3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency
3-hydroxy-3-methylglutaryl-CoA synthase deficiency is an autosomal recessive disorder of hepatic ketone body synthesis caused by biallelic pathogenic variants in HMGCS2. Loss of mitochondrial HMG-CoA synthase 2 activity blocks ketone body biosynthesis during fasting or intercurrent illness, producing acute metabolic decompensation with hypoketotic hypoglycemia, dicarboxylic aciduria, metabolic acidosis, vomiting, lethargy, hepatomegaly, seizures, and in severe cases encephalopathy or coma. Diagnosis is often difficult because routine organic acid and acylcarnitine profiles can be nonspecific outside acute crises; urinary 4-hydroxy-6- methyl-2-pyrone and an elevated plasma C2/C0 acylcarnitine ratio during decompensation improve recognition. Long-term management centers on avoiding fasting, rapid carbohydrate support during illness, and genetic counseling.
Ask OpenScientist
Ask a research question about 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (2 references)
Mechanistic Hypotheses
1Show evidence (12 references)
Pathophysiology
4Show evidence (4 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (3 references)
Pathograph
Phenotypes
25Cardiovascular 1
Show evidence (1 reference)
Digestive 5
Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Metabolism 7
Show evidence (2 references)
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (2 references)
Nervous System 4
Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (2 references)
Respiratory 2
Show evidence (1 reference)
Show evidence (1 reference)
Other 6
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
1Show evidence (3 references)
Medical Actions
5Show evidence (2 references)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Biochemical Markers
4Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Source YAML
click to showname: 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency
creation_date: '2026-05-04T09:20:00Z'
updated_date: '2026-05-21T19:05:00Z'
category: Mendelian
description: >
3-hydroxy-3-methylglutaryl-CoA synthase deficiency is an autosomal
recessive disorder of hepatic ketone body synthesis caused by biallelic
pathogenic variants in HMGCS2. Loss of mitochondrial HMG-CoA synthase 2
activity blocks ketone body biosynthesis during fasting or intercurrent
illness, producing acute metabolic decompensation with hypoketotic
hypoglycemia, dicarboxylic aciduria, metabolic acidosis, vomiting, lethargy,
hepatomegaly, seizures, and in severe cases encephalopathy or coma.
Diagnosis is often difficult because routine organic acid and acylcarnitine
profiles can be nonspecific outside acute crises; urinary 4-hydroxy-6-
methyl-2-pyrone and an elevated plasma C2/C0 acylcarnitine ratio during
decompensation improve recognition. Long-term management centers on avoiding
fasting, rapid carbohydrate support during illness, and genetic counseling.
disease_term:
preferred_term: 3-hydroxy-3-methylglutaryl-CoA synthase deficiency
term:
id: MONDO:0011614
label: 3-hydroxy-3-methylglutaryl-CoA synthase deficiency
synonyms:
- HMG-CoA synthase deficiency
- HMG-CoA synthase-2 deficiency
- HMGCS2 deficiency
- HMGCS2D
- Mitochondrial HMG-CoA synthase deficiency
parents:
- Disorder of Fatty Acid Oxidation and Ketogenesis
- Inborn Error of Metabolism
notes: >-
ORPHA:35701 maps this disorder exactly to MONDO:0011614 and OMIM:605911, and
lists ICD-10:E71.3, ICD-11:5C52.02, and UMLS:C4510940. The ORPHA
definition describes fewer than 20 patients, while a 2025 systematic review
reports 93 published cases plus 2 newly diagnosed patients, so the PubMed
review is used for current case-count and phenotype-scope statements.
inheritance:
- name: Autosomal Recessive
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "Autosomal recessive"
explanation: Orphanet records autosomal recessive inheritance.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "rare, potentially life-threatening autosomal recessive disorder resulting from"
explanation: The 2025 systematic review states the autosomal recessive inheritance pattern.
prevalence:
- population: Worldwide
measure_type: POINT_PREVALENCE
prevalence_class: BELOW_1_IN_1000000
rate_high: 0.1
percentage: <1 per 1,000,000
notes: >
Orphanet records a worldwide point-prevalence estimate below 1 per
1,000,000. The 2025 systematic review identified 93 reported cases plus two
new patients, supporting ultra-rare prevalence.
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "<1 / 1 000 000 | Worldwide | Point prevalence | ORPHANET"
explanation: Orphanet provides the worldwide point-prevalence estimate.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "reported cases and 2 new patients diagnosed based on biochemical findings."
explanation: The systematic review summarizes the published case count and two additional patients.
progression:
- phase: Early childhood onset
age_range: 3 months to 6 years
notes: >
Most reported patients present before age three, usually during a first
catabolic stress episode.
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "Age of onset: Childhood"
explanation: Orphanet records childhood onset.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "mostly before the age of 3."
explanation: The systematic review indicates that most reported onset occurs before age three.
- phase: Acute metabolic decompensation
notes: >
Decompensation is typically triggered by gastroenteritis-like illness,
fasting, poor intake, or other catabolic stress and can progress to altered
consciousness, seizures, encephalopathy, or coma.
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "episodes of decompensation (often associated with gastroenteritis or fasting)"
explanation: Orphanet identifies gastroenteritis and fasting as decompensation triggers.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "of gastroenteritis or gastroenteritis-like symptoms."
explanation: The systematic review identifies gastroenteritis-like episodes as common initial triggers.
- phase: Post-diagnosis relapse prevention
notes: >
After diagnosis, proactive fasting avoidance, illness-period carbohydrate
support, and high-glucose rescue can reduce recurrent acute relapses, and
contemporary cohort follow-up suggests many children have normal physical
development when crises are recognized and prevented.
evidence:
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The implementation of a high glucose infusion and proactive management strategies-such as preventing prolonged fasting and providing enteral carbohydrate/glucose infusion during illness-effectively reduced the rate of acute relapses following accurate diagnosis."
explanation: The Vietnamese cohort reports relapse reduction after diagnosis using high-glucose infusion and fasting-avoidance/carbohydrate strategies.
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Currently, all 19 patients are alive, with ages ranging from 5 months to 14 years, and exhibit normal physical development."
explanation: The same cohort supports favorable observed development and survival after recognition and proactive management.
mechanistic_hypotheses:
- hypothesis_group_id: canonical_hmgcs2_ketogenesis_failure_model
hypothesis_label: Canonical HMGCS2 Ketogenesis Failure Model
status: CANONICAL
description: >
Biallelic HMGCS2 pathogenic variants reduce mitochondrial
hydroxymethylglutaryl-CoA synthase activity in hepatocytes. During fasting
or illness, fatty acid beta-oxidation supplies acetyl-CoA but defective
HMGCS2 blocks conversion of acetyl-CoA and acetoacetyl-CoA into HMG-CoA,
the rate-limiting step in ketone body synthesis. The liver therefore fails
to produce adequate ketone bodies when glucose availability falls, producing
hypoketotic hypoglycemia and a characteristic acute-phase metabolite
pattern including dicarboxylic aciduria, 4HMP excretion, and elevated C2/C0
acylcarnitine ratio.
notes: >-
Retained as CANONICAL. The 2026 openscientist hypothesis-search report
(kb/hypotheses/3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiency/canonical_hmgcs2_ketogenesis_failure_model)
reviewed 41 papers and confirmed the core mechanism with no serious
competing model. Five qualifications refine the canonical description:
(1) hypoglycemia is NOT universal — observed in only ~56% of acute
episodes, with normoglycemic and even severe hyperglycemic
presentations documented; (2) acetyl-CoA accumulation drives secondary
pathology beyond energy failure (mitochondrial protein
hyperacetylation, ACSL1-mediated fatty acid re-esterification, hepatic
steatosis); (3) compensatory acetate production in mouse models may
partially substitute for absent ketogenesis and explain incomplete
penetrance; (4) the phenotypic spectrum has expanded to include
neonatal-onset severe hyperammonemia, metabolic stroke with basal
ganglia lesions, dyslipidemia, and MSUD-mimicking metabolic screening;
and (5) SIRT3-mediated K310 deacetylation may modify residual HMGCS2
activity in patients with hypomorphic variants. The "hypoketotic
hypoglycemia" hallmark is best reframed as "hypoketotic metabolic
decompensation" with hypoglycemia as a frequent but not defining
feature.
evidence:
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: OTHER
snippet: "which is the rate-limiting step of ketone body synthesis"
explanation: This review statement places HMGCS2 at the rate-limiting step of ketogenesis.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "mutations in the HMGCS2 gene, leading to impaired ketogenesis."
explanation: The systematic review links HMGCS2 mutations to impaired ketogenesis.
- reference: PMID:40515583
reference_title: "Mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase deficiency: From metabolism to clinical implications."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "No genotype-phenotype correlation can be established."
explanation: >
Comprehensive 75-patient review confirms HMGCS2D as an autosomal
recessive ketogenesis disorder and documents that no
genotype-phenotype correlation has been established, supporting the
role of modifiers and incomplete penetrance.
- reference: PMID:33619377
reference_title: "Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "acetylome analysis of Hmgcs2 KO cells revealed enhanced acetylation of mitochondrial proteins"
explanation: >
Hmgcs2 knockout mouse model directly demonstrates accumulation of
acetyl-CoA and pathologic hyperacetylation of mitochondrial proteins,
extending the mechanism beyond simple ketone body deficiency into
acetyl-CoA-driven protein modification pathology.
- reference: PMID:40272888
reference_title: "Ketogenesis mitigates metabolic dysfunction-associated steatotic liver disease through mechanisms that extend beyond fat oxidation."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "disruption of mitochondrial HMG-CoA synthase (HMGCS2), the rate-limiting step of ketogenesis, impairs overall hepatic fat oxidation"
explanation: >
Loss-of-function mouse models confirm HMGCS2 as the rate-limiting
ketogenic enzyme and extend the canonical model by showing that loss
of ketogenesis impairs hepatic fat oxidation and induces MASLD-MASH-
like hepatic steatosis.
- reference: PMID:35421611
reference_title: "Hmgcs2-mediated ketogenesis modulates high-fat diet-induced hepatosteatosis."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "Hmgcs2-mediated ketogenesis modulates hepatic lipid regulation"
explanation: >
Genetic gain- and loss-of-function studies show that Hmgcs2-mediated
ketogenesis modulates hepatic lipid regulation, supporting the
canonical model and providing a mechanistic basis for hepatic
steatosis in HMGCS2D.
- reference: PMID:40692014
reference_title: "Hepatic Ketogenesis Regulates Lipid Homeostasis via ACSL1-mediated Fatty Acid Partitioning."
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: "the accumulation of acetyl-CoA because of impaired hepatic ketogenesis drives the elevated translocation of ACSL1 to the ER"
explanation: >
Extends the canonical mechanism with a specific molecular pathway:
acetyl-CoA accumulation downstream of HMGCS2 block drives ACSL1
translocation to the endoplasmic reticulum, increasing fatty acid
re-esterification and promoting hepatic steatosis.
- reference: PMID:38876267
reference_title: "Hepatic ketogenesis is not required for starvation adaptation in mice."
supports: PARTIAL
evidence_source: MODEL_ORGANISM
snippet: "alternative mechanisms drive the increased mortality from forced feeding during illness-induced anorexia"
explanation: >
Qualifies the canonical model by showing that hepatic HMGCS2-deficient
mice maintain blood glucose during prolonged fasting, with
compensatory plasma acetate elevation, suggesting that absent
ketogenesis is not uniformly catastrophic and that alternative fuels
may partially compensate.
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "hypoglycemia (56.3%), and elevated plasma ammonia levels (31.3%)"
explanation: >
Vietnamese cohort of 19 cases shows that hypoglycemia occurred in
only 56.3% of symptomatic acute episodes, qualifying the canonical
"hypoketotic hypoglycemia" hallmark.
- reference: PMID:32905056
reference_title: "Hypoglycemia is not a defining feature of metabolic crisis in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: Further evidence of specific biochemical markers which may aid diagnosis."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "presence of ketonuria does not exclude a diagnosis of a disorder of ketogenesis"
explanation: >
Case report of HMGCS2D presenting with normoglycemia, profound
encephalopathy, and partial ketonuria qualifies the canonical model
by demonstrating that residual ketogenesis and absent hypoglycemia do
not exclude HMGCS2D.
- reference: PMID:40937626
reference_title: "Mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 deficiency with severe hyperglycemia in a child: A rare case report."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "hyperglycemia (25.8 mmol/L), ketonuria (1+), glucosuria (3+), metabolic acidosis"
explanation: >
First reported HMGCS2D case presenting with severe hyperglycemia
(25.8 mmol/L) qualifies the canonical hypoketotic-hypoglycemia model
and broadens the phenotypic spectrum of metabolic decompensation in
HMGCS2D.
