15q11q13 Microduplication Syndrome
15q11q13 microduplication syndrome is a recurrent chromosome 15q11.2-q13.1 copy-number disorder characterized by neurodevelopmental impairment, autistic behavior, hypotonia, language delay, seizures, sleep disturbance, and variable epilepsy severity. The syndrome reflects altered dosage of imprinted and neurodevelopmental genes within 15q11-q13, especially neuronal UBE3A and the GABAA receptor gene cluster (GABRB3, GABRA5, GABRG3), with stronger phenotypic impact for maternally inherited duplications.
Ask OpenScientist
Ask a research question about 15q11q13 Microduplication Syndrome. OpenScientist will conduct autonomous deep research using the Disorder Mechanisms Knowledge Base and PubMed literature (typically 10-30 minutes).
Do not include personal health information in your question. Questions and results are cached in your browser's local storage.
Inheritance
1Show evidence (5 references)
Subtypes
3Pathophysiology
5Show evidence (4 references)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Pathograph
Phenotypes
7Musculoskeletal 1
Show evidence (1 reference)
Nervous System 6
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Genetic Associations
4Show evidence (3 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Medical Actions
6Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Differential Diagnoses
2Conditions with similar clinical presentations that must be differentiated from 15q11q13 Microduplication Syndrome:
Clinical Trials
4Show evidence (2 references)
Show evidence (1 reference)
Show evidence (1 reference)
Show evidence (1 reference)
Source YAML
click to showname: 15q11q13 Microduplication Syndrome
creation_date: "2026-04-15T23:46:24Z"
updated_date: "2026-04-20T00:00:00Z"
synonyms:
- Dup15q syndrome
- Duplication 15q11q13 syndrome
- 15q11-q13 duplication syndrome
- 15q11.2-q13.1 duplication syndrome
- Chromosome 15q11-q13 duplication syndrome
description: >-
15q11q13 microduplication syndrome is a recurrent chromosome 15q11.2-q13.1
copy-number disorder characterized by neurodevelopmental impairment, autistic
behavior, hypotonia, language delay, seizures, sleep disturbance, and variable
epilepsy severity. The syndrome reflects altered dosage of imprinted and
neurodevelopmental genes within 15q11-q13, especially neuronal UBE3A and the
GABAA receptor gene cluster (GABRB3, GABRA5, GABRG3), with stronger
phenotypic impact for maternally inherited duplications.
category: Mendelian
parents:
- hereditary disease
- chromosomal disorder
disease_term:
preferred_term: 15q11q13 microduplication syndrome
term:
id: MONDO:0012081
label: 15q11q13 microduplication syndrome
external_assertions:
- name: OMIM chromosome 15q11-q13 duplication syndrome record
source: OMIM
assertion_type: disease_record
external_id: OMIM:608636
description: >-
OMIM entry for chromosome 15q11-q13 duplication syndrome, cataloguing the
clinical features and molecular basis of Dup15q syndrome.
inheritance:
- name: Maternal parent-of-origin effect with subtype-dependent recurrence risk
inheritance_term:
preferred_term: Autosomal dominant inheritance with maternal imprinting
term:
id: HP:0012275
label: Autosomal dominant inheritance with maternal imprinting
parent_of_origin_effect: >-
The Dup15q clinical phenotype requires at least one extra maternally derived
copy of the 15q11.2-q13.1 Prader-Willi/Angelman critical region; paternal
duplications are usually clinically silent or much milder.
de_novo_rate: >-
Maternal idic(15) has been reported de novo in all affected individuals to
date. Maternal interstitial 15q11.2-q13.1 duplication is de novo in
approximately 85% of probands and maternally inherited in approximately
15%.
description: >-
Recurrence counseling depends on the structural subtype and parental origin.
Unaffected parents of a child with de novo idic(15) generally have low
recurrence risk, with residual concern for maternal germline mosaicism. If a
mother carries an interstitial 15q11.2-q13.1 duplication, each child has a
50% risk of inheriting the duplication; prenatal or preimplantation genetic
testing by chromosomal microarray can detect the duplication but cannot
reliably predict severity.
evidence:
- reference: PMID:27308687
reference_title: Maternal 15q Duplication Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The diagnosis of maternal dup15q is established by detection of at least
one extra maternally derived copy of the Prader-Willi/Angelman critical
region
explanation: >-
GeneReviews defines maternal origin of the duplicated critical-region copy
as central to the Dup15q diagnosis.
- reference: PMID:27308687
reference_title: Maternal 15q Duplication Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: De novo in all affected individuals reported to date
explanation: >-
GeneReviews supports de novo occurrence as the rule for maternal idic(15),
with residual recurrence risk from possible maternal germline mosaicism.
- reference: PMID:27308687
reference_title: Maternal 15q Duplication Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: De novo in approximately 85% of probands and inherited from the mother in approximately 15%
explanation: >-
GeneReviews gives the subtype-specific de novo and inherited proportions
for maternal interstitial 15q11.2-q13.1 duplications.
- reference: PMID:27308687
reference_title: Maternal 15q Duplication Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: the risk to each child of inheriting the duplication is 50%
explanation: >-
GeneReviews supports 50% recurrence risk when the mother carries the
interstitial duplication.
- reference: PMID:27308687
reference_title: Maternal 15q Duplication Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Prenatal testing or preimplantation genetic testing using chromosomal
microarray (CMA) will detect the 15q interstitial duplication
explanation: >-
GeneReviews supports prenatal and preimplantation testing availability
when the familial duplication is known.
has_subtypes:
- name: int15
display_name: Interstitial 15q11-q13 duplication (int15)
description: >-
Interstitial tandem duplication of 15q11-q13 on the maternal chromosome.
Typically milder than idic15, with developmental and behavioral features
but lower burden of refractory epilepsy and SUDEP.
- name: idic15
display_name: Isodicentric 15 (idic15)
description: >-
Supernumerary isodicentric marker chromosome containing an extra copy of
15q11-q13. More strongly associated with severe, refractory epilepsy and
elevated risk of sudden unexpected death in epilepsy (SUDEP).
