, nucleoporin NUP107, or receptor FSHR). (zangen2011xxovariandysgenesis pages 1-2, weinbergshukron2015amutationin pages 1-2, martin2020clinicalandmolecular pages 63-66) 2. Failure of follicle pool establishment/maintenance or gonadotropin response, leading to severely reduced ovarian steroidogenesis. (zangen2011xxovariandysgenesis pages 1-2, martin2020clinicalandmolecular pages 63-66) 3. Low estrogen → loss of negative feedback on hypothalamic-pituitary axis → elevated FSH/LH (hypergonadotropic hypogonadism). (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2, martin2020clinicalandmolecular pages 63-66) 4. Absent/delayed puberty and primary amenorrhea; uterine hypoplasia likely reflects hypoestrogenism during puberty. (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2)
6.2 Ovarian determination pathway concepts (pro‑ovarian vs pro‑testis)
A 2024 non‑CAH 46,XX DSD management review describes that WNT4 and RSPO1 stabilize β‑catenin (CTNNB1), and that in the 46,XX gonad, WNT/RSPO1/CTNNB1/FOXL2/FST promote ovarian development while suppressing testicular pathways (including inhibition of SOX9/FGF9). (abalı2024diagnosisandmanagement pages 6-7)
6.3 Suggested ontology terms
GO biological process (suggested, to be verified in GO): - gonad development; ovarian follicle development; meiotic cell cycle; steroid hormone biosynthetic process; regulation of transcription by nuclear receptor. (zangen2011xxovariandysgenesis pages 1-2, weinbergshukron2015amutationin pages 1-2, luppino2024roleofnr5a1 pages 7-8)
Cell Ontology (CL) likely relevant cell types (suggested): - granulosa cell; theca cell; oocyte; ovarian stromal cells; pituitary gonadotrophs (downstream endocrine response). (abalı2024diagnosisandmanagement pages 6-7, zangen2011xxovariandysgenesis pages 1-2)
UBERON (anatomy) (suggested): - ovary; uterus; hypothalamus; anterior pituitary gland. (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2)
7. Anatomical structures affected
- Primary organs: ovaries/gonads (dysgenetic or absent follicular function). (zangen2011xxovariandysgenesis pages 1-2, weinbergshukron2015amutationin pages 1-2)
- Secondary/related structures: uterus is often hypoplastic (likely secondary to hypoestrogenism). (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2)
- Systems: endocrine/reproductive axis (hypothalamic–pituitary–gonadal). (martin2020clinicalandmolecular pages 63-66)
8. Temporal development
- Typical detection: adolescence (evaluation for absent puberty/primary amenorrhea). (weinbergshukron2015amutationin pages 1-2, martin2020clinicalandmolecular pages 63-66)
- Course: persistent ovarian failure without intervention; POI spectrum may have variable residual function in some genetic contexts. (zangen2011xxovariandysgenesis pages 1-2, cattoni2020thepotentialsynergic pages 1-2)
9. Inheritance and population
9.1 Epidemiology
Direct prevalence/incidence of 46,XX gonadal dysgenesis specifically was not provided in the retrieved evidence set.