- reference: PMID:40548098
reference_title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "severe hyperammonemia is an uncommon phenotype and an exception to neonatal decompensation in HMGCS2 deficiency"
explanation: >
Extends the phenotypic spectrum of HMGCS2D into neonatal-onset
severe hyperammonemia with a metabolic screening profile that
mimics maple syrup urine disease, illustrating the diagnostic
challenge and that the canonical model does not capture all
presentations.
pathophysiology:
- name: HMGCS2 Loss of Function
description: >
Disease-causing germline HMGCS2 variants reduce mitochondrial HMG-CoA
synthase 2 function. Functional studies of patient variants show absent or
markedly reduced enzymatic activity, establishing loss of HMGCS2 catalytic
function as the initiating defect.
gene:
preferred_term: HMGCS2
term:
id: hgnc:5008
label: HMGCS2
molecular_functions:
- preferred_term: hydroxymethylglutaryl-CoA synthase activity
term:
id: GO:0004421
label: hydroxymethylglutaryl-CoA synthase activity
modifier: DECREASED
biological_processes:
- preferred_term: ketone body biosynthetic process
term:
id: GO:0046951
label: ketone body biosynthetic process
modifier: DECREASED
locations:
- preferred_term: mitochondrion
term:
id: GO:0005739
label: mitochondrion
- preferred_term: mitochondrial matrix
term:
id: GO:0005759
label: mitochondrial matrix
cell_types:
- preferred_term: hepatocyte
term:
id: CL:0000182
label: hepatocyte
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "HMGCS2 | 3-hydroxy-3-methylglutaryl-CoA synthase 2 | hgnc:5008 | Disease-causing germline mutation(s) (loss of function) in"
explanation: Orphanet records loss-of-function germline HMGCS2 variants as disease-causing.
- reference: PMID:11479731
reference_title: "Genetic basis of mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Sequencing of the entire coding region of the HMGCS2 gene revealed"
explanation: Human molecular investigation identified HMGCS2 variants in an affected patient.
- reference: PMID:11228257
reference_title: "Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: clinical course and description of causal mutations in two patients."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: "F174L-mHS produces a low level of mHS polypeptide with no detectable activity."
explanation: Functional expression evidence shows loss of HMG-CoA synthase activity for a disease-associated variant.
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: "The other four variants had either no detectable activity or negligible enzymatic activity."
explanation: Functional analysis of patient variants supports absent or near-absent HMGCS2 activity.
downstream:
- target: Impaired Hepatic Ketogenesis
description: HMGCS2 loss blocks mitochondrial ketone body synthesis from acetyl-CoA and acetoacetyl-CoA.
causal_link_type: DIRECT
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "mutations in the HMGCS2 gene, leading to impaired ketogenesis."
explanation: The systematic review directly links HMGCS2 mutations to impaired ketogenesis.
- name: Impaired Hepatic Ketogenesis
description: >
HMGCS2 normally catalyzes formation of HMG-CoA from acetoacetyl-CoA and
acetyl-CoA in the mitochondrial matrix. Deficiency blocks ketone body
biosynthesis during ketogenic stress, leaving affected patients unable to
generate adequate alternative energy substrates during fasting or illness.
biological_processes:
- preferred_term: ketone body biosynthetic process
term:
id: GO:0046951
label: ketone body biosynthetic process
modifier: DECREASED
- preferred_term: ketone body metabolic process
term:
id: GO:1902224
label: ketone body metabolic process
modifier: DECREASED
chemical_entities:
- preferred_term: ketone body
term:
id: CHEBI:73693
label: ketone body
modifier: DECREASED
- preferred_term: acetoacetyl-CoA
term:
id: CHEBI:15345
label: acetoacetyl-CoA
- preferred_term: acetyl-CoA
term:
id: CHEBI:15351
label: acetyl-CoA
cell_types:
- preferred_term: hepatocyte
term:
id: CL:0000182
label: hepatocyte
locations:
- preferred_term: mitochondrial matrix
term:
id: GO:0005759
label: mitochondrial matrix
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "mutations in the HMGCS2 gene, leading to impaired ketogenesis."
explanation: The systematic review directly links HMGCS2 mutation to impaired ketogenesis.
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: OTHER
snippet: "catalyzes the condensation of acetoacetyl-CoA and acetyl-CoA to form"
explanation: This mechanistic statement defines the HMGCS2 reaction in ketone body synthesis.
downstream:
- target: Catabolic Stress Metabolic Decompensation
description: Inadequate ketogenesis during fasting or illness causes energy failure and acute metabolic crises.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Reduced ketone body availability during fasting or illness leaves patients dependent on limited glucose stores.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Most patients presented with acute metabolic decompensation with hypoglycemia, dicarboxyluria and inadequate ketonuria."
explanation: Human case synthesis links impaired ketogenesis to acute metabolic decompensation with inadequate ketone production.
- target: Acute-Phase Acetylcarnitine and Organic Acid Pattern
description: Acetyl-CoA cannot enter ketogenesis normally, contributing to elevated acetylcarnitine, dicarboxylic aciduria, and 4HMP excretion during crises.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Acetyl-CoA diversion to acetylcarnitine and alternative fatty-acid oxidation products during failed ketogenesis.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Dicarboxylic acid levels were elevated in 54/56 cases."
explanation: The systematic review supports the acute organic-acid component of the biochemical pattern.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The plasma C2/C0 acylcarnitine ratio was abnormal in 16/18 (88.9 %) of acute plasma samples"
explanation: The systematic review supports the acute acylcarnitine-ratio component of the biochemical pattern.
- target: Hepatic steatosis
description: Blocked ketogenesis is associated with fatty liver during acute decompensation.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Altered hepatic fatty-acid handling during acute failed ketogenesis.
evidence:
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Fatty liver was identified in three cases, which suggested the unavailability of fatty acids."
explanation: The Japanese patient series links HMGCS2 deficiency crises with fatty liver.
- target: Ketone bodies
description: Decreased ketone bodies are the direct biochemical readout of impaired hepatic ketogenesis.
causal_link_type: DIRECT
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Most patients presented with acute metabolic decompensation with hypoglycemia, dicarboxyluria and inadequate ketonuria."
explanation: Inadequate ketonuria supports decreased ketone bodies as a readout of the ketogenesis block.
- name: Catabolic Stress Metabolic Decompensation
description: >
During fasting, gastroenteritis, poor intake, or illness, impaired
ketogenesis leaves patients dependent on limited glucose availability. The
acute phenotype includes hypoketotic hypoglycemia, metabolic acidosis,
altered consciousness, dyspnea, seizures, hepatomegaly, shock, and
sometimes coma.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "decompensation with hypoglycemia, dicarboxyluria and inadequate ketonuria."
explanation: The systematic review describes the core acute metabolic decompensation pattern.
- reference: PMID:39143735
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency: A Cyclic Vomiting Mimic Without Reliable Biochemical Markers."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "typically present at a young age with hypoketotic hypoglycemia,"
explanation: A recent case report summary describes the typical decompensation phenotype.
downstream:
- target: Hypoglycemia
description: Failure to generate ketone bodies under catabolic stress accompanies hypoglycemia.
causal_link_type: DIRECT
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Most patients presented with acute metabolic decompensation with hypoglycemia, dicarboxyluria and inadequate ketonuria."
explanation: The systematic review identifies hypoglycemia as a core feature of acute decompensation.
- target: Metabolic acidosis
description: Acute decompensation commonly includes severe metabolic acidosis.
causal_link_type: DIRECT
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Each patient (10/10) had a different degree of hepatomegaly and increased aminotransferase, severe metabolic acidosis, and hypofibrinogenemia."
explanation: The Chinese patient series supports metabolic acidosis during acute decompensation.
- target: Seizure
description: Severe acute crises may include seizures.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Neuroglycopenia and acid-base disturbance during severe metabolic decompensation.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Other commonly observed symptoms during the first clinical episode included poor intake, altered consciousness, dyspnea, seizures and hepatomegaly."
explanation: The systematic review lists seizures among common first-episode symptoms.
- target: Hepatomegaly
description: Hepatic metabolic stress during crises is associated with hepatomegaly.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Hepatic metabolic stress and altered fatty-acid handling during acute crises.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Other commonly observed symptoms during the first clinical episode included poor intake, altered consciousness, dyspnea, seizures and hepatomegaly."
explanation: The systematic review lists hepatomegaly among common first-episode symptoms.
- target: Hypoketotic hypoglycemia
description: Ketogenesis failure during catabolic stress produces hypoglycemia with inadequate ketone production.
causal_link_type: DIRECT
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "decompensation with hypoglycemia, dicarboxyluria and inadequate ketonuria."
explanation: The systematic review identifies hypoglycemia with inadequate ketonuria as the core acute decompensation pattern.
- target: Vomiting
description: Catabolic decompensation often presents with vomiting or cyclic-vomiting-like episodes.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:39143735
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency: A Cyclic Vomiting Mimic Without Reliable Biochemical Markers."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "episodes of vomiting and lethargy, often associated with hypoglycemia or"
explanation: The case report links vomiting episodes with hypoglycemia or other metabolic abnormalities in HMGCS2 deficiency.
- target: Diarrhea
description: Gastroenteritis-like acute decompensation may include diarrhea among first-episode symptoms.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "followed by vomiting (8/10), fever (7/10), cough (4/10), diarrhea, and seizures (3/10)."
explanation: The Chinese cohort lists diarrhea among initial HMGCS2D symptoms.
- target: Steatorrhea
description: Acute episodes can include steatorrhea, consistent with crisis-associated dyslipidemia and altered lipid handling.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:33045405
reference_title: "Expanding phenotypic and mutational spectra of mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "During acute episodes, steatorrhea and dyslipidemia occurred"
explanation: The Thai patient report identifies steatorrhea during acute HMGCS2D episodes.
- target: Lethargy
description: Energy failure during acute episodes can manifest as lethargy and altered responsiveness.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Neuroglycopenia and reduced alternative fuel availability during acute decompensation.
evidence:
- reference: PMID:39143735
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency: A Cyclic Vomiting Mimic Without Reliable Biochemical Markers."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "episodes of vomiting and lethargy, often associated with hypoglycemia or"
explanation: The case report documents lethargy during recurrent metabolic episodes.
- target: Encephalopathy
description: Severe decompensation can progress to encephalopathy.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Neuroglycopenia, acid-base disturbance, and severe systemic metabolic stress.
evidence:
- reference: PMID:40548098
reference_title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "hypoketotic hypoglycemia, metabolic acidosis, hepatomegaly, and encephalopathy"
explanation: The neonatal case report states encephalopathy among characteristic severe decompensation features.
- target: Abnormal brain MRI findings
description: Severe metabolic decompensation may be associated with brain MRI abnormalities in a minority of patients.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "Abnormal brain MRI findings were detected in three patients."
explanation: The Vietnamese HMGCS2D cohort documents brain MRI abnormalities, but the specific intermediate injury mechanism is not detailed.
- target: Coma
description: Severe neonatal or childhood crises may progress to coma.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Severe neuroglycopenia and acid-base disturbance during crisis.
evidence:
- reference: PMID:40548098
reference_title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "on day of life 7 with a sepsis-like condition, coma, metabolic acidosis, and"
explanation: The neonatal case presented with coma during metabolic decompensation.
- target: Hypoglycemic coma
description: Severe fasting crises can progress to coma in the setting of hypoglycemia and inadequate ketogenesis.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Neuroglycopenia from hypoglycemia during failed fasting ketogenesis.
evidence:
- reference: PMID:11479731
reference_title: "Genetic basis of mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "children with hypoglycaemic hypoketotic coma during fasting periods"
explanation: The genetic-basis report describes hypoglycemic hypoketotic coma during fasting in affected children.
- target: Hyperammonemia
description: Severe crisis physiology can include hyperammonemia, particularly in neonatal presentations.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "five patients had hyperammonemia, four patients had hyperuricemia"
explanation: The Chinese patient series supports hyperammonemia as a crisis-associated biochemical feature.
- target: Dyspnea
description: Acute metabolic acidosis and decompensation can present with dyspnea.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Respiratory compensation and respiratory distress during acid-base disturbance.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "consciousness, dyspnea, seizures and hepatomegaly."
explanation: The systematic review lists dyspnea among first-episode symptoms.
- target: Tachypnea
description: Acute metabolic acidosis commonly drives rapid breathing as respiratory compensation.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Respiratory compensation for metabolic acidosis during acute crisis.
evidence:
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Clinical manifestations during the first episode were lethargy/coma (81.3%), rapid breathing (68.8%), hepatomegaly (56.3%), shock (37.5%), and seizures (18.8%)."
explanation: The Vietnamese cohort reported rapid breathing during first acute episodes, consistent with tachypnea.
- target: Shock
description: Severe acute metabolic decompensation may include shock during the first clinical episode.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Systemic metabolic acidosis and impaired energy metabolism during severe crisis can compromise tissue perfusion.
evidence:
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Clinical manifestations during the first episode were lethargy/coma (81.3%), rapid breathing (68.8%), hepatomegaly (56.3%), shock (37.5%), and seizures (18.8%)."
explanation: The Vietnamese cohort reported shock during first acute episodes.
- target: Hypofibrinogenemia
description: Acute crises can include coagulation-system disturbance with low fibrinogen.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Each patient (10/10) had a different degree of hepatomegaly and increased aminotransferase, severe metabolic acidosis, and hypofibrinogenemia."
explanation: The Chinese cohort supports hypofibrinogenemia as an acute crisis-associated laboratory phenotype.
- target: Hypophosphatemia
description: Acute decompensation has included hypophosphatemic encephalopathy in reported patients.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:33045405
reference_title: "Expanding phenotypic and mutational spectra of mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Both patients had hypophosphatemic encephalopathy"
explanation: The Thai patient report documents hypophosphatemic encephalopathy in both HMGCS2D patients.