- name: Mosaic idic15
description: >-
Mosaic form of idic15 in which only a subset of cells carry the
supernumerary isodicentric 15 chromosome, producing variable and often
attenuated clinical severity relative to non-mosaic idic15.
prevalence:
- population: Pregnant women undergoing genome-wide NIPS
measure_type: POINT_PREVALENCE
prevalence_class: BAND_1_9_PER_100000
rate_per_100000: 6.9
percentage: 0.0069%
evidence:
- reference: PMID:37029316
reference_title: >-
Population screening for 15q11-q13 duplications: corroboration of the
difference in impact between maternally and paternally inherited alleles.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We detect 23 15q11-q13 duplications in 333,187 pregnant women (0.0069%),
with an approximately equal distribution between maternal and paternal
duplications.
explanation: >-
This gives a genome-wide NIPS detection rate for 15q11-q13 duplications;
it is best interpreted as a screening-cohort estimate rather than a
general-population prevalence.
notes: >-
This is a prenatal screening detection rate and should not be interpreted as
a population prevalence of clinically diagnosed Dup15q syndrome.
progression:
- phase: Neurodevelopmental disorder
age_range: Prenatal origin with infantile or childhood presentation
evidence:
- reference: PMID:39457428
reference_title: >-
Chromosome 15q11-q13 Duplication Syndrome: A Review of the Literature and
14 New Cases.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
15q11.2-q13 duplication syndrome has been associated with neurodevelopmental
disorders (hypotonia, developmental delay, speech delay and seizures) and ASD
but is characterized by variable expressivity and reduced penetrance, features
that make genetic counseling a complex procedure especially in prenatal cases.
explanation: >-
This supports an early neurodevelopmental presentation with variable
expressivity and counseling complexity in prenatal cases.
pathophysiology:
- name: Maternal 15q11-q13 dosage increase
description: >-
Maternal duplication of 15q11-q13 increases dosage of imprinted and
neurodevelopmental genes, with UBE3A overexpression and parent-of-origin
effects emerging as key dosage-sensitive drivers.
genes:
- preferred_term: UBE3A
term:
id: hgnc:12496
label: UBE3A
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
evidence:
- reference: PMID:25884337
reference_title: >-
15q11.2 Duplication Encompassing Only the UBE3A Gene Is Associated with
Developmental Delay and Neuropsychiatric Phenotypes.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Duplications of chromosome region 15q11-q13 with the maternal imprint are
associated with a wide spectrum of neuropsychiatric disorders, including
autism spectrum disorders, developmental delay, learning difficulties,
schizophrenia, and seizures.
explanation: >-
This supports maternal 15q11-q13 dosage increase as the initiating
disease mechanism.
- reference: PMID:41751547
reference_title: >-
Genomics of Complex Neurodevelopmental Disorders with Variable Epilepsy
Phenotypes: A Clinical Review of Dup15q Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Dup15q syndrome demonstrates marked heterogeneity in epilepsy severity and
seizure semiology, reflecting variable gene dosing effects, maternal
imprinting of UBE3A, and altered GABAergic signaling.
explanation: >-
This supports dosage-sensitive imprinting, especially UBE3A, as the core
proximal mechanism.
- reference: PMID:37029316
reference_title: >-
Population screening for 15q11-q13 duplications: corroboration of the
difference in impact between maternally and paternally inherited alleles.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Maternally inherited duplications are always associated with a clinical
phenotype (ranging from learning difficulties to intellectual impairment,
epilepsy and psychiatric disorders), while paternal duplications are normal
or associated with milder phenotypes (mild learning difficulties and dyslexia).
explanation: >-
This directly supports a parent-of-origin effect as a key driver of
phenotypic impact in 15q11-q13 duplications.
- reference: PMID:36898382
reference_title: >-
The role of UBE3A in the autism and epilepsy-related Dup15q syndrome using
patient-derived, CRISPR-corrected neurons.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
UBE3A, which encodes an E3 ubiquitin ligase, is likely a major driver of
Dup15q because UBE3A is the only imprinted gene expressed solely from the
maternal allele.
explanation: >-
Patient-derived neuron experiments support UBE3A as a major imprinted
dosage driver in Dup15q syndrome.
downstream:
- target: UBE3A overexpression-driven neuronal hyperexcitability
description: >-
Increased maternal UBE3A dosage contributes to neuronal hyperexcitability
in patient-derived neurons.
- target: GABAergic signaling disruption
description: >-
Gene-dosage imbalance perturbs inhibitory signaling in cortical networks.
- target: Epileptogenic network dysfunction
description: >-
Dosage imbalance increases susceptibility to abnormal neuronal firing and
epilepsy.
- name: UBE3A overexpression-driven neuronal hyperexcitability
description: >-
Patient-derived Dup15q neurons show hyperexcitability that is largely
prevented by normalizing UBE3A dosage, supporting UBE3A overexpression as a
key cellular mechanism downstream of maternal 15q11-q13 copy-number gain.
genes:
- preferred_term: UBE3A
term:
id: hgnc:12496
label: UBE3A
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: regulation of membrane potential
term:
id: GO:0042391
label: regulation of membrane potential
modifier: INCREASED
- preferred_term: chemical synaptic transmission
term:
id: GO:0007268
label: chemical synaptic transmission
evidence:
- reference: PMID:36898382
reference_title: >-
The role of UBE3A in the autism and epilepsy-related Dup15q syndrome
using patient-derived, CRISPR-corrected neurons.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
Dup15q neurons exhibited hyperexcitability compared with control neurons,
and this phenotype was generally prevented by normalizing UBE3A levels
using antisense oligonucleotides. Overexpression of UBE3A resulted in a
profile similar to that of Dup15q neurons except for synaptic phenotypes.
These results indicate that UBE3A overexpression is necessary for most
Dup15q cellular phenotypes but also suggest a role for other genes in the
duplicated region.
explanation: >-
This patient-derived, CRISPR-corrected neuron study directly links UBE3A
overexpression to Dup15q neuronal hyperexcitability while acknowledging
that other duplicated genes may also contribute.
downstream:
- target: Epileptogenic network dysfunction
description: >-
Increased neuronal excitability provides a cellular substrate for the
downstream epilepsy-prone network state.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- UBE3A overexpression increases neuronal excitability in patient-derived neurons.
evidence:
- reference: PMID:36898382
reference_title: >-
The role of UBE3A in the autism and epilepsy-related Dup15q syndrome
using patient-derived, CRISPR-corrected neurons.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
Dup15q neurons exhibited hyperexcitability compared with control
neurons, and this phenotype was generally prevented by normalizing UBE3A
levels using antisense oligonucleotides.
explanation: >-
This supports the mechanistic edge from UBE3A dosage normalization to
reduced neuronal hyperexcitability.