However, adjacent epidemiologic context from related 46,XX DSD conditions: - 46,XX testicular DSD prevalence estimated ~1:20,000, and ~90% are due to SRY translocation (contextual, not XX‑GD). (stancampiano202446xxdifferencesof pages 4-5) - For men with non‑CAH 46,XX DSD in Denmark, national estimate 3.5–4.7 per 100,000 (contextual, reflects XX male/testicular/ovotesticular DSD and related entities rather than ovarian dysgenesis). (grouthier2024longtermoutcomesin pages 2-3)
For POI (broader umbrella that includes ovarian dysgenesis presentations), one case-based synthesis reports age‑stratified prevalence (1:10,000 before 20; 1:1,000 before 30; 1:100 before 40). (cattoni2020thepotentialsynergic pages 1-2)
9.2 Inheritance
- AR inheritance is supported by multiple severe XX‑GD genes (NUP107, PSMC3IP, FSHR). (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2, martin2020clinicalandmolecular pages 63-66)
- AD/heterozygous contribution is supported for NR5A1-related POI and gonadal development disorders. (luppino2024roleofnr5a1 pages 7-8)
- X-linked is noted for BMP15 in the XX‑GD landscape. (weinbergshukron2015amutationin pages 1-2)
10. Diagnostics
10.1 Clinical presentation prompting workup
- Primary amenorrhea with absent/delayed puberty and hypergonadotropic pattern. (weinbergshukron2015amutationin pages 1-2, martin2020clinicalandmolecular pages 63-66)
10.2 Core laboratory evaluation (supported)
- Gonadotropins: FSH and LH are elevated (hypergonadotropic hypogonadism). (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2, martin2020clinicalandmolecular pages 63-66)
- Sex steroids: low estrogen is implied/typical in XX‑GD definitions. (zangen2011xxovariandysgenesis pages 1-2)
10.3 Imaging and anatomic evaluation (supported)
- Pelvic ultrasound and/or MRI often show uterine hypoplasia and may show ovaries not visualized in severe cases. (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2)
10.4 Genetic testing strategy (current implementation trend)
A 2023 clinical approach review for DSD emphasizes that increased availability of next‑generation sequencing has led to recommendations for earlier integration of genetic testing into DSD diagnostic pathways and highlights that establishing a molecular diagnosis can affect individualized management and monitoring. (stancampiano202446xxdifferencesof pages 4-5)
For DSD workups more broadly, a 2021 prospective series used a stepwise genetic protocol including karyotype + SRY testing, followed by targeted gene testing for common etiologies, chromosomal microarray, and NGS panels, with yields varying by technique. (nistal2015perspectivesinpediatric pages 15-16)
For suspected XX‑GD/ovarian dysgenesis, high‑confidence Mendelian diagnoses have been achieved using homozygosity mapping + WES (PSMC3IP) and WES with functional validation (FSHR). (zangen2011xxovariandysgenesis pages 1-2, martin2020clinicalandmolecular pages 63-66)
10.5 Differential diagnosis (contextual)
In primary amenorrhea, XX‑GD/ovarian dysgenesis must be distinguished from other major causes such as Müllerian agenesis (MRKH), central (hypogonadotropic) causes, and other DSDs; the key distinguishing laboratory feature for ovarian dysgenesis is typically hypergonadotropic hypogonadism. (martin2020clinicalandmolecular pages 63-66)
11. Outcomes / prognosis
11.1 Fertility
Fertility is typically severely impaired in non‑CAH 46,XX DSD overall, and for ovarian dysgenesis specifically, ovarian function is absent or markedly reduced. (grouthier2024longtermoutcomesin pages 2-3, zangen2011xxovariandysgenesis pages 1-2)
11.2 Long-term health outcomes and QoL (what is known vs unknown)
A 2024 review on long-term outcomes in non‑CAH 46,XX DSD highlights that long‑term follow-up data are scarce, with limited adult QoL data, and emphasizes needs in bone/cardiometabolic monitoring, cancer risk, and mortality assessment. (grouthier2024longtermoutcomesin pages 2-3)
For POI more broadly, clinical impact includes fertility, psychological/sexual quality of life, and long-term bone/cardiovascular health consequences of hypoestrogenism; this context is emphasized in a 2020 report framing POI diagnostic criteria and prevalence. (cattoni2020thepotentialsynergic pages 1-2)
12. Treatment
12.1 Core management principles (evidence-supported high level)