- target: Hypertriglyceridemia
description: Acute metabolic crises may include increased circulating triglycerides.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Blocked ketogenesis and altered hepatic fatty-acid handling can divert lipid metabolism toward triglyceride accumulation during crisis.
evidence:
- reference: PMID:32905056
reference_title: "Hypoglycemia is not a defining feature of metabolic crisis in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: Further evidence of specific biochemical markers which may aid diagnosis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "We propose that elevated acetylcarnitine, triglycerides, and 3HG are additional biochemical features during acute presentations."
explanation: This case report proposes elevated triglycerides as an additional acute biochemical feature.
- target: Elevated hepatic transaminase
description: Hepatic stress during acute crises can include increased aminotransferases.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Hepatic cellular stress during acute metabolic decompensation.
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "different degree of hepatomegaly and increased aminotransferase, severe"
explanation: The Chinese patient series documents increased aminotransferase during severe acute presentations.
- target: Abnormal metabolism or homeostasis
description: Acute decompensation represents the systemic metabolic-homeostasis phenotype caused by failed fasting ketogenesis.
causal_link_type: DIRECT
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "HP:0001939 | Abnormality of metabolism/homeostasis | Very frequent (99-80%)"
explanation: Orphanet records abnormal metabolism/homeostasis as a very frequent phenotype.
- name: Acute-Phase Acetylcarnitine and Organic Acid Pattern
description: >
HMGCS2 deficiency can be difficult to recognize biochemically because
routine organic acid and acylcarnitine abnormalities may be nonspecific.
During acute episodes, dicarboxylic aciduria, urinary 4-hydroxy-6-
methyl-2-pyrone, and increased plasma C2/C0 acylcarnitine ratio support the
diagnosis.
chemical_entities:
- preferred_term: O-acetylcarnitine
term:
id: CHEBI:73024
label: O-acetylcarnitine
modifier: INCREASED
- preferred_term: acetyl-CoA
term:
id: CHEBI:15351
label: acetyl-CoA
modifier: INCREASED
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Dicarboxylic acid levels were elevated in 54/56 cases."
explanation: The systematic review quantifies dicarboxylic aciduria in acute samples.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "4-hydroxy-6-methyl-2-pyrone (4HMP) was detected in 33/35 urine samples taken"
explanation: The systematic review supports 4HMP as a frequent acute-phase diagnostic marker.
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "acetyl-CoA is accumulated and produces acetylcarnitine (C2) in hepatocytes."
explanation: The Japanese case series explains why C2 acetylcarnitine rises during ketogenic stress.
downstream:
- target: Dicarboxylic aciduria
description: Acute organic acid profiles commonly show dicarboxylic aciduria.
causal_link_type: DIRECT
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Dicarboxylic acid levels were elevated in 54/56 cases."
explanation: The systematic review supports dicarboxylic aciduria as part of the acute organic-acid pattern.
- target: Ketonuria
description: Some acute episodes show partial or marked urinary ketones despite the underlying ketogenesis defect.
causal_link_type: INDIRECT_UNKNOWN_INTERMEDIATES
evidence:
- reference: PMID:32905056
reference_title: "Hypoglycemia is not a defining feature of metabolic crisis in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: Further evidence of specific biochemical markers which may aid diagnosis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "He had marked ketonuria at the time of presentation"
explanation: This HMGCS2D crisis case demonstrates that urinary ketones can be present during acute presentation.
- target: 4-hydroxy-6-methyl-2-pyrone
description: Urinary 4HMP is a frequent acute-phase readout of the HMGCS2 deficiency organic-acid pattern.
causal_link_type: DIRECT
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "4-hydroxy-6-methyl-2-pyrone (4HMP) was detected in 33/35 urine samples taken"
explanation: The systematic review supports 4HMP as a frequent acute-phase biochemical readout.
- target: Dicarboxylic acids
description: Elevated dicarboxylic acids are the biochemical readout corresponding to dicarboxylic aciduria during acute crises.
causal_link_type: DIRECT
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Dicarboxylic acid levels were elevated in 54/56 cases."
explanation: The systematic review quantifies elevated dicarboxylic acids during acute episodes.
- target: Plasma C2/C0 acylcarnitine ratio
description: The plasma C2/C0 acylcarnitine ratio reports the acute acetylcarnitine pattern caused by blocked ketogenesis.
causal_link_type: DIRECT
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The plasma C2/C0 acylcarnitine ratio was abnormal in 16/18 (88.9 %) of acute plasma samples"
explanation: The systematic review supports increased C2/C0 ratio as an acute-phase biochemical readout.
phenotypes:
- name: Hypoglycemia
frequency: VERY_FREQUENT
description: >
Hypoglycemia is a cardinal acute finding, often with inadequate ketone
production during fasting or illness.
phenotype_term:
preferred_term: Hypoglycemia
term:
id: HP:0001943
label: Hypoglycemia
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "HP:0001943 | Hypoglycemia | Very frequent (99-80%)"
explanation: Orphanet records hypoglycemia as very frequent.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "decompensation with hypoglycemia, dicarboxyluria and inadequate ketonuria."
explanation: The systematic review describes hypoglycemia with inadequate ketonuria during acute crises.
- name: Hypoketotic hypoglycemia
frequency: VERY_FREQUENT
description: >
Inappropriately low ketone production during hypoglycemia distinguishes
HMGCS2 deficiency from many other causes of fasting intolerance.
phenotype_term:
preferred_term: Hypoketotic hypoglycemia
term:
id: HP:0001985
label: Hypoketotic hypoglycemia
evidence:
- reference: PMID:11228257
reference_title: "Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: clinical course and description of causal mutations in two patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "episodes of severe hypoketotic hypoglycemia."
explanation: Early clinical follow-up identifies severe hypoketotic hypoglycemia as a defining feature.
- reference: PMID:39143735
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency: A Cyclic Vomiting Mimic Without Reliable Biochemical Markers."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "individuals typically present at a young age with hypoketotic hypoglycemia,"
explanation: Recent case report summary supports hypoketotic hypoglycemia as typical.
- name: Abnormal metabolism or homeostasis
frequency: VERY_FREQUENT
description: >
Acute crises reflect abnormal systemic energy metabolism and homeostasis
caused by impaired ketogenesis.
phenotype_term:
preferred_term: Abnormality of metabolism/homeostasis
term:
id: HP:0001939
label: Abnormality of metabolism/homeostasis
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "HP:0001939 | Abnormality of metabolism/homeostasis | Very frequent (99-80%)"
explanation: Orphanet records abnormal metabolism/homeostasis as very frequent.
- name: Dicarboxylic aciduria
frequency: VERY_FREQUENT
description: >
Dicarboxylic aciduria is a common acute-phase biochemical phenotype.
phenotype_term:
preferred_term: Dicarboxylic aciduria
term:
id: HP:0003215
label: Dicarboxylic aciduria
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Dicarboxylic acid levels were elevated in 54/56 cases."
explanation: The systematic review supports a very frequent acute-phase frequency.
- name: Metabolic acidosis
frequency: VERY_FREQUENT
description: >
Severe metabolic acidosis is commonly reported during decompensation.
phenotype_term:
preferred_term: Metabolic acidosis
term:
id: HP:0001942
label: Metabolic acidosis
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "metabolic acidosis, and hypofibrinogenemia."
explanation: All ten patients in this case series had severe metabolic acidosis during crisis.
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Hepatomegaly and severe metabolic acidosis were observed in all cases."
explanation: The Japanese case series supports metabolic acidosis as a common crisis finding.
- name: Seizure
frequency: OCCASIONAL
description: >
Seizures occur during a subset of severe metabolic crises; recent cohort
data suggest a lower frequency than the older Orphanet estimate.
phenotype_term:
preferred_term: Seizure
term:
id: HP:0001250
label: Seizure
evidence:
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Clinical manifestations during the first episode were lethargy/coma (81.3%), rapid breathing (68.8%), hepatomegaly (56.3%), shock (37.5%), and seizures (18.8%)."
explanation: Seizures occurred in 18.8% of symptomatic first episodes in the Vietnamese cohort, which falls in the OCCASIONAL band.
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "followed by vomiting (8/10), fever (7/10), cough (4/10), diarrhea, and seizures (3/10)."
explanation: >
In the Chinese cohort, seizures occurred in 3/10 patients. Combined with
the Vietnamese symptomatic cohort rate of 18.8% (3/16), the two recent
cohorts total 6/26 patients with seizures, which falls in the OCCASIONAL
band and is clearly lower than VERY_FREQUENT.
- name: Hepatomegaly
description: >
Hepatomegaly is observed during acute metabolic decompensation and may
accompany fatty liver or elevated liver enzymes.
phenotype_term:
preferred_term: Hepatomegaly
term:
id: HP:0002240
label: Hepatomegaly
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "vomiting, lethargy, hepatomegaly, non ketotic hypoglycemia"
explanation: Orphanet lists hepatomegaly among characteristic decompensation features.
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Hepatomegaly and severe metabolic acidosis were observed in all cases."
explanation: The Japanese case series documents hepatomegaly in all four reported patients.
- name: Hepatic steatosis
frequency: OCCASIONAL
description: >
Fatty liver is reported during acute decompensation, consistent with
impaired hepatic ketogenesis and altered fatty-acid handling.
phenotype_term:
preferred_term: Hepatic steatosis
term:
id: HP:0001397
label: Hepatic steatosis
evidence:
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "liver was identified in three cases, which suggested the unavailability of fatty acids."
explanation: The Japanese case series identified fatty liver in three of four patients.
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "hepatomegaly or fatty liver with elevated liver enzymes"
explanation: The full-text summary lists fatty liver among acute episode liver findings.
- name: Elevated hepatic transaminase
frequency: VERY_FREQUENT
description: >
Elevated hepatic transaminases accompany acute metabolic crises in reported
patients.
phenotype_term:
preferred_term: Elevated circulating hepatic transaminase concentration
term:
id: HP:0002910
label: Elevated circulating hepatic transaminase concentration
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "different degree of hepatomegaly and increased aminotransferase, severe"
explanation: The Chinese case series reports increased aminotransferase in all ten patients.
- name: Vomiting
description: >
Vomiting is a common decompensation symptom and may lead to misdiagnosis as
cyclic vomiting syndrome.
phenotype_term:
preferred_term: Vomiting
term:
id: HP:0002013
label: Vomiting
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "that present with vomiting, lethargy, hepatomegaly"
explanation: Orphanet lists vomiting among characteristic decompensation features.
- reference: PMID:39143735
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency: A Cyclic Vomiting Mimic Without Reliable Biochemical Markers."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "episodes of vomiting and lethargy, often associated with hypoglycemia or"
explanation: This case report broadens the presentation to cyclic-vomiting-like episodes.
- name: Diarrhea
description: >
Diarrhea can occur during first acute HMGCS2D presentations in the context
of gastroenteritis-like metabolic decompensation.
phenotype_term:
preferred_term: Diarrhea
term:
id: HP:0002014
label: Diarrhea
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "followed by vomiting (8/10), fever (7/10), cough (4/10), diarrhea, and seizures (3/10)."
explanation: The Chinese cohort lists diarrhea among initial clinical symptoms.
- name: Steatorrhea
description: >
Steatorrhea has been reported during acute HMGCS2D episodes, expanding the
phenotype beyond the classic hypoketotic-hypoglycemia presentation.
phenotype_term:
preferred_term: Steatorrhea
term:
id: HP:0002570
label: Steatorrhea
evidence:
- reference: PMID:33045405
reference_title: "Expanding phenotypic and mutational spectra of mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "During acute episodes, steatorrhea and dyslipidemia occurred"
explanation: The Thai patient report documents steatorrhea during acute episodes.
- name: Lethargy
description: >
Lethargy occurs during acute crises and may progress to altered
consciousness or encephalopathy.
phenotype_term:
preferred_term: Lethargy
term:
id: HP:0001254
label: Lethargy
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "that present with vomiting, lethargy, hepatomegaly"
explanation: Orphanet lists lethargy among characteristic decompensation features.
- reference: PMID:39143735
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency: A Cyclic Vomiting Mimic Without Reliable Biochemical Markers."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "episodes of vomiting and lethargy, often associated with hypoglycemia or"
explanation: The case report documents recurrent lethargy with vomiting and glycemic abnormalities.
- name: Encephalopathy
description: >
Encephalopathy and altered consciousness can occur during severe episodes.
phenotype_term:
preferred_term: Encephalopathy
term:
id: HP:0001298
label: Encephalopathy
evidence:
- reference: PMID:40548098
reference_title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "hypoketotic hypoglycemia, metabolic acidosis, hepatomegaly, and encephalopathy"
explanation: The 2025 case report states encephalopathy as part of the characteristic phenotype.
- name: Abnormal brain MRI findings
description: >
Brain MRI abnormalities were detected in a minority of patients in the
Vietnamese cohort, although the abstract does not specify the imaging
pattern.
phenotype_term:
preferred_term: Abnormal brain MRI findings
term:
id: HP:0012443
label: Abnormal brain morphology
evidence:
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Abnormal brain MRI findings were detected in three patients."
explanation: The Vietnamese cohort supports brain MRI abnormalities as a reported neurological imaging finding.