- target: Autistic behavior
description: >-
UBE3A dosage-dependent neuronal dysfunction contributes to autism-related
neurodevelopmental phenotypes in Dup15q syndrome.
- name: GABAergic signaling disruption
description: >-
Dup15q disrupts GABAergic signaling in cortical networks, with the duplicated
15q11.2-q13.1 interval including a GABAA receptor gene cluster (GABRB3,
GABRA5, GABRG3) that contributes to beta-band EEG abnormalities and
refractory epilepsy.
genes:
- preferred_term: GABRB3
term:
id: hgnc:4083
label: GABRB3
- preferred_term: GABRA5
term:
id: hgnc:4079
label: GABRA5
- preferred_term: GABRG3
term:
id: hgnc:4088
label: GABRG3
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
biological_processes:
- preferred_term: gamma-aminobutyric acid signaling pathway
term:
id: GO:0007214
label: gamma-aminobutyric acid signaling pathway
evidence:
- reference: PMID:41751547
reference_title: >-
Genomics of Complex Neurodevelopmental Disorders with Variable Epilepsy
Phenotypes: A Clinical Review of Dup15q Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Dup15q syndrome demonstrates marked heterogeneity in epilepsy severity and
seizure semiology, reflecting variable gene dosing effects, maternal
imprinting of UBE3A, and altered GABAergic signaling.
explanation: >-
This supports GABAergic signaling disruption as a downstream disease
mechanism.
- reference: PMID:32791992
reference_title: Properties of beta oscillations in Dup15q syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The 15q region harbors genes critical for brain development, particularly
UBE3A and a cluster of gamma-aminobutyric acid type A receptor (GABAAR)
genes.
explanation: >-
This supports the GABAA receptor gene cluster in the duplicated region as
part of the mechanistic basis for altered GABAergic signaling in Dup15q.
downstream:
- target: Seizure
description: >-
GABAergic signaling disruption increases seizure susceptibility.
- target: Autistic behavior
description: >-
Circuit-level inhibitory imbalance contributes to autistic behavior.
- target: Sleep disturbance
description: >-
Altered inhibitory signaling is associated with abnormal sleep physiology
and beta EEG activity in Dup15q syndrome.
- name: Sodium channel dysfunction
description: >-
Dup15q neurons show abnormal sodium-channel kinetics with increased sodium
current density and altered inactivation.
cell_types:
- preferred_term: neuron
term:
id: CL:0000540
label: neuron
evidence:
- reference: PMID:37329181
reference_title: >-
Dysfunctional sodium channel kinetics as a novel epilepsy mechanism in
chromosome 15q11-q13 duplication syndrome.
supports: SUPPORT
evidence_source: IN_VITRO
snippet: >-
Dup15q neurons showed increased sodium current density and a depolarizing
shift in steady-state inactivation.
explanation: >-
This directly supports sodium-channel dysfunction in Dup15q neurons.
downstream:
- target: Seizure
description: >-
Abnormal sodium-channel kinetics increase seizure susceptibility.
- name: Epileptogenic network dysfunction
description: >-
Network-level dysregulation in Dup15q syndrome produces early-onset
neurodevelopmental impairment and refractory epilepsy.
evidence:
- reference: PMID:41751547
reference_title: >-
Genomics of Complex Neurodevelopmental Disorders with Variable Epilepsy
Phenotypes: A Clinical Review of Dup15q Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
While idic15 is more strongly associated with refractory epilepsy and
SUDEP, both idic15 and int15 subtypes show overlapping developmental and
behavioral phenotypes.
explanation: >-
This supports a downstream epileptogenic network phenotype with
developmental consequences.
downstream:
- target: Global developmental delay
description: >-
Early network dysfunction manifests as global developmental delay.
- target: Delayed speech and language development
description: >-
Neurodevelopmental network dysfunction contributes to delayed speech and
language acquisition in Dup15q syndrome.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Maternal 15q11-q13 dosage increase perturbs UBE3A/GABAergic neuronal networks during development.
evidence:
- reference: PMID:24239951
reference_title: >-
Duplication of the 15q11-q13 region: clinical and genetic study of 30
new cases.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
includes central hypotonia, developmental delay, speech delay, seizure,
explanation: >-
The Dup15q clinical series identifies speech delay as part of the
neurodevelopmental phenotype downstream of the 15q11-q13 duplication.
- target: Intellectual disability
description: >-
Developmental network dysfunction produces broader cognitive impairment,
ranging from learning difficulties to intellectual impairment.
causal_link_type: INDIRECT_KNOWN_INTERMEDIATES
intermediate_mechanisms:
- Maternal 15q11-q13 dosage increase disrupts neurodevelopmental circuits through UBE3A dosage and GABAergic signaling effects.
evidence:
- reference: PMID:37029316
reference_title: >-
Population screening for 15q11-q13 duplications: corroboration of the
difference in impact between maternally and paternally inherited alleles.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
ranging from learning difficulties to intellectual impairment
explanation: >-
Maternal 15q11-q13 duplications are reported to cause cognitive
phenotypes spanning learning difficulties to intellectual impairment.
- target: Autistic behavior
description: >-
Circuit dysfunction contributes to autistic behavior.
- target: Hypotonia
description: >-
Abnormal neuronal network development contributes to hypotonia.
- target: Seizure
description: >-
Epileptogenic network dysfunction manifests as seizures of variable
severity.
phenotypes:
- name: Global developmental delay
description: >-
Developmental delay is a core manifestation of Dup15q syndrome.
phenotype_term:
preferred_term: Global developmental delay
term:
id: HP:0001263
label: Global developmental delay
evidence:
- reference: PMID:25884337
reference_title: >-
15q11.2 Duplication Encompassing Only the UBE3A Gene Is Associated with
Developmental Delay and Neuropsychiatric Phenotypes.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Duplications of chromosome region 15q11-q13 with the maternal imprint are
associated with a wide spectrum of neuropsychiatric disorders, including
autism spectrum disorders, developmental delay, learning difficulties,
schizophrenia, and seizures.
explanation: >-
This directly supports developmental delay as a core phenotype.