Because the core endocrine defect is ovarian estrogen deficiency with hypergonadotropic hypogonadism, treatment typically involves: - Sex hormone replacement to induce/maintain secondary sexual development and mitigate hypoestrogenism sequelae (supported as a key management topic in 46,XX DSD reviews; specific regimens not detailed in the retrieved excerpt-level evidence). (grouthier2024longtermoutcomesin pages 2-3, stancampiano202446xxdifferencesof pages 4-5) - Fertility counseling and consideration of fertility preservation approaches where applicable; fertility is generally impaired in non‑CAH 46,XX DSD. (grouthier2024longtermoutcomesin pages 2-3)
12.2 Experimental / clinical trial activity (real-world implementation)
ClinicalTrials.gov NCT06518746 (University of Colorado, Denver; interventional pilot; posted as 2021 record) evaluates gonadal tissue cryopreservation in patients with gonadal dysgenesis/DSD undergoing clinically indicated gonadectomy or at risk of POI. The study processes gonadal tissue removed at surgery, histologically examines it, and cryopreserves tissue if viable germ cells are present and no tumor is found; outcomes include sample viability and adverse events. (NCT06518746 chunk 1, NCT06518746 chunk 2)
12.3 Suggested MAXO terms (non-exhaustive; to be verified)
- Hormone replacement therapy (MAXO concept) (grouthier2024longtermoutcomesin pages 2-3)
- Fertility preservation procedure / cryopreservation of gonadal tissue (MAXO concept; aligned with NCT06518746 intervention). (NCT06518746 chunk 1)
- Genetic counseling (MAXO concept). (stancampiano202446xxdifferencesof pages 4-5)
13. Prevention
Primary prevention of Mendelian XX‑GD is not generally feasible, but secondary/tertiary prevention focuses on: - Genetic counseling and family-based risk assessment, particularly for recessive forms in consanguineous families. (zangen2011xxovariandysgenesis pages 1-2, weinbergshukron2015amutationin pages 1-2) - Consideration of early molecular diagnosis to guide individualized monitoring and management planning in DSD conditions. (stancampiano202446xxdifferencesof pages 4-5)
14. Other species / natural disease
A specific naturally occurring veterinary analog was not identified in the retrieved evidence set. However, the NUP107 study provides direct functional evidence in Drosophila that Nup107 is required in somatic gonadal cells for female fertility, supporting evolutionary conservation of aspects of ovarian development mechanisms. (weinbergshukron2015amutationin pages 1-2)
15. Model organisms
- Drosophila model (functional genomics): Nup107 knockdown in somatic gonadal cells causes female sterility, and the human-corresponding mutant allele leads to near complete sterility and ovarian/egg chamber defects, supporting NUP107 causality in XX‑GD. (weinbergshukron2015amutationin pages 1-2)
- Cell models (in vitro): FSHR mutant functional testing in HEK293T cells and quantitative flow cytometry demonstrates impaired receptor surface localization and signaling (cAMP response), supporting pathogenicity. (martin2020clinicalandmolecular pages 63-66)
Recent developments and 2023–2024 highlights (prioritized)
- Expanded and clinically oriented reviews for non‑CAH 46,XX DSD (2024): Multiple Frontiers in Endocrinology reviews synthesize etiology, diagnostic and management challenges, and long-term outcomes, emphasizing rarity and limited adult outcome datasets. (abalı2024diagnosisandmanagement pages 1-2, grouthier2024longtermoutcomesin pages 2-3)
- Updated gene/pathway framing (2024): Reviews emphasize gene networks in ovarian determination and maintenance (WNT4/RSPO1/β‑catenin/FOXL2), and highlight ongoing uncertainty about genotype–phenotype correlation in DSD. (abalı2024diagnosisandmanagement pages 6-7)
- NR5A1 relevance to ovarian dysfunction (2024): A 2024 review summarizes reported frequencies of NR5A1 variants in POI-related cohorts and reinforces that NR5A1 contributes to ovarian failure phenotypes (amenorrhea, elevated gonadotropins, infertility) and may warrant early genetic testing and fertility considerations. (luppino2024roleofnr5a1 pages 7-8)
- Long-term outcome knowledge gaps (2024): Long-term data for non‑CAH 46,XX DSD (including XX gonadal dysgenesis/POI) remain sparse, motivating multicenter longitudinal follow-up studies. (grouthier2024longtermoutcomesin pages 2-3)