- name: Coma
description: >
Coma is a severe but uncommon crisis manifestation, including reported
neonatal hyperammonemic coma.
phenotype_term:
preferred_term: Coma
term:
id: HP:0001259
label: Coma
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "and, in rare cases, coma."
explanation: Orphanet records coma as a rare severe manifestation.
- reference: PMID:40548098
reference_title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "on day of life 7 with a sepsis-like condition, coma, metabolic acidosis, and"
explanation: A neonatal case presented with coma during metabolic decompensation.
- name: Hypoglycemic coma
description: >
Severe fasting-triggered crises can produce hypoglycemic hypoketotic coma.
phenotype_term:
preferred_term: Hypoglycemic coma
term:
id: HP:0001325
label: Hypoglycemic coma
evidence:
- reference: PMID:11479731
reference_title: "Genetic basis of mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "children with hypoglycaemic hypoketotic coma during fasting periods"
explanation: The report describes fasting-associated hypoglycemic hypoketotic coma in affected children.
- name: Hyperammonemia
frequency: OCCASIONAL
description: >
Hyperammonemia can occur during severe acute crises, including neonatal
hyperammonemic coma, but is not part of every presentation.
phenotype_term:
preferred_term: Hyperammonemia
term:
id: HP:0001987
label: Hyperammonemia
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "five patients had hyperammonemia, four patients had hyperuricemia"
explanation: The Chinese series reported hyperammonemia in five of ten patients.
- reference: PMID:40548098
reference_title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "severe hyperammonemia is"
explanation: A neonatal case report emphasizes that severe hyperammonemia can occur.
- name: Hypophosphatemia
description: >
Hypophosphatemia has been reported as hypophosphatemic encephalopathy
during acute HMGCS2D presentation.
phenotype_term:
preferred_term: Hypophosphatemia
term:
id: HP:0002148
label: Hypophosphatemia
evidence:
- reference: PMID:33045405
reference_title: "Expanding phenotypic and mutational spectra of mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Both patients had hypophosphatemic encephalopathy"
explanation: The Thai patient report documents hypophosphatemic encephalopathy in both affected children.
- name: Dyspnea
description: >
Dyspnea can occur during the first clinical episode, often alongside
metabolic acidosis and altered consciousness.
phenotype_term:
preferred_term: Dyspnea
term:
id: HP:0002094
label: Dyspnea
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "consciousness, dyspnea, seizures and hepatomegaly."
explanation: The systematic review lists dyspnea among common first-episode symptoms.
- name: Tachypnea
frequency: FREQUENT
description: >
Rapid breathing is a common first-episode sign and likely reflects
respiratory compensation for metabolic acidosis.
phenotype_term:
preferred_term: Tachypnea
term:
id: HP:0002789
label: Tachypnea
evidence:
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Clinical manifestations during the first episode were lethargy/coma (81.3%), rapid breathing (68.8%), hepatomegaly (56.3%), shock (37.5%), and seizures (18.8%)."
explanation: Rapid breathing occurred in 68.8% of symptomatic first episodes in the Vietnamese cohort, which falls in the FREQUENT band.
- name: Shock
frequency: FREQUENT
description: >
Shock can occur during severe first metabolic crises, particularly when
acidosis and systemic energy failure are profound.
phenotype_term:
preferred_term: Shock
term:
id: HP:0031273
label: Shock
evidence:
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Clinical manifestations during the first episode were lethargy/coma (81.3%), rapid breathing (68.8%), hepatomegaly (56.3%), shock (37.5%), and seizures (18.8%)."
explanation: Shock occurred in 37.5% of symptomatic first episodes in this Vietnamese cohort, which falls in the FREQUENT band; cohort-specific frequency may not generalize to all HMGCS2D patients.
- name: Hypofibrinogenemia
description: >
Hypofibrinogenemia has been reported during acute crises in a Chinese
patient series, consistent with transient severe systemic metabolic stress.
phenotype_term:
preferred_term: Hypofibrinogenemia
term:
id: HP:0011900
label: Hypofibrinogenemia
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Each patient (10/10) had a different degree of hepatomegaly and increased aminotransferase, severe metabolic acidosis, and hypofibrinogenemia."
explanation: The Chinese case series reported hypofibrinogenemia during acute metabolic crisis; no whole-disease frequency is assigned because this is a small severe cohort.
- name: Hypertriglyceridemia
frequency: FREQUENT
description: >
Hypertriglyceridemia may appear during acute presentations, reflecting
altered lipid handling when hepatic ketogenesis is blocked.
phenotype_term:
preferred_term: Hypertriglyceridemia
term:
id: HP:0002155
label: Hypertriglyceridemia
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Five patients had hypocalcemia, five patients had hyperammonemia, four patients had hyperuricemia, and three had hypertriglyceridemia."
explanation: Three of ten patients in the Chinese cohort had hypertriglyceridemia, which is 30% and falls in the FREQUENT band; the estimate is cohort-specific.
- reference: PMID:32905056
reference_title: "Hypoglycemia is not a defining feature of metabolic crisis in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: Further evidence of specific biochemical markers which may aid diagnosis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "the sample was markedly lipemic, with significant hypertriglyceridemia detected."
explanation: A normoglycemic crisis case independently supports hypertriglyceridemia during acute HMGCS2D presentation.
- name: Ketonuria
description: >
Although inadequate ketone production is typical, some HMGCS2D crises show
partial or marked ketonuria; urinary ketones therefore do not exclude a
ketogenesis disorder.
phenotype_term:
preferred_term: Ketonuria
term:
id: HP:0002919
label: Ketonuria
evidence:
- reference: PMID:32905056
reference_title: "Hypoglycemia is not a defining feature of metabolic crisis in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: Further evidence of specific biochemical markers which may aid diagnosis."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "He had marked ketonuria at the time of presentation"
explanation: This case report documents marked ketonuria during an HMGCS2D metabolic crisis.
biochemical:
- name: Ketone bodies
presence: DECREASED
context: >
Ketone production is inappropriately low during hypoglycemia and catabolic
stress.
readouts:
- target: Impaired Hepatic Ketogenesis
relationship: READOUT_OF
direction: NEGATIVE
endpoint_context: DIAGNOSTIC
interpretation: Inadequate ketone body production reports the hepatic ketogenesis block during fasting or illness.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "decompensation with hypoglycemia, dicarboxyluria and inadequate ketonuria."
explanation: Inadequate ketonuria directly supports decreased ketone bodies as a readout of impaired ketogenesis.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "decompensation with hypoglycemia, dicarboxyluria and inadequate ketonuria."
explanation: Inadequate ketonuria supports decreased ketone body production.
- name: 4-hydroxy-6-methyl-2-pyrone
presence: INCREASED
context: >
Urinary 4HMP is a useful acute-phase diagnostic marker, although it may be
detected retrospectively.
readouts:
- target: Acute-Phase Acetylcarnitine and Organic Acid Pattern
relationship: READOUT_OF
direction: POSITIVE
endpoint_context: DIAGNOSTIC
interpretation: Urinary 4HMP is an acute-phase biochemical marker that reports the HMGCS2 deficiency organic-acid pattern.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "4-hydroxy-6-methyl-2-pyrone (4HMP) was detected in 33/35 urine samples taken"
explanation: The systematic review supports 4HMP as a frequent acute-phase readout.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "4-hydroxy-6-methyl-2-pyrone (4HMP) was detected in 33/35 urine samples taken"
explanation: The systematic review supports increased urinary 4HMP during acute episodes.
- name: Dicarboxylic acids
presence: INCREASED
context: >
Urinary dicarboxylic acid levels are commonly elevated during metabolic
decompensation.
readouts:
- target: Acute-Phase Acetylcarnitine and Organic Acid Pattern
relationship: READOUT_OF
direction: POSITIVE
endpoint_context: DIAGNOSTIC
interpretation: Dicarboxylic aciduria reports the acute organic-acid pattern seen during HMGCS2 decompensation.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Dicarboxylic acid levels were elevated in 54/56 cases."
explanation: The systematic review quantifies dicarboxylic acid elevation during acute episodes.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Dicarboxylic acid levels were elevated in 54/56 cases."
explanation: The systematic review quantifies elevated dicarboxylic acids.
- name: Plasma C2/C0 acylcarnitine ratio
presence: INCREASED
context: >
An increased plasma acetylcarnitine to free carnitine ratio can help
identify HMGCS2 deficiency during acute decompensation.
readouts:
- target: Acute-Phase Acetylcarnitine and Organic Acid Pattern
relationship: READOUT_OF
direction: POSITIVE
endpoint_context: DIAGNOSTIC
interpretation: An increased plasma C2/C0 ratio reports acetylcarnitine accumulation during the acute-phase HMGCS2 biochemical pattern.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "increased plasma C2/C0 acylcarnitine ratio to facilitate the"
explanation: The systematic review recommends the C2/C0 ratio as a diagnostic readout.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "increased plasma C2/C0 acylcarnitine ratio to facilitate the"
explanation: The systematic review recommends C2/C0 ratio as an acute-phase diagnostic clue.
genetic:
- name: HMGCS2 pathogenic variants
gene_term:
preferred_term: HMGCS2
term:
id: hgnc:5008
label: HMGCS2
association: Causative biallelic pathogenic variants
relationship_type: CAUSATIVE
variant_origin: GERMLINE
inheritance:
- name: Autosomal recessive inheritance
inheritance_term:
preferred_term: Autosomal recessive inheritance
term:
id: HP:0000007
label: Autosomal recessive inheritance
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "Autosomal recessive"
explanation: Orphanet records autosomal recessive inheritance.
variants:
- name: Biallelic HMGCS2 pathogenic variants
description: >
Reported pathogenic HMGCS2 variants include missense, splice, truncating,
and deletion alleles. Functional assays show that selected missense
variants have absent, negligible, or reduced HMGCS2 enzyme activity.
Biallelic truncating variants may indicate more severe disease in some
cohorts, but a larger review did not establish a definitive
genotype-phenotype correlation.
gene:
preferred_term: HMGCS2
term:
id: hgnc:5008
label: HMGCS2
clinical_significance: PATHOGENIC
type: loss_of_function_variant
functional_effects:
- function: hydroxymethylglutaryl-CoA synthase activity
description: Patient variants reduce or abolish mitochondrial HMG-CoA synthase enzymatic activity.
type: loss-of-function
evidence:
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Molecular analysis identified 15 variants in the HMGCS2 gene, of which 10 were"
explanation: The Chinese case series expands the HMGCS2 variant spectrum.
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: IN_VITRO
snippet: "In conclusion, in vitro analysis has shown that the p.G219E,"
explanation: In vitro analysis supports pathogenicity of multiple patient HMGCS2 variants.
- reference: PMID:35308163
reference_title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: "We found that patients with biallelic truncation mutation appeared to show a more severe clinical condition through a literature review."
explanation: The Chinese cohort review suggests greater severity with biallelic truncating variants, but this remains qualified by broader review evidence.
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: PARTIAL
evidence_source: OTHER
snippet: "HMGCS2 | 3-hydroxy-3-methylglutaryl-CoA synthase 2 | hgnc:5008 | Disease-causing germline mutation(s) (loss of function) in"
explanation: Orphanet records HMGCS2 as the disease-causing germline gene.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "mutations in the HMGCS2 gene, leading to impaired ketogenesis."
explanation: The systematic review supports HMGCS2 mutations as causative.
- reference: CGGV:assertion_b3f49254-4635-4961-83b9-31c7ebc7f159-2018-05-22T160000.000Z
reference_title: "HMGCS2 / 3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Definitive)"
supports: SUPPORT
evidence_source: OTHER
snippet: "HMGCS2 | HGNC:5008 | 3-hydroxy-3-methylglutaryl-CoA synthase deficiency | MONDO:0011614 | AR | Definitive"
explanation: ClinGen classifies the HMGCS2-3-hydroxy-3-methylglutaryl-CoA synthase deficiency gene-disease relationship as definitive with autosomal recessive inheritance.
treatments:
- name: Fasting avoidance and sick-day carbohydrate support
description: >
Avoiding prolonged fasting and giving rapid carbohydrate support during
poor intake or intercurrent illness reduces risk of recurrent hypoglycemic
metabolic decompensation.
treatment_term:
preferred_term: dietary intervention
term:
id: MAXO:0000088
label: dietary intervention
target_mechanisms:
- target: Catabolic Stress Metabolic Decompensation
treatment_effect: INHIBITS
description: Avoiding fasting reduces the catabolic state that unmasks ketogenesis failure.
evidence:
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "each patient was advised to avoid prolonged fasting"
explanation: Patients were advised to avoid fasting after metabolic crises.
evidence:
- reference: PMID:40548098
reference_title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "fasting avoidance with or without l-carnitine during intercurrent illness should"
explanation: The 2025 case report recommends preemptive fasting avoidance during illness.
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "preventing prolonged fasting and providing enteral carbohydrate/glucose infusion during illness"
explanation: The Vietnamese cohort supports fasting avoidance and illness-period enteral carbohydrate/glucose as relapse-prevention strategies.