- name: Autistic behavior
description: >-
Autistic behavior and autism-spectrum features are frequent.
phenotype_term:
preferred_term: Autistic behavior
term:
id: HP:0000729
label: Autistic behavior
evidence:
- reference: PMID:25884337
reference_title: >-
15q11.2 Duplication Encompassing Only the UBE3A Gene Is Associated with
Developmental Delay and Neuropsychiatric Phenotypes.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Duplications of chromosome region 15q11-q13 with the maternal imprint are
associated with a wide spectrum of neuropsychiatric disorders, including
autism spectrum disorders, developmental delay, learning difficulties,
schizophrenia, and seizures.
explanation: >-
This directly supports autistic behavior / autism-spectrum features.
- name: Hypotonia
description: >-
Hypotonia is common, especially in infancy.
phenotype_term:
preferred_term: Hypotonia
term:
id: HP:0001252
label: hypotonia
evidence:
- reference: PMID:24239951
reference_title: >-
Duplication of the 15q11-q13 region: clinical and genetic study of 30 new cases.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Duplications are clinically characterized by a variable phenotype that
includes central hypotonia, developmental delay, speech delay, seizure,
minor dysmorphic features and autism.
explanation: >-
This directly supports hypotonia in Dup15q syndrome.
- name: Delayed speech and language development
description: >-
Language delay is a prominent feature.
phenotype_term:
preferred_term: Delayed speech and language development
term:
id: HP:0000750
label: delayed speech and language development
evidence:
- reference: PMID:25884337
reference_title: >-
15q11.2 Duplication Encompassing Only the UBE3A Gene Is Associated with
Developmental Delay and Neuropsychiatric Phenotypes.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Duplications of chromosome region 15q11-q13 with the maternal imprint are
associated with a wide spectrum of neuropsychiatric disorders, including
autism spectrum disorders, developmental delay, learning difficulties,
schizophrenia, and seizures.
explanation: >-
This supports the language-delayed neurodevelopmental phenotype.
- name: Intellectual disability
description: >-
Intellectual disability or broader learning difficulties are part of the
recurrent neurodevelopmental phenotype of Dup15q syndrome.
phenotype_term:
preferred_term: Intellectual disability
term:
id: HP:0001249
label: Intellectual disability
evidence:
- reference: PMID:25884337
reference_title: >-
15q11.2 Duplication Encompassing Only the UBE3A Gene Is Associated with
Developmental Delay and Neuropsychiatric Phenotypes.
supports: PARTIAL
evidence_source: HUMAN_CLINICAL
snippet: >-
Duplications of chromosome region 15q11-q13 with the maternal imprint are
associated with a wide spectrum of neuropsychiatric disorders, including
autism spectrum disorders, developmental delay, learning difficulties,
schizophrenia, and seizures.
explanation: >-
This provides partial support for intellectual disability by directly
documenting recurrent learning difficulties within the Dup15q
neurodevelopmental phenotype.
- name: Seizure
description: >-
Seizures are a core feature, often refractory and clinically variable.
phenotype_term:
preferred_term: Seizure
term:
id: HP:0001250
label: seizure
evidence:
- reference: PMID:41751547
reference_title: >-
Genomics of Complex Neurodevelopmental Disorders with Variable Epilepsy
Phenotypes: A Clinical Review of Dup15q Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
RESULTS: Dup15q syndrome demonstrates marked heterogeneity in epilepsy
severity and seizure semiology, reflecting variable gene dosing effects,
maternal imprinting of UBE3A, and altered GABAergic signaling.
explanation: >-
This directly supports seizures as a major and variable phenotype.
- name: Sleep disturbance
description: >-
Sleep disruption is a recognized comorbidity in Dup15q syndrome, with
clinical EEG studies showing elevated beta power, reduced spindle density,
and reduced or absent slow-wave sleep.
phenotype_term:
preferred_term: Sleep disturbance
term:
id: HP:0002360
label: Sleep disturbance
evidence:
- reference: PMID:34344470
reference_title: >-
Abnormal sleep physiology in children with 15q11.2-13.1 duplication
(Dup15q) syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Children with Dup15q syndrome showed abnormal sleep physiology with
elevated beta power, reduced spindle density, and reduced or absent SWS
compared to age-matched neurotypical controls.
explanation: >-
This directly supports sleep disturbance as a Dup15q phenotype and
provides the observed quantitative EEG sleep abnormalities.
biochemical: []
genetic:
- name: Maternal 15q11-q13 duplication
association: Causal chromosomal duplication
notes: >-
Dup15q syndrome is typically caused by a maternally derived duplication or
idic(15) involving 15q11-q13. Recurrent breakpoints in the low-copy-repeat-rich
15q11.2-q13 region generate variable duplication sizes, and parental origin
influences penetrance and severity.
evidence:
- reference: PMID:41751547
reference_title: >-
Genomics of Complex Neurodevelopmental Disorders with Variable Epilepsy
Phenotypes: A Clinical Review of Dup15q Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We synthesized current literature on genomic mechanisms underlying
complex neurodevelopmental disorders focusing on Dup15q syndrome and its
subtypes-int15, idic15, and mosaic idic15.
explanation: >-
This directly supports the chromosomal duplication basis of the disorder.
- reference: PMID:39457428
reference_title: >-
Chromosome 15q11-q13 Duplication Syndrome: A Review of the Literature and
14 New Cases.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The 15q11.2q13 chromosomal region is particularly susceptible to
chromosomal rearrangements due to low-copy repeats (LCRs) located inside
this area.
explanation: >-
This supports the LCR-mediated mechanism responsible for recurrent
15q11-q13 duplications.