Key abstract-supported quotes (verbatim excerpts available in retrieved text)
- FSHR-WES study (Human Reproduction 2016) states: “FSHR mutations are an extremely rare cause of 46, XX gonadal dysgenesis with primary amenorrhea due to hypergonadotropic ovarian failure.” (martin2020clinicalandmolecular pages 63-66)
- PSMC3IP (Am J Hum Genet 2011) functional conclusion: mutation “abolished PSMC3IP activation of estrogen-driven transcription.” (zangen2011xxovariandysgenesis pages 1-2)
Visual evidence (extracted tables/figures)
Gene/phenotype tables relevant to 46,XX DSD etiologies (including gonadal dysgenesis/POI context) were extracted from Stancampiano et al. 2024 (Frontiers in Endocrinology). (stancampiano202446xxdifferencesof media 3d672b2a, stancampiano202446xxdifferencesof media 5ebb60ba, stancampiano202446xxdifferencesof media 7a210e9a)
Structured gene summary artifact
The following table compiles the principal evidence-supported genetic etiologies and quantitative data points relevant to 46,XX gonadal dysgenesis / XX ovarian dysgenesis and closely related POI presentations:
| Gene (HGNC symbol) | Typical inheritance pattern (AR/AD/X-linked or reported) | Molecular mechanism | Key clinical features | Key study evidence | Key quantitative data | URL |
|---|---|---|---|---|---|---|
| NUP107 | Autosomal recessive / recessive reported | Missense loss of function affecting nucleoporin function; ovarian development defect supported by functional model data | 46,XX gonadal dysgenesis with lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, hypergonadotropic hypogonadism; ovaries not visualized on imaging in reported cases (weinbergshukron2015amutationin pages 1-2) | Weinberg-Shukron 2015, J Clin Invest (weinbergshukron2015amutationin pages 1-2) | Example values reported: LH 38–60 IU/L, FSH 50–92 IU/L; variant absent from databases and 150 ethnically matched controls (weinbergshukron2015amutationin pages 1-2) | https://doi.org/10.1172/JCI83553 |
| PSMC3IP (HOP2) | Often autosomal recessive / often AR in unexplained XX-GD families | Loss of function; meiotic recombination defect and abolished coactivation of estrogen-driven transcription | Rare 46,XX gonadal dysgenesis with absent spontaneous puberty, primary amenorrhea, uterine hypoplasia, streak gonads, hypergonadotropic hypogonadism (zangen2011xxovariandysgenesis pages 1-2) | Zangen 2011, Am J Hum Genet (zangen2011xxovariandysgenesis pages 1-2) | Homozygous 3-bp deletion p.Glu201del identified in consanguineous family; functional assay showed mutation abolished estrogen-driven transcriptional coactivation (zangen2011xxovariandysgenesis pages 1-2) | https://doi.org/10.1016/j.ajhg.2011.09.006 |
| FSHR | Autosomal recessive / AR reported | Receptor resistance / inactivating loss of function in FSH signaling | Primary amenorrhea due to hypergonadotropic ovarian failure; 46,XX gonadal dysgenesis / ovarian dysgenesis phenotype with absent puberty or delayed puberty and high gonadotropins (weinbergshukron2015amutationin pages 1-2, martin2020clinicalandmolecular pages 63-66) | Bramble 2016, Hum Reprod; cited in reviews of XX-GD/amenorrhea (weinbergshukron2015amutationin pages 1-2, martin2020clinicalandmolecular pages 63-66) | Described as an “extremely rare” cause; novel p.Asp408Tyr showed ~48% reduction in cell-surface signal and ~50% reduction in FSH-stimulated cAMP (martin2020clinicalandmolecular pages 63-66) | https://doi.org/10.1093/humrep/dew025 |
| BMP15 | X-linked recessive reported; heterozygous and homozygous variants reported | Oocyte-derived growth factor dysfunction / impaired ovarian growth and maturation | Hypergonadotropic ovarian failure; ovarian dysgenesis/POI with primary amenorrhea possible, including severe ovarian dysgenesis phenotypes (weinbergshukron2015amutationin pages 1-2, yatsenko2024primaryamenorrheaand pages 16-17) | Di Pasquale 2004, Am J Hum Genet; summarized in later reviews (weinbergshukron2015amutationin pages 1-2, yatsenko2024primaryamenorrheaand pages 16-17) | Ovarian dysgenesis accounts for about half of primary amenorrhea cases in older review context; BMP15 variants reported in 1.5%–15% of POI in review summary (yatsenko2024primaryamenorrheaand pages 16-17) | https://doi.org/10.1086/422103 |