- name: Intravenous glucose during acute illness
description: >
Intravenous glucose is used during acute decompensation or anorexia to
correct hypoglycemia and suppress catabolism.
treatment_term:
preferred_term: Pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
therapeutic_agent:
- preferred_term: glucose
term:
id: CHEBI:17234
label: glucose
target_mechanisms:
- target: Catabolic Stress Metabolic Decompensation
treatment_effect: INHIBITS
description: Glucose support supplies carbohydrate and reduces reliance on failed ketone body synthesis.
evidence:
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Hypoglycemia was immediately corrected by glucose infusion."
explanation: Acute case management corrected hypoglycemia with glucose infusion.
evidence:
- reference: PMID:32952630
reference_title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "to receive glucose infusion prophylactically during anorexia"
explanation: Patients were advised to receive prophylactic glucose infusion during anorexia.
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "The implementation of a high glucose infusion and proactive management strategies-such as preventing prolonged fasting and providing enteral carbohydrate/glucose infusion during illness-effectively reduced the rate of acute relapses following accurate diagnosis."
explanation: Cohort follow-up supports high-glucose infusion as part of relapse-reducing proactive management after diagnosis.
- name: Dietary fat moderation
description: >
Moderating dietary fat intake has been used as a long-term dietary
intervention in reported HMGCS-deficiency patients, alongside fasting
avoidance and sick-day carbohydrate planning.
treatment_term:
preferred_term: dietary intervention
term:
id: MAXO:0000088
label: dietary intervention
target_mechanisms:
- target: Catabolic Stress Metabolic Decompensation
treatment_effect: MODULATES
description: Dietary fat moderation is a supportive metabolic strategy reported in long-term follow-up of ketogenesis disorders.
evidence:
- reference: PMID:38567177
reference_title: "Inborn Errors of Ketogenesis: Novel Variants, Clinical Presentation, and Follow-Up in a Series of Four Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Diet with moderation of fat intake was followed in two individuals with HMGCS deficiency."
explanation: Two individuals with HMGCS deficiency in this ketogenesis-disorder series followed dietary fat moderation.
evidence:
- reference: PMID:38567177
reference_title: "Inborn Errors of Ketogenesis: Novel Variants, Clinical Presentation, and Follow-Up in a Series of Four Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Diet with moderation of fat intake was followed in two individuals with HMGCS deficiency."
explanation: This directly supports dietary fat moderation as a reported long-term management strategy for HMGCS deficiency.
- name: Carnitine supplementation during illness
description: >
L-carnitine may be used during intercurrent illness, especially in patients
with abnormal acylcarnitine handling, although evidence is based on case
reports rather than controlled trials.
treatment_term:
preferred_term: carnitine supplementation
term:
id: MAXO:0010006
label: carnitine supplementation
therapeutic_agent:
- preferred_term: carnitine
term:
id: CHEBI:17126
label: carnitine
target_mechanisms:
- target: Acute-Phase Acetylcarnitine and Organic Acid Pattern
treatment_effect: MODULATES
description: Carnitine supplementation may support acylcarnitine handling during illness.
evidence:
- reference: PMID:40548098
reference_title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "fasting avoidance with or without l-carnitine during intercurrent illness should"
explanation: The case report recommends fasting avoidance with or without L-carnitine during illness.
evidence:
- reference: PMID:40548098
reference_title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "fasting avoidance with or without l-carnitine during intercurrent illness should"
explanation: This supports carnitine as an optional illness-period intervention.
- name: Genetic counseling
description: >
Genetic counseling is indicated for families because the disorder is caused
by autosomal recessive germline HMGCS2 pathogenic variants.
treatment_term:
preferred_term: Genetic Counseling
term:
id: NCIT:C15240
label: Genetic Counseling
target_mechanisms:
- target: HMGCS2 Loss of Function
treatment_effect: MODULATES
description: Counseling addresses the autosomal recessive HMGCS2 cause and recurrence risk rather than directly changing metabolism.
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "Autosomal recessive"
explanation: Autosomal recessive inheritance supports genetics-informed counseling and recurrence-risk management.
evidence:
- reference: ORPHA:35701
reference_title: "3-hydroxy-3-methylglutaryl-CoA synthase deficiency (Orphanet structured-database record)"
supports: SUPPORT
evidence_source: OTHER
snippet: "Autosomal recessive"
explanation: Autosomal recessive inheritance supports genetic counseling and recurrence-risk counseling.
diagnosis:
- name: HMGCS2 molecular testing
diagnosis_term:
preferred_term: genetic testing
term:
id: MAXO:0000127
label: genetic testing
description: >
Sequencing or gene-panel testing for biallelic HMGCS2 pathogenic variants
confirms the diagnosis, particularly when enzyme testing is difficult and
routine biochemical markers are unreliable.
results: Biallelic pathogenic HMGCS2 variants support diagnosis.
evidence:
- reference: PMID:11479731
reference_title: "Genetic basis of mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Molecular studies may facilitate or confirm future diagnoses"
explanation: Molecular studies are recommended to facilitate or confirm diagnosis.
- reference: PMID:39143735
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency: A Cyclic Vomiting Mimic Without Reliable Biochemical Markers."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "highlights the importance of molecular genetic testing in"
explanation: Molecular testing was important in a cyclic-vomiting-like presentation without specific metabolic markers.
- reference: PMID:40004108
reference_title: "Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Due to the absence of reliable biochemical markers, genetic testing has become the definitive method for diagnosing HMGCS2D."
explanation: The Vietnamese cohort explicitly supports genetic testing as the definitive diagnostic method when biochemical markers are unreliable.
- name: Acute-phase biochemical testing
diagnosis_term:
preferred_term: disease screening
term:
id: MAXO:0000124
label: disease screening
description: >
Urinary organic acid analysis for 4HMP and dicarboxylic acids, plus plasma
C2/C0 acylcarnitine ratio during acute decompensation, helps distinguish
HMGCS2 deficiency from fatty acid oxidation disorders.
results: 4HMP, dicarboxylic aciduria, and elevated C2/C0 ratio during crisis support diagnosis.
evidence:
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "Laboratories should look for 4HMP in urinary organic acid"
explanation: The systematic review recommends targeted 4HMP testing in urine organic acids.
- reference: PMID:39798988
reference_title: "Mitochondrial HMG-CoA synthase deficiency."
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: "increased plasma C2/C0 acylcarnitine ratio to facilitate the"
explanation: The review recommends plasma C2/C0 ratio to facilitate diagnosis.
references:
- reference: ORPHA:35701
title: 3-hydroxy-3-methylglutaryl-CoA synthase deficiency
- reference: PMID:11228257
title: "Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: clinical course and description of causal mutations in two patients."
- reference: PMID:11479731
title: Genetic basis of mitochondrial HMG-CoA synthase deficiency.
- reference: PMID:32952630
title: "Japanese patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: In vitro functional analysis of five novel HMGCS2 mutations."
- reference: PMID:32905056
title: "Hypoglycemia is not a defining feature of metabolic crisis in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: Further evidence of specific biochemical markers which may aid diagnosis."
- reference: PMID:33045405
title: Expanding phenotypic and mutational spectra of mitochondrial HMG-CoA synthase deficiency.
- reference: PMID:35308163
title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients."
- reference: PMID:38567177
title: "Inborn Errors of Ketogenesis: Novel Variants, Clinical Presentation, and Follow-Up in a Series of Four Patients."
- reference: PMID:39143735
title: "Mitochondrial HMG-CoA Synthase Deficiency: A Cyclic Vomiting Mimic Without Reliable Biochemical Markers."
- reference: PMID:39798988
title: Mitochondrial HMG-CoA synthase deficiency.
- reference: PMID:40004108
title: Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients.
- reference: PMID:40548098
title: "HMG-CoA Synthase-2 Deficiency: Neonatal Hyperammonemic Coma and Abnormal Metabolic Screening Resembling Maple Syrup Urine Disease."
- reference: DOI:10.3390/ijms26041644
title: Mitochondrial HMG-CoA Synthase Deficiency in Vietnamese Patients
found_in:
- 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiency-deep-research-falcon.md
findings: []
- reference: DOI:10.1002/jmd2.12146
title: "Hypoglycemia is not a defining feature of metabolic crisis in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: Further evidence of specific biochemical markers which may aid diagnosis"
found_in:
- 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiency-deep-research-falcon.md
findings: []
- reference: DOI:10.3390/biom15040580
title: "Not Just an Alternative Energy Source: Diverse Biological Functions of Ketone Bodies and Relevance of HMGCS2 to Health and Disease"
found_in:
- 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiency-deep-research-falcon.md
findings: []
- reference: DOI:10.3389/fgene.2021.816779
title: "Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients"
found_in:
- 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiency-deep-research-falcon.md
findings: []
- reference: DOI:10.1177/03000605251375537
title: "Mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 deficiency with severe hyperglycemia in a child: A rare case report"
found_in:
- 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiency-deep-research-falcon.md
findings: []
- reference: DOI:10.1002/ajmg.a.61590
title: "Expanding the clinical spectrum of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency with Turkish cases harboring novel HMGCS2 gene mutations and literature review"
found_in:
- 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiency-deep-research-falcon.md
findings: []
- reference: DOI:10.1055/s-0042-1749362
title: "Inborn Errors of Ketogenesis: Novel Variants, Clinical Presentation, and Follow-Up in a Series of Four Patients"
found_in:
- 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiency-deep-research-falcon.md
findings: []
References & Deep Research
References
19Deep Research
21. Disease Information
1.1 Overview / definition
Mitochondrial 3‑hydroxy‑3‑methylglutaryl‑CoA synthase deficiency (HMGCS2D) is a rare inborn error of ketone body synthesis (ketogenesis) caused by loss of function of mitochondrial HMG‑CoA synthase (encoded by HMGCS2), leading to inability to appropriately generate ketone bodies during fasting or illness and resulting in episodic metabolic decompensation. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, conlon2020hypoglycemiaisnot pages 1-3, suresh2025notjustan pages 22-23)
Direct abstract quote (2025 Vietnamese case series): “Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency (HMGCS2D) is a rare metabolic disorder that impairs the body’s ability to produce ketone bodies and regulate energy metabolism.” (Nguyen et al., 2025; https://doi.org/10.3390/ijms26041644; published Feb 2025) (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
1.2 Key identifiers
- OMIM (disease): 605911 (reported explicitly in multiple sources) (nguyen2025mitochondrialhmgcoasynthase pages 1-2, conlon2020hypoglycemiaisnot pages 1-3)
- OMIM (gene HMGCS2): 600234 (nguyen2025mitochondrialhmgcoasynthase pages 2-3)
- Orphanet / ICD‑10 / ICD‑11 / MeSH / MONDO: Not identified in the retrieved full‑text evidence in this run; therefore not reported here.