- reference: PMID:39457428
reference_title: >-
Chromosome 15q11-q13 Duplication Syndrome: A Review of the Literature and
14 New Cases.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
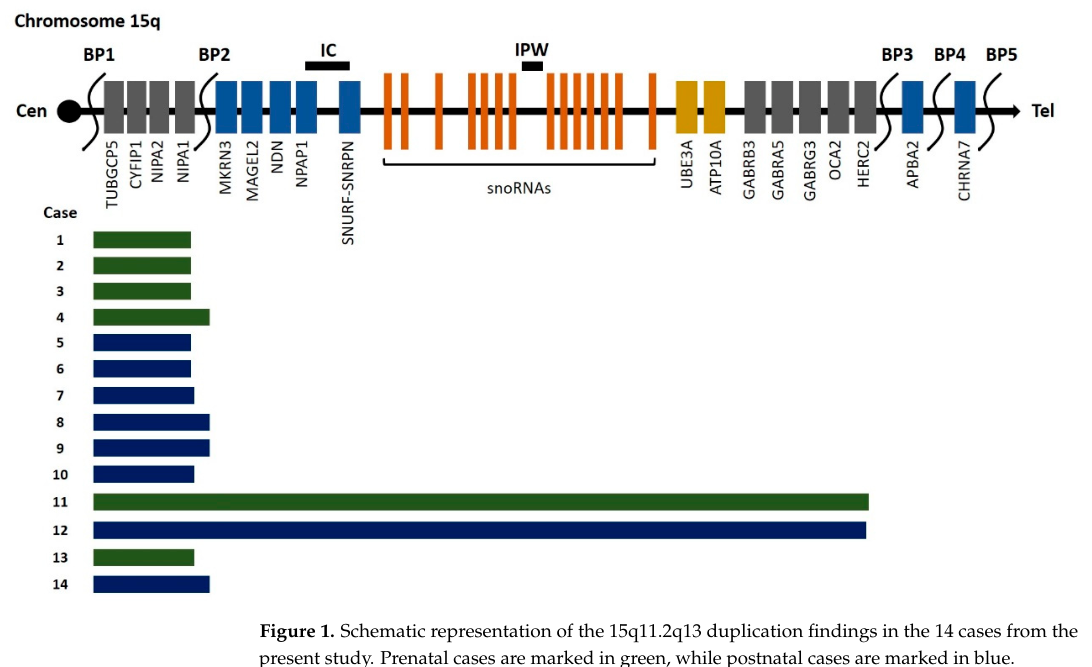
Specific breakpoints (BP1-BP5) that lead to deletions and duplications of
variable size have been identified.
explanation: >-
This supports named LCR breakpoints as generating variable duplication
sizes in Dup15q syndrome.
- name: GABRB3 duplicated-region dosage
gene_term:
preferred_term: GABRB3
term:
id: hgnc:4083
label: GABRB3
association: Duplicated GABA-A receptor subunit gene in the 15q11.2-q13.1 region
notes: >-
GABRB3 is part of the non-imprinted GABA-A receptor subunit gene cluster
within the duplicated region and is implicated in the elevated beta EEG
phenotype and GABAergic signaling disruption.
evidence:
- reference: PMID:34344470
reference_title: >-
Abnormal sleep physiology in children with 15q11.2-13.1 duplication
(Dup15q) syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
a cluster of gamma-aminobutyric acid type A receptor (GABAAR) genes,
GABRB3, GABRA5, and GABRG3, which encode the β3, α5 and γ3 receptor
subunits, respectively.
explanation: >-
This identifies GABRB3 as one of the duplicated GABA-A receptor subunit
genes in the 15q11.2-q13.1 critical region.
- name: GABRA5 duplicated-region dosage
gene_term:
preferred_term: GABRA5
term:
id: hgnc:4079
label: GABRA5
association: Duplicated GABA-A receptor subunit gene in the 15q11.2-q13.1 region
notes: >-
GABRA5 is part of the non-imprinted GABA-A receptor subunit gene cluster
within the duplicated region and is implicated in the elevated beta EEG
phenotype and GABAergic signaling disruption.
evidence:
- reference: PMID:34344470
reference_title: >-
Abnormal sleep physiology in children with 15q11.2-13.1 duplication
(Dup15q) syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
a cluster of gamma-aminobutyric acid type A receptor (GABAAR) genes,
GABRB3, GABRA5, and GABRG3, which encode the β3, α5 and γ3 receptor
subunits, respectively.
explanation: >-
This identifies GABRA5 as one of the duplicated GABA-A receptor subunit
genes in the 15q11.2-q13.1 critical region.
- name: GABRG3 duplicated-region dosage
gene_term:
preferred_term: GABRG3
term:
id: hgnc:4088
label: GABRG3
association: Duplicated GABA-A receptor subunit gene in the 15q11.2-q13.1 region
notes: >-
GABRG3 is part of the non-imprinted GABA-A receptor subunit gene cluster
within the duplicated region and is implicated in the elevated beta EEG
phenotype and GABAergic signaling disruption.
evidence:
- reference: PMID:34344470
reference_title: >-
Abnormal sleep physiology in children with 15q11.2-13.1 duplication
(Dup15q) syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
a cluster of gamma-aminobutyric acid type A receptor (GABAAR) genes,
GABRB3, GABRA5, and GABRG3, which encode the β3, α5 and γ3 receptor
subunits, respectively.
explanation: >-
This identifies GABRG3 as one of the duplicated GABA-A receptor subunit
genes in the 15q11.2-q13.1 critical region.
environmental: []
treatments:
- name: Supportive care
description: >-
Multidisciplinary supportive care is central, especially for developmental,
feeding, communication, educational, and behavioral management. Core
services include physical therapy for hypotonia and gross motor delay,
occupational therapy for fine-motor and adaptive skills, speech-language
therapy with augmentative and alternative communication when needed, feeding
therapy, behavioral therapy such as applied behavior analysis, and
psychotropic medication when behavioral manifestations require medication.
treatment_term:
preferred_term: Supportive Care
term:
id: NCIT:C15747
label: Supportive Care
evidence:
- reference: PMID:27308687
reference_title: Maternal 15q Duplication Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Supportive care may include: feeding therapy, occupational and physical
therapy, alternative and augmentative communication, behavioral therapy
(e.g., applied behavioral analysis therapy), psychotropic medications
for behavioral manifestations
explanation: >-
GeneReviews supports the specific developmental, communication, feeding,
behavioral, and psychotropic components of supportive management.