| NR5A1 | Heterozygous variants reported; AD/reported | Loss of function affecting ovarian steroidogenic/gonadal developmental transcriptional regulation | POI or 46,XX DSD with primary or secondary amenorrhea, estrogen deficiency, elevated gonadotropins, infertility; can overlap with ovarian dysgenesis spectrum (luppino2024roleofnr5a1 pages 7-8) | Luppino 2024, Curr Issues Mol Biol; Jaillard 2020, Maturitas summarized therein (luppino2024roleofnr5a1 pages 7-8) | NR5A1 variants found in 2.8% of 142 women with ovarian deficiency/DOR/unexplained infertility; pathogenic variants reported in 0.26%–8% of sporadic POI (luppino2024roleofnr5a1 pages 7-8) | https://doi.org/10.3390/cimb46050274 |
| FIGLA | Reported; autosomal recessive possible in some families, but not specified here | Transcription factor defect in ovarian maturation / folliculogenesis | Ovarian dysgenesis or POI spectrum with primary amenorrhea and reduced/absent ovarian function (martin2020clinicalandmolecular pages 63-66, cattoni2020thepotentialsynergic pages 1-2, yatsenko2024primaryamenorrheaand pages 16-17) | Cattoni 2020, Front Endocrinol; review summaries (martin2020clinicalandmolecular pages 63-66, cattoni2020thepotentialsynergic pages 1-2, yatsenko2024primaryamenorrheaand pages 16-17) | FIGLA variants found in 4% of one Chinese sporadic POI series (martin2020clinicalandmolecular pages 63-66) | https://doi.org/10.3389/fendo.2020.540683 |
| NOBOX | Reported; autosomal recessive possible in some families, but not specified here | Loss of function in ovarian developmental transcription factor | Ovarian dysgenesis/POI with primary amenorrhea possible; impaired ovarian function spectrum (martin2020clinicalandmolecular pages 63-66, cattoni2020thepotentialsynergic pages 1-2, yatsenko2024primaryamenorrheaand pages 16-17) | Cattoni 2020, Front Endocrinol; review summaries (martin2020clinicalandmolecular pages 63-66, cattoni2020thepotentialsynergic pages 1-2, yatsenko2024primaryamenorrheaand pages 16-17) | Loss-of-function NOBOX variants accounted for 6.2%, 6.5%, and 5.6% in three POI cohorts (martin2020clinicalandmolecular pages 63-66) | https://doi.org/10.3389/fendo.2020.540683 |
| FOXL2 | Autosomal dominant in BPES syndromic context; heterozygous and homozygous variants reported in ovarian dysgenesis/POI | Transcription factor dysfunction affecting granulosa/overy maintenance | Ovarian dysgenesis/POI with delayed puberty, primary amenorrhea, or POI; can be syndromic (BPES) or isolated ovarian insufficiency (yatsenko2024primaryamenorrheaand pages 16-17) | Yatsenko 2024, Endocrinol Metab Clin N Am; Luo 2023, J Ovarian Res summarized in gathered evidence (yatsenko2024primaryamenorrheaand pages 16-17) | In 500 POI patients, FOXL2 had the highest occurrence frequency at 3.2% (16/500); p.R349G accounted for 2.6% in that cohort (yatsenko2024primaryamenorrheaand pages 16-17) | https://doi.org/10.1016/j.ecl.2024.01.009 |
| WNT4 | Not specified in gathered evidence | Pro-ovarian developmental pathway defect / ovarian determination | Included among known causes or candidate genes for XX-GD/ovarian development disorders; detailed XX-GD phenotype specifics not provided in gathered evidence (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) | Summarized in Weinberg-Shukron 2015, J Clin Invest and Zangen 2011, Am J Hum Genet (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) | Not specified | https://doi.org/10.1172/JCI83553 |
| RSPO1 | Not specified in gathered evidence | Pro-ovarian developmental pathway defect / ovarian determination | Included among genes implicated in ovarian development disorders and 46,XX gonadal development pathways; detailed pure XX-GD quantitative data not specified here (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) | Summarized in Weinberg-Shukron 2015, J Clin Invest and Zangen 2011, Am J Hum Genet (weinbergshukron2015amutationin pages 1-2, zangen2011xxovariandysgenesis pages 1-2) | Not specified | https://doi.org/10.1172/JCI83553 |
Table: This table summarizes evidence-supported genes implicated in 46,XX gonadal dysgenesis / XX ovarian dysgenesis and closely related primary ovarian insufficiency presenting with primary amenorrhea. It highlights inheritance, mechanisms, hallmark phenotypes, and key quantitative findings to support disease knowledge base curation.