1.3 Synonyms / alternative names
Common names in the literature include: - “Mitochondrial HMG‑CoA synthase deficiency” - “mHS deficiency” (mitochondrial HMG‑CoA synthase deficiency) - “HMGCS2 deficiency” - “Mitochondrial 3‑hydroxy‑3‑methylglutaryl‑CoA synthase deficiency” (conlon2020hypoglycemiaisnot pages 1-3, suresh2025notjustan pages 22-23)
1.4 Evidence source type
Evidence in this report is primarily derived from aggregated disease-level clinical resources in peer‑reviewed case series and case reports (human clinical evidence), plus mechanistic interpretation from reviews. The quantitative phenotype and laboratory frequencies below come mainly from two retrospective patient series: a Vietnamese cohort (n=19) and a Chinese cohort (n=10). (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3)
2. Etiology
2.1 Disease causal factors
Genetic cause (primary): biallelic pathogenic variants in HMGCS2 cause autosomal recessive mitochondrial HMG‑CoA synthase deficiency. (conlon2020hypoglycemiaisnot pages 1-3, suresh2025notjustan pages 22-23, dong2025mitochondrial3hydroxy3methylglutarylcoenzymea pages 1-3)
Direct abstract quote (2020 JIMD Reports): “Mitochondrial 3‐hydroxy‐3‐methylglutaryl‐CoA (HMG Co‐A) synthase (mHS) deficiency is an autosomal recessive disorder of ketone body synthesis…” (Conlon et al., 2020; https://doi.org/10.1002/jmd2.12146; published Jun 2020) (conlon2020hypoglycemiaisnot pages 1-3)
Mechanistic cause: deficiency of the mitochondrial ketogenesis enzyme leads to inadequate ketone availability under catabolic stress and downstream biochemical derangements (see Section 6). (suresh2025notjustan pages 22-23, kılıc2020expandingtheclinical pages 6-7)
2.2 Risk factors
- Genetic: having biallelic loss‑of‑function (especially truncating) variants in HMGCS2 increases risk of severe presentations; aggregated analysis suggests severity correlates with the number of truncating alleles. (wu2022clinicalbiochemicalmolecular pages 8-9, wu2022clinicalbiochemicalmolecular media 0a979ec7)
- Environmental/physiologic triggers (for crises rather than disease occurrence): fasting/prolonged reduced intake and intercurrent infections/illness are common crisis precipitants. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, conlon2020hypoglycemiaisnot pages 1-3, suresh2025notjustan pages 22-23)
In the Vietnamese series, the most common triggers for the initial crisis were poor feeding/fasting (93.8%), vomiting (56.3%), diarrhea (25.0%), and fever (18.8%). (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
2.3 Protective factors
No validated genetic protective variants or environmental protective exposures were identified in the retrieved evidence. Clinically, prevention of prolonged fasting and proactive carbohydrate administration during illness function as protective management strategies against acute decompensation (Section 12/13), but these are not “protective factors” for disease occurrence. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
2.4 Gene–environment interaction
The dominant gene–environment interaction is that HMGCS2 loss of function may be clinically silent until a catabolic state (fasting/illness) increases reliance on ketogenesis, precipitating crisis. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, suresh2025notjustan pages 22-23)
3. Phenotypes
3.1 Core phenotype pattern
HMGCS2 deficiency typically manifests as episodic metabolic decompensation in infancy/early childhood, often with encephalopathy (lethargy, coma), respiratory compensation for acidosis, and hepatic involvement (hepatomegaly, transaminitis). (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3)
A key clinical point is that hypoglycemia is common but not universal; cases with normal glucose at presentation are documented. (conlon2020hypoglycemiaisnot pages 1-3, wu2022clinicalbiochemicalmolecular pages 1-2)
3.2 Age of onset, severity, and course
- Onset: typically within the first year; Chinese series first crisis at 5–12 months. (wu2022clinicalbiochemicalmolecular pages 2-3)
- Range: Vietnamese series first acute episode at 10 days to 28 months. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Course: episodic; crises precipitated by fasting/illness; many laboratory abnormalities normalize between crises. (wu2022clinicalbiochemicalmolecular pages 1-2)
3.3 Symptom/sign frequencies (recent patient series)
Vietnamese cohort (n=16 symptomatic of 19 total): - Lethargy/coma 81.3% - Rapid breathing 68.8% - Hepatomegaly 56.3% - Shock 37.5% - Seizures 18.8% (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
Chinese cohort (n=10): - Anorexia 10/10 - Dyspnea 10/10 - Disturbance of consciousness 10/10 - Vomiting 8/10 - Fever 7/10 - Cough 4/10 - Diarrhea 3/10 - Seizures 3/10 - Hepatomegaly 10/10 (wu2022clinicalbiochemicalmolecular pages 2-3, wu2022clinicalbiochemicalmolecular pages 1-2)
3.4 Laboratory abnormalities (phenotype‑linked)
Common acute‑episode laboratory findings include metabolic acidosis, hypoglycemia (variable), elevated aminotransferases, and sometimes hyperammonemia and hypertriglyceridemia. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 1-2)
Notably, Conlon et al. emphasize that hypoglycemia is not mandatory for crisis presentation. (conlon2020hypoglycemiaisnot pages 1-3)
3.5 Neuroimaging findings
In the Chinese series, brain MRI abnormalities were variably present, including widened sulci/subarachnoid spaces, basal ganglia signal abnormalities, and delayed myelination in subsets of imaged patients. (wu2022clinicalbiochemicalmolecular pages 2-3)
3.6 Suggested HPO terms (non-exhaustive)
Based on the reported clinical features and labs: - Hypoglycemia (HP:0001943) (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 1-2) - Metabolic acidosis (HP:0001942) (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3) - Encephalopathy / altered mental status (HP:0001298) (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3) - Lethargy (HP:0001254) / Coma (HP:0001259) (nguyen2025mitochondrialhmgcoasynthase pages 1-2) - Hepatomegaly (HP:0002240) (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3) - Elevated hepatic transaminases (HP:0002910) (nguyen2025mitochondrialhmgcoasynthase pages 1-2) - Hyperammonemia (HP:0001987) (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 1-2) - Seizures (HP:0001250) (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3) - Shock (HP:0030148) (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
(Exact HPO IDs are provided as standard ontology mappings; the clinical evidence for each term is as cited.)
4. Genetic / Molecular Information
4.1 Causal gene
- Gene: HMGCS2 (mitochondrial 3‑hydroxy‑3‑methylglutaryl‑CoA synthase)
- Disease mechanism: generally loss of function (LOF) of mitochondrial ketogenesis enzyme activity (nguyen2025mitochondrialhmgcoasynthase pages 2-3, suresh2025notjustan pages 22-23)
4.2 Pathogenic variants and variant classes
Across clinical series and reviews, pathogenic variants include missense, nonsense, frameshift, and splice‑site changes. (suresh2025notjustan pages 22-23, wu2022clinicalbiochemicalmolecular pages 1-2)
Examples from recent cohorts/case reports: - Vietnamese series identified a novel c.407A>T (p.D136V) and recurrent variants c.559+1G>A and c.1090T>A (p.F364I), each present in >50% of the 19 cases (57.9% and 55.5%, respectively). (nguyen2025mitochondrialhmgcoasynthase pages 1-2) - Chinese cohort identified 15 variants (10 novel) including missense, frameshift, nonsense, and splice variants; and highlighted c.1201G>T (p.E401*) as a possible hotspot in Chinese patients (6/40 mutated alleles, 15.0% in their combined dataset of Chinese patients). (wu2022clinicalbiochemicalmolecular pages 1-2)
4.3 Genotype–phenotype correlation (severity)
An aggregated analysis (Wu et al., combining their cases with literature) reported that severity correlates with truncating alleles; mortality was highest in the group with biallelic truncating variants (25% in their grouped analysis). (wu2022clinicalbiochemicalmolecular pages 8-9, wu2022clinicalbiochemicalmolecular media ec3f9f64)
4.4 Population genetics / epidemiology (limited)
- A Vietnamese series/review states an estimated incidence <1/1,000,000 (rare disease). (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Population‑wide prevalence and carrier frequency were not found in the retrieved evidence.
4.5 Somatic vs germline
All cited disease-causing variants are inherited germline variants in HMGCS2 causing an autosomal recessive metabolic disease. (suresh2025notjustan pages 22-23, dong2025mitochondrial3hydroxy3methylglutarylcoenzymea pages 1-3)
4.6 Modifier genes / epigenetics / chromosomal abnormalities
No validated modifier genes, epigenetic signatures, or chromosomal abnormalities specific to HMGCS2 deficiency were identified in the retrieved evidence.
5. Environmental Information
This is a Mendelian disorder; environmental factors primarily influence crisis occurrence rather than disease causation.
5.1 Key environmental/physiologic precipitants
- Fasting / reduced intake (“poor feeding”) is a major trigger. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Intercurrent illness (e.g., febrile infections, gastroenteritis) is a major trigger. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, conlon2020hypoglycemiaisnot pages 1-3)
5.2 Infectious triggers
Fever/illness is repeatedly reported as a precipitant of metabolic crises. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, conlon2020hypoglycemiaisnot pages 1-3)
6. Mechanism / Pathophysiology
6.1 Key pathway and biochemical defect
HMGCS2 is the mitochondrial HMG‑CoA synthase, a rate‑limiting ketogenesis enzyme. Loss-of-function variants block ketone body synthesis, particularly critical during fasting/illness when ketones act as alternative fuels. (suresh2025notjustan pages 22-23, kılıc2020expandingtheclinical pages 6-7)
6.2 Causal chain (clinical mechanism narrative)
- Trigger: fasting/reduced intake or intercurrent illness increases catabolic demand and lipolysis. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, conlon2020hypoglycemiaisnot pages 1-3)
- Primary defect: impaired mitochondrial ketogenesis due to HMGCS2 deficiency limits ketone production. (suresh2025notjustan pages 22-23)
- Biochemical consequences: inadequate ketone availability leads to metabolic crisis characterized by severe metabolic acidosis and “hypoketotic” patterns; urine organic acids may show dicarboxylic aciduria and proposed markers such as 4HMP and 3‑hydroxyglutarate in some cases. (conlon2020hypoglycemiaisnot pages 1-3, wu2022clinicalbiochemicalmolecular pages 1-2)
- Clinical manifestations: encephalopathy (lethargy/coma), respiratory compensation (tachypnea), hepatic stress (hepatomegaly/transaminitis), and in severe cases shock, seizures, and multi‑organ dysfunction. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3)
6.3 Proposed diagnostic biomarkers (expert interpretation)
A recurring expert opinion across case-based literature is that biochemical markers can be inconsistent and therefore diagnosis is frequently delayed unless samples are captured during crisis. - Conlon et al. propose additional acute clues including elevated acetylcarnitine, triglycerides, and 3‑hydroxyglutarate. (conlon2020hypoglycemiaisnot pages 1-3) - Wu et al. propose an elevated acetylcarnitine/free carnitine ratio as an additional signature. (wu2022clinicalbiochemicalmolecular pages 1-2) - Nguyen et al. note 4‑hydroxy‑6‑methyl‑2‑pyrone (4HMP) as a recently reported biomarker but not routine. (nguyen2025mitochondrialhmgcoasynthase pages 2-3)
6.4 Suggested ontology mappings
- Pathway: ketone body synthesis / ketogenesis (supported conceptually by disease definition and review) (suresh2025notjustan pages 22-23)
- GO biological processes (suggested): “ketone body biosynthetic process”, “fatty acid beta-oxidation” (secondary metabolic interplay), “response to starvation”. Evidence for ketogenesis impairment is explicit; downstream processes are inferred as standard biology of fasting dependence on ketones. (suresh2025notjustan pages 22-23, kılıc2020expandingtheclinical pages 6-7)
- Cell types (CL, suggested): hepatocyte (primary site of hepatic ketogenesis and prominent liver phenotype), supported by clinical liver involvement and ketogenesis biology described in reviews. (suresh2025notjustan pages 22-23, wu2022clinicalbiochemicalmolecular pages 2-3)
7. Anatomical Structures Affected
7.1 Primary organs/systems
- Liver: hepatomegaly and elevated transaminases are highly frequent in patient series. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3)
- Central nervous system: encephalopathy, seizures, coma during crises; MRI abnormalities reported in subsets. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3)
7.2 Suggested UBERON terms (examples)
- Liver (UBERON:0002107) (wu2022clinicalbiochemicalmolecular pages 2-3)
- Brain (UBERON:0000955) (wu2022clinicalbiochemicalmolecular pages 2-3)
7.3 Subcellular localization
The affected enzyme is mitochondrial; the disorder is a mitochondrial ketogenesis defect. (conlon2020hypoglycemiaisnot pages 1-3, suresh2025notjustan pages 22-23)
8. Temporal Development
8.1 Onset and pattern
- Typical onset in infancy/early childhood, with episodes after fasting/illness. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, suresh2025notjustan pages 22-23)
8.2 Progression/course
- Often episodic with inter-episode normalization of many laboratory abnormalities. (wu2022clinicalbiochemicalmolecular pages 1-2)
- Acute episodes can be severe/life-threatening; severe genotype groups show higher mortality. (wu2022clinicalbiochemicalmolecular pages 8-9)
9. Inheritance and Population
9.1 Inheritance
Autosomal recessive inheritance is consistently reported. (conlon2020hypoglycemiaisnot pages 1-3, suresh2025notjustan pages 22-23)
9.2 Epidemiology
- A Vietnamese series/review reports estimated incidence <1/1,000,000. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Robust population prevalence, incidence, and carrier frequency estimates were not identified in the retrieved evidence.
9.3 Population-specific variants
- Chinese cohort: c.1201G>T (p.E401*) represented 15.0% of mutated alleles in the reported unrelated Chinese patients and was not reported in other populations in that analysis, suggesting a population hotspot. (wu2022clinicalbiochemicalmolecular pages 1-2)
- Vietnamese cohort: recurrent variants c.559+1G>A and c.1090T>A (p.F364I) were present in ~56% of cases in a single-center cohort. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
10. Diagnostics
10.1 Clinical and biochemical testing
During acute crisis, testing commonly includes: - Blood gas (metabolic acidosis), plasma glucose, ammonia - Liver enzymes (ALT/AST), coagulation markers (e.g., fibrinogen) - Plasma acylcarnitines (may show elevated acetylcarnitine and/or low free carnitine; and proposed elevated C2/C0 ratio) - Urine organic acids (dicarboxylic aciduria; potential presence of 4HMP, elevated glutarate/3HG in some cases) (conlon2020hypoglycemiaisnot pages 1-3, wu2022clinicalbiochemicalmolecular pages 1-2)
Quantitative series examples: - Vietnamese cohort: elevated transaminases 100%, metabolic acidosis 75%, hypoglycemia 56.3%, elevated ammonia 31.3%. (nguyen2025mitochondrialhmgcoasynthase pages 1-2) - Chinese cohort: severe metabolic acidosis 10/10; hypoglycemia 9/10; hyperammonemia 5/10; hypertriglyceridemia 3/10; hypofibrinogenemia 10/10. (wu2022clinicalbiochemicalmolecular pages 2-3, wu2022clinicalbiochemicalmolecular pages 1-2)
10.2 Genetic testing (definitive diagnosis)
Multiple sources emphasize that due to the lack of reliable biochemical markers and limitations of enzyme assays, genetic testing is considered definitive.