- name: Antiseizure medication
description: >-
Antiseizure medications are used for seizure control, although response is
variable and seizures may be refractory.
notes: >-
No prospective or randomized controlled trial data establish an antiseizure
medication hierarchy for Dup15q; therapy should be individualized by seizure
type, syndrome subtype, tolerability, and epilepsy specialist judgment.
treatment_term:
preferred_term: Pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
evidence:
- reference: PMID:41751547
reference_title: >-
Genomics of Complex Neurodevelopmental Disorders with Variable Epilepsy
Phenotypes: A Clinical Review of Dup15q Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
There is a well-known differential response to anti-seizure medications
and emerging evidence for neurostimulation and precision medicine.
explanation: >-
This directly supports antiseizure medication use in the syndrome.
- name: Seizure trigger avoidance and surveillance
description: >-
Families should receive counseling on avoidable seizure triggers such as
sleep deprivation and stress, and clinical follow-up should monitor for new
seizures, changes in seizure type, and nutritional or growth issues that can
affect epilepsy management.
treatment_term:
preferred_term: supportive care
term:
id: MAXO:0000950
label: supportive care
target_phenotypes:
- preferred_term: Seizure
term:
id: HP:0001250
label: seizure
evidence:
- reference: PMID:27308687
reference_title: Maternal 15q Duplication Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Agents/circumstances to avoid: Seizure triggers (e.g., sleep deprivation,
stress).
explanation: >-
GeneReviews explicitly identifies seizure-trigger avoidance as management
guidance for maternal Dup15q.
- name: Genetic counseling
description: >-
Genetic counseling should address maternal parent-of-origin effects,
subtype-specific de novo versus inherited recurrence risk, maternal
interstitial duplication transmission risk, testing of at-risk siblings, and
prenatal or preimplantation testing options.
treatment_term:
preferred_term: Genetic Counseling
term:
id: NCIT:C15240
label: Genetic Counseling
evidence:
- reference: PMID:27308687
reference_title: Maternal 15q Duplication Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Evaluation of relatives at risk: Consider genetic testing of sibs of a
proband
explanation: >-
GeneReviews supports genetic counseling and testing for relatives at risk
of inherited maternal interstitial 15q11.2-q13.1 duplication.
- reference: PMID:37029316
reference_title: >-
Population screening for 15q11-q13 duplications: corroboration of the
difference in impact between maternally and paternally inherited alleles.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We recommend reporting 15q11-q13 duplications identified during genome-wide
NIPS with appropriate genetic counselling for these pregnant women in the
interest of both mothers and future children.
explanation: >-
This supports the need for genetic counseling when 15q11-q13 duplications
are detected, especially in prenatal screening contexts.
- name: Investigational soticlestat
description: >-
Soticlestat (TAK-935/OV935), a cholesterol 24-hydroxylase inhibitor, was
evaluated as an adjunctive pharmacotherapy for motor seizure frequency in
participants with Dup15q syndrome in the phase II ARCADE trial. The trial
found increased motor seizure frequency in the Dup15q subgroup.
treatment_term:
preferred_term: pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
therapeutic_agent:
- preferred_term: soticlestat
term:
id: CHEBI:233158
label: soticlestat
target_phenotypes:
- preferred_term: Seizure
term:
id: HP:0001250
label: Seizure
evidence:
- reference: PMID:37011526
reference_title: >-
Efficacy, safety, and tolerability of soticlestat as adjunctive therapy
for the treatment of seizures in patients with Dup15q syndrome or CDKL5
deficiency disorder in an open-label signal-finding phase II study (ARCADE).
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Soticlestat treatment was associated with an increase in motor seizure
frequency in patients with Dup15q syndrome.
explanation: >-
Published ARCADE results document soticlestat was not effective in the
Dup15q subgroup; this evidence prevents overstating treatment benefit.
- name: Investigational all-trans-retinoic acid
description: >-
All-trans-retinoic acid (ATRA) is under investigation as a pharmacotherapy
for autism-related outcomes in Dup15q syndrome, with phase II clinical
trials assessing ADOS-2 outcomes in affected children.
treatment_term:
preferred_term: pharmacotherapy
term:
id: NCIT:C15986
label: Pharmacotherapy
therapeutic_agent:
- preferred_term: all-trans-retinoic acid
term:
id: CHEBI:15367
label: all-trans-retinoic acid
target_phenotypes:
- preferred_term: Autistic behavior
term:
id: HP:0000729
label: Autistic behavior
evidence:
- reference: clinicaltrials:NCT07079696
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
This study aims to evaluate ATRA treatment in children with Dup15q
syndrome-related autism , assessing changes in their ADOS-2 scores , to
potentially provide a novel therapeutic approach for autism treatment.
explanation: >-
Phase II trial registry record supports ATRA as an investigational
therapy for Dup15q-related autistic behavior.
diagnosis:
- name: Chromosomal copy-number testing
description: >-
Chromosomal copy-number testing is used to identify the recurrent
15q11-q13 duplication.
diagnosis_term:
preferred_term: genetic testing
term:
id: MAXO:0000127
label: genetic testing
evidence:
- reference: PMID:41751547
reference_title: >-
Genomics of Complex Neurodevelopmental Disorders with Variable Epilepsy
Phenotypes: A Clinical Review of Dup15q Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We synthesized current literature on genomic mechanisms underlying
complex neurodevelopmental disorders focusing on Dup15q syndrome and its
subtypes-int15, idic15, and mosaic idic15.
explanation: >-
This supports chromosomal testing to define the Dup15q subtype.
- reference: PMID:39457428
reference_title: >-
Chromosome 15q11-q13 Duplication Syndrome: A Review of the Literature and
14 New Cases.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
In the present study, a total of 14 pre- and postnatal cases were
diagnosed as 15q11.2q13 duplication carriers using Affymetrix CytoScan 750
K array-CGH, and our analysis combined these with 120 cases existing in
the literature.
explanation: >-
This directly supports chromosomal microarray/array-CGH copy-number
testing as a diagnostic method for Dup15q.