References (URLs and dates)
The citations embedded above include publication year/month and URLs where available, notably: - Stancampiano et al., 2024-05, Frontiers in Endocrinology. https://doi.org/10.3389/fendo.2024.1402579 (stancampiano202446xxdifferencesof pages 4-5) - Abalı & Guran, 2024-05, Frontiers in Endocrinology. https://doi.org/10.3389/fendo.2024.1354759 (abalı2024diagnosisandmanagement pages 1-2, abalı2024diagnosisandmanagement pages 6-7) - Grouthier & Bachelot, 2024-04, Frontiers in Endocrinology. https://doi.org/10.3389/fendo.2024.1372887 (grouthier2024longtermoutcomesin pages 2-3) - Luppino et al., 2024-05, Current Issues in Molecular Biology. https://doi.org/10.3390/cimb46050274 (luppino2024roleofnr5a1 pages 7-8) - Yatsenko et al., 2024-06, Endocrinology and Metabolism Clinics of North America. https://doi.org/10.1016/j.ecl.2024.01.009 (yatsenko2024primaryamenorrheaand pages 16-17) - Weinberg‑Shukron et al., 2015-11, J Clin Invest. https://doi.org/10.1172/JCI83553 (weinbergshukron2015amutationin pages 1-2) - Zangen et al., 2011-10, Am J Hum Genet. https://doi.org/10.1016/j.ajhg.2011.09.006 (zangen2011xxovariandysgenesis pages 1-2) - Bramble et al., 2016-02, Human Reproduction. https://doi.org/10.1093/humrep/dew025 (martin2020clinicalandmolecular pages 63-66) - ClinicalTrials.gov NCT06518746 (record year 2021). https://clinicaltrials.gov/study/NCT06518746 (NCT06518746 chunk 1, NCT06518746 chunk 2)
References
-
(weinbergshukron2015amutationin pages 1-2): Ariella Weinberg-Shukron, Paul Renbaum, Rachel Kalifa, Sharon Zeligson, Ziva Ben-Neriah, Amatzia Dreifuss, Amal Abu-Rayyan, Noa Maatuk, Nilly Fardian, Dina Rekler, Moien Kanaan, Abraham O. Samson, Ephrat Levy-Lahad, Offer Gerlitz, and David Zangen. A mutation in the nucleoporin-107 gene causes xx gonadal dysgenesis. The Journal of clinical investigation, 125 11:4295-304, Nov 2015. URL: https://doi.org/10.1172/jci83553, doi:10.1172/jci83553. This article has 111 citations.
-
(zangen2011xxovariandysgenesis pages 1-2): David Zangen, Yotam Kaufman, Sharon Zeligson, Shira Perlberg, Hila Fridman, Moein Kanaan, Maha Abdulhadi-Atwan, Abdulsalam Abu Libdeh, Ayal Gussow, Irit Kisslov, Liran Carmel, Paul Renbaum, and Ephrat Levy-Lahad. Xx ovarian dysgenesis is caused by a psmc3ip/hop2 mutation that abolishes coactivation of estrogen-driven transcription. American journal of human genetics, 89 4:572-9, Oct 2011. URL: https://doi.org/10.1016/j.ajhg.2011.09.006, doi:10.1016/j.ajhg.2011.09.006. This article has 140 citations and is from a highest quality peer-reviewed journal.
-
(martin2020clinicalandmolecular pages 63-66): I Martínez de la Piscina Martín. Clinical and molecular characterization of dsd patients: impact of next generation sequencing in diagnosis. Unknown journal, 2020.
-
(luppino2024roleofnr5a1 pages 7-8): Giovanni Luppino, Malgorzata Wasniewska, Roberto Coco, Giorgia Pepe, Letteria Anna Morabito, Alessandra Li Pomi, Domenico Corica, and Tommaso Aversa. Role of nr5a1 gene mutations in disorders of sex development: molecular and clinical features. Current Issues in Molecular Biology, 46:4519-4532, May 2024. URL: https://doi.org/10.3390/cimb46050274, doi:10.3390/cimb46050274. This article has 24 citations.
-
(yatsenko2024primaryamenorrheaand pages 16-17): Svetlana A. Yatsenko, Selma F. Witchel, and Catherine M. Gordon. Primary amenorrhea and premature ovarian insufficiency. Endocrinology and Metabolism Clinics of North America, 53:293-305, Jun 2024. URL: https://doi.org/10.1016/j.ecl.2024.01.009, doi:10.1016/j.ecl.2024.01.009. This article has 22 citations and is from a peer-reviewed journal.
-
(abalı2024diagnosisandmanagement pages 1-2): Zehra Yavas Abalı and Tulay Guran. Diagnosis and management of non-cah 46,xx disorders/differences in sex development. Frontiers in Endocrinology, May 2024. URL: https://doi.org/10.3389/fendo.2024.1354759, doi:10.3389/fendo.2024.1354759. This article has 11 citations.