Direct abstract quote (2025 Vietnamese case series): “Due to the absence of reliable biochemical markers, genetic testing has become the definitive method for diagnosing HMGCS2D.” (Nguyen et al., 2025; https://doi.org/10.3390/ijms26041644; published Feb 2025) (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
10.3 Enzyme assay limitations (expert opinion)
Enzyme assays are described as limited by challenges in distinguishing mitochondrial vs cytosolic HMG‑CoA synthase in practice, reinforcing reliance on molecular diagnosis. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, nguyen2025mitochondrialhmgcoasynthase pages 2-3)
10.4 Differential diagnosis
Clinical presentations overlap with fatty acid oxidation disorders (FAODs) due to fasting intolerance and hypoketotic crisis patterns. (kılıc2020expandingtheclinical pages 6-7)
11. Outcome / Prognosis
11.1 Overall prognosis with diagnosis and prevention
With timely diagnosis and preventive management (fasting avoidance, sick-day carbohydrate plans), outcomes can be favorable. - Vietnamese cohort: “Currently, all 19 patients are alive… and exhibit normal physical development.” (Nguyen et al., 2025; https://doi.org/10.3390/ijms26041644; published Feb 2025) (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
11.2 Risk of severe outcomes
Severe metabolic crises can require intensive care measures (mechanical ventilation, CRRT) and deaths have occurred in reported cohorts; aggregated genotype–phenotype analysis suggests higher mortality with biallelic truncating variants. (wu2022clinicalbiochemicalmolecular pages 3-4, wu2022clinicalbiochemicalmolecular pages 8-9, wu2022clinicalbiochemicalmolecular media 0a979ec7)
12. Treatment
12.1 Acute management (real-world implementation)
Management is largely supportive and aims to reverse catabolism: - High glucose/dextrose infusion is emphasized as a key acute therapy in series and reports. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3) - Correction of metabolic acidosis (e.g., bicarbonate) and supportive critical care when required. (wu2022clinicalbiochemicalmolecular pages 2-3) - Adjuncts used in some reports/series include carnitine supplementation and organ-support therapies (mechanical ventilation, CRRT/hemodialysis for severe crises). (wu2022clinicalbiochemicalmolecular pages 3-4, wu2022clinicalbiochemicalmolecular pages 2-3)
12.2 Long-term management
- Avoidance of prolonged fasting is consistently recommended after diagnosis. (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3)
- Dietary strategies in case series include moderation/restriction of fat intake and planning enteral carbohydrate provision during illness (“sick day” management). (nguyen2025mitochondrialhmgcoasynthase pages 1-2, sait2024inbornerrorsof pages 1-2)
The Vietnamese cohort specifically reports that “high glucose infusion” combined with proactive strategies (prevent prolonged fasting; enteral carbohydrate/glucose infusion during illness) reduced acute relapse rates. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
12.3 Experimental/advanced therapeutics
No gene therapy, RNA therapy, or targeted molecular therapy trials specific to HMGCS2 deficiency were identified in the retrieved evidence for this run.
12.4 Suggested MAXO terms (examples)
- Intravenous glucose administration (acute crisis) (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Avoidance of fasting (preventive management) (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Dietary fat modification / low-fat diet (long-term management in cohorts) (wu2022clinicalbiochemicalmolecular pages 2-3)
- Continuous renal replacement therapy / hemodialysis (severe crises) (wu2022clinicalbiochemicalmolecular pages 3-4)
13. Prevention
13.1 Primary prevention
Not applicable in the classical public-health sense (genetic disease), but risk reduction for crises includes: - Avoid prolonged fasting - Provide early carbohydrate during intercurrent illness - Care pathways for vomiting/poor intake (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3)
13.2 Secondary prevention
- Genetic testing in suspected cases to establish diagnosis early and implement fasting-avoidance plans. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
13.3 Newborn screening
HMGCS2 deficiency is described as not detectable via standard newborn screening dried blood spot approaches in at least one literature review excerpt; the condition lacks a consistently reliable routine biochemical marker. (kılıc2020expandingtheclinical pages 6-7)
14. Other Species / Natural Disease
No naturally occurring veterinary disease analogs were identified in the retrieved evidence.
15. Model Organisms
No model organism specifically established to recapitulate human HMGCS2 deficiency was identified in the retrieved evidence for this run. (Note: retrieved papers included HMGCS2 biology in mice and other disease contexts, but not a dedicated HMGCS2-deficiency Mendelian disease model relevant to the human inborn error.) (suresh2025notjustan pages 22-23)
Recent developments and latest research emphasis (2023–2025)
- Larger contemporary cohorts with systematic frequencies and variant spectra: The 2025 Vietnamese case series (n=19, Oct 2018–Oct 2024) provides detailed trigger frequencies, clinical presentation frequencies, and recurrent variants in that population, and reports favorable outcomes with proactive management. (Nguyen et al., Feb 2025; https://doi.org/10.3390/ijms26041644) (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Ongoing refinement of diagnostic biomarkers beyond “hypoketotic hypoglycemia”: Case-based evidence emphasizes that hypoglycemia may be absent, and proposes additional acute-phase biomarkers (e.g., 4HMP, 3HG, hypertriglyceridemia, acetylcarnitine patterns and C2/C0 ratio). (conlon2020hypoglycemiaisnot pages 1-3, wu2022clinicalbiochemicalmolecular pages 1-2, nguyen2025mitochondrialhmgcoasynthase pages 2-3)
- Genotype–phenotype correlation analyses: Aggregated analysis suggests truncating variant burden influences severity and mortality risk, supporting more genotype-informed counseling and risk stratification. (wu2022clinicalbiochemicalmolecular pages 8-9, wu2022clinicalbiochemicalmolecular media ec3f9f64)
Key statistics (selected)
- Triggers at first crisis (Vietnamese cohort): poor feeding 93.8%, vomiting 56.3%, diarrhea 25.0%, fever 18.8%. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Presenting features (Vietnamese cohort): lethargy/coma 81.3%, rapid breathing 68.8%, hepatomegaly 56.3%, shock 37.5%, seizures 18.8%. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Acute labs (Vietnamese cohort): transaminases 100%, metabolic acidosis 75%, hypoglycemia 56.3%, hyperammonemia 31.3%. (nguyen2025mitochondrialhmgcoasynthase pages 1-2)
- Presenting features (Chinese cohort): anorexia/dyspnea/disturbance of consciousness 10/10; vomiting 8/10; fever 7/10; seizures 3/10. (wu2022clinicalbiochemicalmolecular pages 2-3)
- Acute labs (Chinese cohort): hypoglycemia 9/10; hyperammonemia 5/10; hypertriglyceridemia 3/10; hypofibrinogenemia 10/10. (wu2022clinicalbiochemicalmolecular pages 2-3, wu2022clinicalbiochemicalmolecular pages 1-2)
Summary table (knowledge-base ready)
The following table consolidates identifiers, phenotype frequencies, diagnostic markers, and management.
| Domain | Summary | Key details / frequencies |
|---|---|---|
| Identifiers | Disease: mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency / HMGCS2 deficiency; OMIM disease #605911; causal gene HMGCS2 (OMIM gene #600234) (nguyen2025mitochondrialhmgcoasynthase pages 1-2, nguyen2025mitochondrialhmgcoasynthase pages 2-3, conlon2020hypoglycemiaisnot pages 1-3) | Rare ketogenesis disorder with estimated incidence <1/1,000,000 in the 2025 Vietnamese series/review (nguyen2025mitochondrialhmgcoasynthase pages 1-2) |
| Synonyms | Mitochondrial HMG-CoA synthase deficiency; mHS deficiency; HMGCS2 deficiency; mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency (conlon2020hypoglycemiaisnot pages 1-3, suresh2025notjustan pages 22-23) | Defect of hepatic ketone-body synthesis due to loss of mitochondrial HMG-CoA synthase activity (nguyen2025mitochondrialhmgcoasynthase pages 1-2, suresh2025notjustan pages 22-23) |
| Inheritance | Autosomal recessive; caused by biallelic pathogenic variants in HMGCS2 (conlon2020hypoglycemiaisnot pages 1-3, suresh2025notjustan pages 22-23, dong2025mitochondrial3hydroxy3methylglutarylcoenzymea pages 1-3) | Missense, nonsense, splice, and frameshift variants reported; biallelic truncating variants are associated with more severe disease/higher mortality in aggregated analyses (suresh2025notjustan pages 22-23, wu2022clinicalbiochemicalmolecular pages 8-9, nguyen2025mitochondrialhmgcoasynthase pages 2-3) |
| Typical onset / triggers | Usually infancy to early childhood; first crisis often in the first year of life and may occur from 10 days to 28 months (nguyen2025mitochondrialhmgcoasynthase pages 1-2, suresh2025notjustan pages 22-23, wu2022clinicalbiochemicalmolecular pages 2-3) | Vietnamese series: poor feeding 93.8%, vomiting 56.3%, diarrhea 25.0%, fever 18.8% as triggers (nguyen2025mitochondrialhmgcoasynthase pages 1-2, nguyen2025mitochondrialhmgcoasynthase pages 2-3); crises typically follow fasting or intercurrent illness (conlon2020hypoglycemiaisnot pages 1-3, suresh2025notjustan pages 22-23, sait2024inbornerrorsof pages 1-2) |
| Key clinical features | Acute metabolic decompensation with encephalopathy and liver involvement (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3) | Nguyen 2025: lethargy/coma 81.3%, rapid breathing 68.8%, hepatomegaly 56.3%, shock 37.5%, seizures 18.8% (nguyen2025mitochondrialhmgcoasynthase pages 1-2, nguyen2025mitochondrialhmgcoasynthase pages 2-3); Wu 2022: anorexia 10/10, dyspnea 10/10, disturbance of consciousness 10/10, vomiting 8/10, fever 7/10, cough 4/10, diarrhea 3/10, seizures 3/10, hepatomegaly 10/10 (wu2022clinicalbiochemicalmolecular pages 2-3, wu2022clinicalbiochemicalmolecular pages 1-2) |
| Key lab findings | Hallmark pattern is impaired ketogenesis with severe metabolic crisis; findings may be nonspecific and can normalize between episodes (nguyen2025mitochondrialhmgcoasynthase pages 2-3, wu2022clinicalbiochemicalmolecular pages 1-2) | Metabolic acidosis: 75% in Nguyen 2025; 10/10 in Wu 2022 (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3). Hypoglycemia: 56.3% in Nguyen 2025; 9/10 in Wu 2022, but absence of hypoglycemia does not exclude disease (nguyen2025mitochondrialhmgcoasynthase pages 1-2, conlon2020hypoglycemiaisnot pages 1-3, wu2022clinicalbiochemicalmolecular pages 1-2). Elevated transaminases: 100% in Nguyen 2025 and 10/10 in Wu 2022 (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3). Hyperammonemia: 31.3% in Nguyen 2025 and 5/10 in Wu 2022 (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 1-2). Hypofibrinogenemia: 10/10 in Wu 2022 (wu2022clinicalbiochemicalmolecular pages 2-3, wu2022clinicalbiochemicalmolecular pages 1-2). Urinary dicarboxylic acids: reported during crises, often prominent, with only mild/absent ketones (conlon2020hypoglycemiaisnot pages 1-3, wu2022clinicalbiochemicalmolecular pages 3-4, wu2022clinicalbiochemicalmolecular pages 1-2). Acetylcarnitine/free carnitine signal: elevated C2/C0 ratio proposed by Wu 2022; low free carnitine and/or elevated acetylcarnitine variably observed across patients (wu2022clinicalbiochemicalmolecular pages 3-4, wu2022clinicalbiochemicalmolecular pages 1-2) |
| Proposed biomarkers | No single routine biomarker is fully reliable; several candidate acute-phase markers have been proposed (nguyen2025mitochondrialhmgcoasynthase pages 2-3, conlon2020hypoglycemiaisnot pages 1-3, kılıc2020expandingtheclinical pages 6-7) | 4-hydroxy-6-methyl-2-pyrone (4HMP) proposed as a novel marker (conlon2020hypoglycemiaisnot pages 1-3, nguyen2025mitochondrialhmgcoasynthase pages 2-3); 3-hydroxyglutarate (3HG) and markedly elevated glutarate may support diagnosis (conlon2020hypoglycemiaisnot pages 1-3); hypertriglyceridemia proposed as an additional acute clue (conlon2020hypoglycemiaisnot pages 1-3, wu2022clinicalbiochemicalmolecular pages 1-2); raised acetylcarnitine (C2) and elevated acetylcarnitine/free carnitine ratio proposed as signatures in some patients (conlon2020hypoglycemiaisnot pages 1-3, wu2022clinicalbiochemicalmolecular pages 1-2) |
| Diagnostic approach | Diagnosis requires integration of acute biochemical testing with molecular confirmation; genetic testing is now considered definitive because enzyme assay and routine biochemical markers can be unreliable (nguyen2025mitochondrialhmgcoasynthase pages 1-2, nguyen2025mitochondrialhmgcoasynthase pages 2-3) | Suggested workup during crisis: blood gas, glucose, ammonia, liver enzymes, free fatty acids/ketones if available, plasma acylcarnitines, and urine organic acids/GC-MS (conlon2020hypoglycemiaisnot pages 1-3, kılıc2020expandingtheclinical pages 6-7, wu2022clinicalbiochemicalmolecular pages 2-3). Typical clues: hypoketotic or relatively low-ketone crisis, dicarboxylic aciduria, raised C2 or C2/C0 ratio, normal/nonspecific TMS, and sometimes 4HMP/3HG (conlon2020hypoglycemiaisnot pages 1-3, sait2024inbornerrorsof pages 1-2, wu2022clinicalbiochemicalmolecular pages 1-2). Confirm by Sanger, targeted NGS, WES or similar sequencing of HMGCS2 (nguyen2025mitochondrialhmgcoasynthase pages 2-3, dong2025mitochondrial3hydroxy3methylglutarylcoenzymea pages 1-3) |
| Management / treatment | Acute care centers on reversal of catabolism with high-glucose/dextrose infusion, correction of acidosis, and supportive intensive care as needed; long-term care focuses on fasting avoidance and sick-day carbohydrate plans (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3) | Reported acute measures: IV glucose/dextrose, sodium bicarbonate, carnitine, liver-protective/supportive therapy, mechanical ventilation, and CRRT/hemodialysis for severe crises (sait2024inbornerrorsof pages 1-2, wu2022clinicalbiochemicalmolecular pages 3-4, wu2022clinicalbiochemicalmolecular pages 2-3). Vietnamese series noted that high-glucose infusion plus prevention of prolonged fasting and enteral carbohydrate/glucose during illness reduced relapses (nguyen2025mitochondrialhmgcoasynthase pages 1-2). Long-term diet: avoidance of prolonged fasting and often low/moderate-fat intake (sait2024inbornerrorsof pages 1-2, wu2022clinicalbiochemicalmolecular pages 2-3) |
| Prognosis / outcomes | Prognosis is often good after diagnosis with preventive management, but acute episodes can be life-threatening and fatalities are reported (nguyen2025mitochondrialhmgcoasynthase pages 1-2, wu2022clinicalbiochemicalmolecular pages 8-9) | Nguyen 2025: all 19 patients alive at follow-up (ages 5 months-14 years) with normal physical development after proactive management (nguyen2025mitochondrialhmgcoasynthase pages 1-2). Wu 2022/literature aggregation: most recover fully and maintain normal growth; only limited neurologic sequelae reported, but genotype severity matters; mortality highest in the biallelic truncating group (25%) (wu2022clinicalbiochemicalmolecular pages 8-9). Individual severe cases can progress to multiorgan failure and death (dong2025mitochondrial3hydroxy3methylglutarylcoenzymea pages 1-3, wu2022clinicalbiochemicalmolecular pages 3-4) |
Table: This table condenses identifiers, phenotype frequencies, biochemical clues, diagnostic strategy, management, and prognosis for mitochondrial HMG-CoA synthase deficiency. It is designed as a disease knowledge base quick-reference using recent case-series evidence plus key prior reports.