- reference: PMID:27308687
reference_title: Maternal 15q Duplication Syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The diagnosis of maternal dup15q is established by detection of at least
one extra maternally derived copy of the Prader-Willi/Angelman critical
region, a region approximately 5 Mb long within chromosome region
15q11.2-q13.1.
explanation: >-
GeneReviews supports detection of the extra maternally derived
Prader-Willi/Angelman critical-region copy as the diagnostic basis of
maternal Dup15q.
- name: Prenatal genome-wide NIPS detection
description: >-
Genome-wide cell-free DNA sequencing during non-invasive prenatal screening
can detect 15q11-q13 duplications, but positive screening findings require
genetic counseling and confirmatory clinical interpretation.
diagnosis_term:
preferred_term: prenatal genome-wide NIPS
term:
id: MAXO:0000127
label: genetic testing
evidence:
- reference: PMID:37029316
reference_title: >-
Population screening for 15q11-q13 duplications: corroboration of the
difference in impact between maternally and paternally inherited alleles.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
We recommend reporting 15q11-q13 duplications identified during
genome-wide NIPS with appropriate genetic counselling for these pregnant
women in the interest of both mothers and future children.
explanation: >-
This supports genome-wide NIPS as a prenatal screening route for detecting
15q11-q13 duplications, paired with genetic counseling rather than treated
as a standalone postnatal diagnosis.
- name: Electroencephalography
description: >-
EEG is used to characterize seizure type and severity.
diagnosis_term:
preferred_term: electroencephalography
term:
id: MAXO:0000932
label: electroencephalography
evidence:
- reference: PMID:24239951
reference_title: >-
Duplication of the 15q11-q13 region: clinical and genetic study of 30 new cases.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
18 patients had an abnormal EEG with a typical, recognizable pattern of
excessive diffuse rapid spikes in the waking record, similar to the
pattern observed after benzodiazepine exposure.
explanation: >-
This directly supports EEG as an abnormal diagnostic finding.
- name: EEG beta oscillation biomarker
description: >-
Quantitative EEG assessment of elevated beta power can serve as a
reproducible Dup15q biomarker with strong translational validity and may
support clinical-trial stratification or target-engagement readouts.
diagnosis_term:
preferred_term: electroencephalography
term:
id: MAXO:0000932
label: electroencephalography
evidence:
- reference: PMID:32791992
reference_title: Properties of beta oscillations in Dup15q syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
an electrophysiological biomarker of the syndrome, characterized by
excessive beta oscillations (12-30 Hz), resembling electroencephalogram
(EEG) changes induced by allosteric modulation of GABAARs.
explanation: >-
This defines the characteristic Dup15q beta oscillation EEG biomarker
and links it to GABAAR allosteric modulation.
- reference: PMID:32791992
reference_title: Properties of beta oscillations in Dup15q syndrome.
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Beta power and peak frequency showed high stability across repeated visits
(beta power ICC = 0.93, BPF ICC = 0.92). A reproducibility analysis
revealed that beta power estimates are comparable between research and
clinical EEG (ICC = 0.94).
explanation: >-
This supports elevated beta oscillations as a stable and clinically
reproducible EEG biomarker in Dup15q syndrome.
- reference: PMID:39014349
reference_title: >-
Sleep EEG signatures in mouse models of 15q11.2-13.1 duplication
(Dup15q) syndrome.
supports: SUPPORT
evidence_source: MODEL_ORGANISM
snippet: >-
that the beta EEG biomarker has strong translational validity
explanation: >-
Mouse-model EEG data support the translational validity of the Dup15q beta
EEG biomarker as a pharmacodynamic measure.
differential_diagnoses:
- name: Angelman syndrome
description: >-
Angelman syndrome overlaps because it involves the same imprinted region
but typically reflects loss of UBE3A function rather than duplication.
disease_term:
preferred_term: Angelman syndrome
term:
id: MONDO:0007113
label: Angelman syndrome
- name: Autism spectrum disorder
description: >-
Autism spectrum disorder is part of the Dup15q phenotype and may dominate
the presentation before the chromosomal duplication is recognized.
disease_term:
preferred_term: autism spectrum disorder
term:
id: MONDO:0005258
label: autism spectrum disorder
clinical_trials:
- name: NCT03694275
phase: PHASE_II
status: COMPLETED
description: >-
ARCADE was an open-label phase II soticlestat signal-finding study in
participants with Dup15q syndrome or CDKL5 deficiency disorder and motor
seizures.
target_phenotypes:
- preferred_term: Seizure
term:
id: HP:0001250
label: seizure
evidence:
- reference: clinicaltrials:NCT03694275
reference_title: >-
A Multicenter, Open-label, Pilot Study of TAK-935 (OV935) in Patients
With 15Q Duplication Syndrome or CDKL5 Deficiency Disorder (ARCADE Study)
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
The purpose of this study is to investigate the effect of soticlestat on
the frequency of motor seizures for participants with Dup15q or CDD during
the Maintenance Period.
explanation: >-
ClinicalTrials.gov documents a completed soticlestat trial targeting motor
seizure frequency in Dup15q syndrome.
- reference: PMID:37011526
reference_title: >-
Efficacy, safety, and tolerability of soticlestat as adjunctive therapy
for the treatment of seizures in patients with Dup15q syndrome or CDKL5
deficiency disorder in an open-label signal-finding phase II study
(ARCADE).
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Soticlestat treatment was associated with an increase in motor seizure
frequency in patients with Dup15q syndrome.
explanation: >-
The published ARCADE results show that this completed trial did not reduce
motor seizure frequency in the Dup15q subgroup, preventing the trial entry
from overstating treatment benefit.
- name: NCT05281965
phase: PHASE_I
status: UNKNOWN
description: >-
Early-phase randomized crossover study evaluating retinoic acid in patients
with 15q11-q13 duplication syndrome.
target_phenotypes:
- preferred_term: Autistic behavior
term:
id: HP:0000729
label: Autistic behavior
evidence:
- reference: clinicaltrials:NCT05281965
reference_title: >-
A Clinical Study Evaluating the Efficacy and Safety of Retinoic Acid in
Patients With 15q11-q13 Duplication Syndrome
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Therefore, retinoic acid supplementation in the treatment of dup15q
syndrome is a potential therapeutic target.
explanation: >-
ClinicalTrials.gov documents an early-phase retinoic-acid trial motivated
by UBE3A-related Dup15q biology.