-
(grouthier2024longtermoutcomesin pages 2-3): Virginie Grouthier and Anne Bachelot. Long-term outcomes in non-cah 46,xx dsd. Frontiers in Endocrinology, Apr 2024. URL: https://doi.org/10.3389/fendo.2024.1372887, doi:10.3389/fendo.2024.1372887. This article has 6 citations.
-
(cattoni2020thepotentialsynergic pages 1-2): Alessandro Cattoni, Alice Spano, Anna Tulone, Annalisa Boneschi, Nicoletta Masera, Silvia Maitz, Anna Maria Di Blasio, Luca Persani, Fabiana Guizzardi, and Raffaella Rossetti. The potential synergic effect of a complex pattern of multiple inherited genetic variants as a pathogenic factor for ovarian dysgenesis: a case report. Frontiers in Endocrinology, Sep 2020. URL: https://doi.org/10.3389/fendo.2020.540683, doi:10.3389/fendo.2020.540683. This article has 11 citations.
-
(abalı2024diagnosisandmanagement pages 6-7): Zehra Yavas Abalı and Tulay Guran. Diagnosis and management of non-cah 46,xx disorders/differences in sex development. Frontiers in Endocrinology, May 2024. URL: https://doi.org/10.3389/fendo.2024.1354759, doi:10.3389/fendo.2024.1354759. This article has 11 citations.
-
(stancampiano202446xxdifferencesof pages 4-5): Marianna Rita Stancampiano, Silvia Laura Carla Meroni, Carmen Bucolo, and Gianni Russo. 46,xx differences of sex development outside congenital adrenal hyperplasia: pathogenesis, clinical aspects, puberty, sex hormone replacement therapy and fertility outcomes. Frontiers in Endocrinology, May 2024. URL: https://doi.org/10.3389/fendo.2024.1402579, doi:10.3389/fendo.2024.1402579. This article has 9 citations.
-
(nistal2015perspectivesinpediatric pages 15-16): Manuel Nistal, Ricardo Paniagua, Pilar González-Peramato, and Miguel Reyes-Múgica. Perspectives in pediatric pathology, chapter 5. gonadal dysgenesis. Pediatric and Developmental Pathology, 18:259-278, Jul 2015. URL: https://doi.org/10.2350/14-04-1471-pb.1, doi:10.2350/14-04-1471-pb.1. This article has 22 citations and is from a peer-reviewed journal.
-
(NCT06518746 chunk 1): Gonadal Dysgenesis Tissue Cryopreservation for Fertility Preservation. University of Colorado, Denver. 2021. ClinicalTrials.gov Identifier: NCT06518746
-
(NCT06518746 chunk 2): Gonadal Dysgenesis Tissue Cryopreservation for Fertility Preservation. University of Colorado, Denver. 2021. ClinicalTrials.gov Identifier: NCT06518746
-
(stancampiano202446xxdifferencesof media 3d672b2a): Marianna Rita Stancampiano, Silvia Laura Carla Meroni, Carmen Bucolo, and Gianni Russo. 46,xx differences of sex development outside congenital adrenal hyperplasia: pathogenesis, clinical aspects, puberty, sex hormone replacement therapy and fertility outcomes. Frontiers in Endocrinology, May 2024. URL: https://doi.org/10.3389/fendo.2024.1402579, doi:10.3389/fendo.2024.1402579. This article has 9 citations.
-
(stancampiano202446xxdifferencesof media 5ebb60ba): Marianna Rita Stancampiano, Silvia Laura Carla Meroni, Carmen Bucolo, and Gianni Russo. 46,xx differences of sex development outside congenital adrenal hyperplasia: pathogenesis, clinical aspects, puberty, sex hormone replacement therapy and fertility outcomes. Frontiers in Endocrinology, May 2024. URL: https://doi.org/10.3389/fendo.2024.1402579, doi:10.3389/fendo.2024.1402579. This article has 9 citations.
-
(stancampiano202446xxdifferencesof media 7a210e9a): Marianna Rita Stancampiano, Silvia Laura Carla Meroni, Carmen Bucolo, and Gianni Russo. 46,xx differences of sex development outside congenital adrenal hyperplasia: pathogenesis, clinical aspects, puberty, sex hormone replacement therapy and fertility outcomes. Frontiers in Endocrinology, May 2024. URL: https://doi.org/10.3389/fendo.2024.1402579, doi:10.3389/fendo.2024.1402579. This article has 9 citations.