Visual evidence from tables (original sources)
The extracted tables below contain the underlying cohort-level clinical and genotype–phenotype correlation information discussed in this report.
- Patient clinical/laboratory/outcome summary table crop (Wu et al., 2022). (wu2022clinicalbiochemicalmolecular media 0a979ec7)
- Genotype–phenotype correlation table crop (Wu et al., 2022). (wu2022clinicalbiochemicalmolecular media ec3f9f64)
References
-
(nguyen2025mitochondrialhmgcoasynthase pages 1-2): Khanh Ngoc Nguyen, Tran Minh Dien, Thi Bich Ngoc Can, Bui Phuong Thao, Thi Kim Giang Dang, Ngoc Lan Nguyen, Van Khanh Tran, Thuy Thu Nguyen, Tran Thi Quynh Trang, Le Thi Phuong, Phan Long Nguyen, Thinh Huy Tran, Nguyen Huu Tu, and Chi Dung Vu. Mitochondrial hmg-coa synthase deficiency in vietnamese patients. International Journal of Molecular Sciences, Feb 2025. URL: https://doi.org/10.3390/ijms26041644, doi:10.3390/ijms26041644. This article has 2 citations.
-
(conlon2020hypoglycemiaisnot pages 1-3): Tracey A. Conlon, Patricia E. Fitzsimons, Ingrid Borovickova, Fidelma Kirby, Sinéad Murphy, Ina Knerr, and Ellen Crushell. Hypoglycemia is not a defining feature of metabolic crisis in mitochondrial 3‐hydroxy‐3‐methylglutaryl‐coa synthase deficiency: further evidence of specific biochemical markers which may aid diagnosis. JIMD Reports, 55:26-31, Jun 2020. URL: https://doi.org/10.1002/jmd2.12146, doi:10.1002/jmd2.12146. This article has 11 citations and is from a peer-reviewed journal.
-
(suresh2025notjustan pages 22-23): Varshini V. Suresh, Sathish Sivaprakasam, Yangzom D. Bhutia, Puttur D. Prasad, Muthusamy Thangaraju, and Vadivel Ganapathy. Not just an alternative energy source: diverse biological functions of ketone bodies and relevance of hmgcs2 to health and disease. Biomolecules, 15:580, Apr 2025. URL: https://doi.org/10.3390/biom15040580, doi:10.3390/biom15040580. This article has 17 citations.
-
(nguyen2025mitochondrialhmgcoasynthase pages 2-3): Khanh Ngoc Nguyen, Tran Minh Dien, Thi Bich Ngoc Can, Bui Phuong Thao, Thi Kim Giang Dang, Ngoc Lan Nguyen, Van Khanh Tran, Thuy Thu Nguyen, Tran Thi Quynh Trang, Le Thi Phuong, Phan Long Nguyen, Thinh Huy Tran, Nguyen Huu Tu, and Chi Dung Vu. Mitochondrial hmg-coa synthase deficiency in vietnamese patients. International Journal of Molecular Sciences, Feb 2025. URL: https://doi.org/10.3390/ijms26041644, doi:10.3390/ijms26041644. This article has 2 citations.
-
(wu2022clinicalbiochemicalmolecular pages 2-3): Shengnan Wu, Linghua Shen, Qiong Chen, Chunxiu Gong, Yanling Yang, Haiyan Wei, Bingyan Cao, and Yongxing Chen. Clinical, biochemical, molecular, and outcome features of mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase deficiency in 10 chinese patients. Frontiers in Genetics, Mar 2022. URL: https://doi.org/10.3389/fgene.2021.816779, doi:10.3389/fgene.2021.816779. This article has 13 citations and is from a peer-reviewed journal.
-
(dong2025mitochondrial3hydroxy3methylglutarylcoenzymea pages 1-3): Chang Dong, Tiantian Lu, Yazhou Jiang, Zihao Yan, and Suyue Zhu. Mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme a synthase 2 deficiency with severe hyperglycemia in a child: a rare case report. Sep 2025. URL: https://doi.org/10.1177/03000605251375537, doi:10.1177/03000605251375537. This article has 0 citations and is from a peer-reviewed journal.
-
(kılıc2020expandingtheclinical pages 6-7): Mustafa Kılıç, Sevil Dorum, Ali Topak, Mutlu U. Yazıcı, Fatih S. Ezgu, and Turgay Coskun. Expanding the clinical spectrum of mitochondrial 3‐hydroxy‐3‐methylglutaryl‐coa synthase deficiency with turkish cases harboring novel hmgcs2 gene mutations and literature review. American Journal of Medical Genetics Part A, 182:1608-1614, Apr 2020. URL: https://doi.org/10.1002/ajmg.a.61590, doi:10.1002/ajmg.a.61590. This article has 15 citations.
-
(wu2022clinicalbiochemicalmolecular pages 8-9): Shengnan Wu, Linghua Shen, Qiong Chen, Chunxiu Gong, Yanling Yang, Haiyan Wei, Bingyan Cao, and Yongxing Chen. Clinical, biochemical, molecular, and outcome features of mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase deficiency in 10 chinese patients. Frontiers in Genetics, Mar 2022. URL: https://doi.org/10.3389/fgene.2021.816779, doi:10.3389/fgene.2021.816779. This article has 13 citations and is from a peer-reviewed journal.
-
(wu2022clinicalbiochemicalmolecular media 0a979ec7): Shengnan Wu, Linghua Shen, Qiong Chen, Chunxiu Gong, Yanling Yang, Haiyan Wei, Bingyan Cao, and Yongxing Chen. Clinical, biochemical, molecular, and outcome features of mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase deficiency in 10 chinese patients. Frontiers in Genetics, Mar 2022. URL: https://doi.org/10.3389/fgene.2021.816779, doi:10.3389/fgene.2021.816779. This article has 13 citations and is from a peer-reviewed journal.
-
(wu2022clinicalbiochemicalmolecular pages 1-2): Shengnan Wu, Linghua Shen, Qiong Chen, Chunxiu Gong, Yanling Yang, Haiyan Wei, Bingyan Cao, and Yongxing Chen. Clinical, biochemical, molecular, and outcome features of mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase deficiency in 10 chinese patients. Frontiers in Genetics, Mar 2022. URL: https://doi.org/10.3389/fgene.2021.816779, doi:10.3389/fgene.2021.816779. This article has 13 citations and is from a peer-reviewed journal.
-
(wu2022clinicalbiochemicalmolecular media ec3f9f64): Shengnan Wu, Linghua Shen, Qiong Chen, Chunxiu Gong, Yanling Yang, Haiyan Wei, Bingyan Cao, and Yongxing Chen. Clinical, biochemical, molecular, and outcome features of mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase deficiency in 10 chinese patients. Frontiers in Genetics, Mar 2022. URL: https://doi.org/10.3389/fgene.2021.816779, doi:10.3389/fgene.2021.816779. This article has 13 citations and is from a peer-reviewed journal.
-
(wu2022clinicalbiochemicalmolecular pages 3-4): Shengnan Wu, Linghua Shen, Qiong Chen, Chunxiu Gong, Yanling Yang, Haiyan Wei, Bingyan Cao, and Yongxing Chen. Clinical, biochemical, molecular, and outcome features of mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase deficiency in 10 chinese patients. Frontiers in Genetics, Mar 2022. URL: https://doi.org/10.3389/fgene.2021.816779, doi:10.3389/fgene.2021.816779. This article has 13 citations and is from a peer-reviewed journal.
-
(sait2024inbornerrorsof pages 1-2): Haseena Sait, Somya Srivastava, Somesh Kumar, Bijo Varughese, Manmohan Pandey, Manjunath Venkatramaiah, Parul Chaudhary, Amita Moirangthem, Kausik Mandal, and Seema Kapoor. Inborn errors of ketogenesis: novel variants, clinical presentation, and follow-up in a series of four patients. Journal of Pediatric Genetics, 13:022-028, Jul 2024. URL: https://doi.org/10.1055/s-0042-1749362, doi:10.1055/s-0042-1749362. This article has 6 citations and is from a peer-reviewed journal.
Artifacts
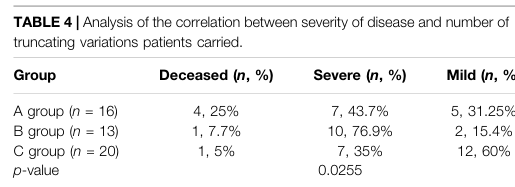
3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency Deep Research Fallback
Provider Attempts
- 2026-05-04T09:20Z:
timeout 120 just research-disorder falcon 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiencyfailed before provider execution because the disorder YAML did not yet exist. - 2026-05-04T09:21Z:
timeout 120 just research-disorder falcon 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiencytimed out with exit code 124 after the provider command was terminated bytimeout. - 2026-05-04T09:24Z:
timeout 120 just research-disorder openai 3-Hydroxy-3-Methylglutaryl-CoA_Synthase_Deficiencytimed out with exit code 124 after the provider command was terminated bytimeout.
No provider-generated deep-research narrative was available within the bounded
runtime. Curation therefore proceeded from generated structured Orphanet
evidence and fetched PubMed caches, without hand-editing any
references_cache/*.md files.
Evidence Scope Used For Curation
- ORPHA:35701 structured record for disease definition, MONDO/OMIM exact mappings, autosomal recessive inheritance, worldwide point-prevalence band, HMGCS2 disease-gene association, childhood onset, and Orphanet HPO phenotypes.
- PMID:39798988 for the 2025 systematic review of 93 reported cases plus two new patients, current clinical spectrum, onset range, acute crisis triggers, dicarboxylic aciduria frequency, 4HMP detection, and C2/C0 acylcarnitine diagnostic recommendation.
- PMID:11228257 and PMID:11479731 for early molecular diagnosis papers and functional confirmation that HMGCS2 variants can abolish mitochondrial HMG-CoA synthase activity.
- PMID:32952630 for Japanese patient cases, in vitro functional analysis of five novel HMGCS2 mutations, ketogenesis biochemistry, acute-phase acetylcarnitine rationale, and practical fasting/glucose management.
- PMID:35308163 for a 10-patient Chinese case series covering crisis phenotype, molecular spectrum, and common biochemical abnormalities.
- PMID:39143735 for cyclic-vomiting-like presentation and the importance of molecular testing when biochemical markers are nonspecific.
- PMID:40548098 for neonatal hyperammonemic coma, intrafamilial variability, 4HMP after fasting, and fasting avoidance with or without L-carnitine during intercurrent illness.
Curation Conclusions
The accepted disease model is biallelic HMGCS2 loss of function causing deficient mitochondrial HMG-CoA synthase 2 activity in hepatocytes. This blocks ketone body biosynthesis from acetyl-CoA and acetoacetyl-CoA during fasting or illness, producing hypoketotic hypoglycemia and acute metabolic decompensation. The acute biochemical pattern can include dicarboxylic aciduria, urinary 4-hydroxy-6-methyl-2-pyrone, and elevated plasma C2/C0 acylcarnitine ratio. Management is preventive and emergency-focused: avoid prolonged fasting, provide carbohydrate/glucose support during poor intake or illness, consider L-carnitine during intercurrent illness where clinically appropriate, and offer genetic counseling for autosomal recessive recurrence risk.