Artifacts
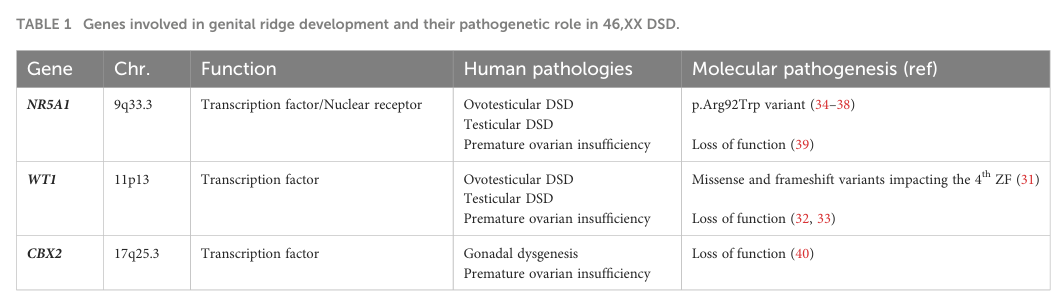
Summary
MONDO:0009299 provides a dedicated disease-level anchor for 46 XX gonadal
dysgenesis. The literature consistently treats this as a rare, genetically
heterogeneous ovarian developmental failure state in phenotypically female
individuals with a normal 46,XX karyotype, with the core phenotype defined by
streak or underdeveloped ovaries, absent spontaneous puberty, primary
amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism.
Disease-Anchor Decision
- Anchor chosen:
46 XX gonadal dysgenesis(MONDO:0009299) - Why this anchor: the disease has its own MONDO term and multiple primary human papers describe it as a coherent clinical entity rather than only as a phenotype mention inside broader DSD literature.
- Why not a generic
46,XX DSDbucket: the core disease definition here is ovarian developmental failure with phenotypically female external genitalia, not generic 46,XX sex-development variation such as prenatal androgen excess. - Why not lump fully with broader primary ovarian insufficiency:
NR5A1demonstrates overlap with46,XX primary ovarian insufficiency, but the46,XX gonadal dysgenesisliterature is narrower and emphasizes congenital streak/underdeveloped ovaries, absent spontaneous puberty, and uterine hypoplasia. - Why not split into single-gene disorders for this issue:
NUP107,PSMC3IP,FSHR, andNR5A1all support the same disease-level anchor, and the issue explicitly asked for a disease-level dismech entry rather than a single-gene subtype.
Mechanistic Themes
- Shared proximal theme: disrupted female gonad development.
- Shared intermediate theme: impaired ovarian hormonal signaling and folliculogenesis.
- Shared downstream theme: streak or severely underdeveloped ovaries leading to hypoestrogenic hypergonadotropic ovarian failure.
Papers Used for YAML Evidence
| PMID | Use in YAML | Key contribution |
|---|---|---|
| 23087880 | prevalence framing | Rare genetically heterogeneous disease framing |
| 26485283 | case definition, pathophysiology, phenotypes, genetics | Strong disease-definition abstract plus ovarian-development mechanism |
| 21963259 | progression, pathophysiology, phenotypes, genetics | Streak gonads, estrogenic signaling, follicular-pool mechanism |
| 7553856 | pathophysiology, genetics | Direct FSHR signaling defect causing ovarian dysgenesis |
| 8855829 | pathophysiology, phenotypes | Elevated FSH/LH and amenorrhea in mechanistically defined subset |
| 19246354 | pathophysiology, genetics | Overlap boundary with broader ovarian insufficiency |
| 35142292 | treatment | Hormone replacement therapy for secondary sexual characteristics and osteoporosis prevention |
| 31809259 | PR framing / differential context | MRKH misdiagnosis pitfall and estrogen-dependent uterine visualization |
Curation Notes
- Evidence snippets were restricted to exact quoted text from the cached PMID records.
- The YAML intentionally avoids generic DSD-management statements as primary treatment evidence; management is limited to hormone replacement therapy with direct PMID-backed wording.
- The YAML intentionally avoids assigning phenotype frequency bands because the available abstracts support association but not robust quantitative frequency.