- name: NCT07079696
phase: PHASE_II
status: NOT_RECRUITING
description: >-
Phase II single-group all-trans retinoic acid study in children ages 3 to 7
years with Dup15q syndrome-related autism. ClinicalTrials.gov currently
lists the trial as not yet recruiting; NOT_RECRUITING is the closest schema
status.
target_phenotypes:
- preferred_term: Autistic behavior
term:
id: HP:0000729
label: Autistic behavior
evidence:
- reference: clinicaltrials:NCT07079696
reference_title: >-
Investigating the Therapeutic Efficacy of All-trans Retinoic Acid in
Autism Spectrum Disorder Patients With 15q11-13 Duplication Syndrome
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
This study aims to evaluate ATRA treatment in children with Dup15q
syndrome-related autism , assessing changes in their ADOS-2 scores , to
potentially provide a novel therapeutic approach for autism treatment.
explanation: >-
ClinicalTrials.gov documents a larger phase II all-trans retinoic acid
study targeting Dup15q-related autistic behavior.
- name: NCT05307679
phase: PHASE_II
status: TERMINATED
description: >-
Phase II randomized study of basmisanil, a GABAA receptor negative
allosteric modulator, in children and adolescents with Dup15q syndrome.
evidence:
- reference: clinicaltrials:NCT05307679
reference_title: >-
A Phase II, Randomized, Double-Blind, Placebo-Controlled, Parallel Group
Study to Evaluate the Safety, Efficacy, and Pharmacodynamics of 52 Weeks
of Treatment With Basmisanil in Participants Aged 2 to 14 Years Old With
Dup15q Syndrome Followed by a 2-Year Optional Open-Label Extension
supports: SUPPORT
evidence_source: HUMAN_CLINICAL
snippet: >-
Part 1 will test the hypothesis that negative allosteric modulation of a
GABAA receptor subtype can address excessive receptor function and
positively impact core neurodevelopmental disease feature in individuals
with Dup15q syndrome.
explanation: >-
ClinicalTrials.gov documents a terminated phase II basmisanil trial based
on the GABAA receptor mechanism implicated in Dup15q syndrome.
datasets: []
notes: >-
PubMed-driven curation was expanded with Falcon deep research on 2026-05-30.
Falcon findings were treated as leads and only incorporated when independently
verified against fetched reference caches.
references:
- reference: PMID:27308687
title: Maternal 15q Duplication Syndrome.
tags:
- GeneReviews
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: PMID:36898382
title: >-
The role of UBE3A in the autism and epilepsy-related Dup15q syndrome using
patient-derived, CRISPR-corrected neurons.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: PMID:37029316
title: >-
Population screening for 15q11-q13 duplications: corroboration of the
difference in impact between maternally and paternally inherited alleles.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: PMID:37011526
title: >-
Efficacy, safety, and tolerability of soticlestat as adjunctive therapy for
the treatment of seizures in patients with Dup15q syndrome or CDKL5
deficiency disorder in an open-label signal-finding phase II study (ARCADE).
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: PMID:39457428
title: >-
Chromosome 15q11-q13 Duplication Syndrome: A Review of the Literature and
14 New Cases.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: PMID:32791992
title: Properties of beta oscillations in Dup15q syndrome.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: PMID:34344470
title: >-
Abnormal sleep physiology in children with 15q11.2-13.1 duplication
(Dup15q) syndrome.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: PMID:39014349
title: >-
Sleep EEG signatures in mouse models of 15q11.2-13.1 duplication (Dup15q)
syndrome.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: clinicaltrials:NCT03694275
title: >-
A Multicenter, Open-label, Pilot Study of TAK-935 (OV935) in Patients With
15Q Duplication Syndrome or CDKL5 Deficiency Disorder (ARCADE Study)
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: clinicaltrials:NCT05281965
title: >-
A Clinical Study Evaluating the Efficacy and Safety of Retinoic Acid in
Patients With 15q11-q13 Duplication Syndrome
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: clinicaltrials:NCT05307679
title: >-
A Phase II, Randomized, Double-Blind, Placebo-Controlled, Parallel Group
Study to Evaluate the Safety, Efficacy, and Pharmacodynamics of 52 Weeks of
Treatment With Basmisanil in Participants Aged 2 to 14 Years Old With
Dup15q Syndrome Followed by a 2-Year Optional Open-Label Extension
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: clinicaltrials:NCT07079696
title: >-
Investigating the Therapeutic Efficacy of All-trans Retinoic Acid in
Autism Spectrum Disorder Patients With 15q11-13 Duplication Syndrome
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: DOI:10.1002/brb3.3437
title: >-
Expanding deep phenotypic spectrum associated with atypical pathogenic
structural variations overlapping 15q11-q13 imprinting region.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: DOI:10.1016/j.stemcr.2023.02.002
title: >-
The role of UBE3A in the autism and epilepsy-related Dup15q syndrome using
patient-derived, CRISPR-corrected neurons.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: DOI:10.1038/s41431-023-01336-6
title: >-
Population screening for 15q11-q13 duplications: corroboration of the
difference in impact between maternally and paternally inherited alleles.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: DOI:10.1177/26330040241254122
title: >-
Linking Angelman and dup15q data for expanded research (LADDER) database:
a model for advancing research, clinical guidance, and therapeutic
development for rare conditions.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: DOI:10.1186/s11689-020-09326-1
title: Properties of beta oscillations in Dup15q syndrome.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: DOI:10.1186/s11689-024-09556-7
title: >-
Sleep EEG signatures in mouse models of 15q11.2-13.1 duplication (Dup15q)
syndrome.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: DOI:10.1186/s13229-021-00460-8
title: >-
Abnormal sleep physiology in children with 15q11.2-13.1 duplication
(Dup15q) syndrome.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: DOI:10.32345/2664-4738.2.2023.08
title: Inherited 15q duplication in three not related Ukrainian families.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
- reference: DOI:10.3390/genes15101304
title: >-
Chromosome 15q11-q13 Duplication Syndrome: A Review of the Literature and
14 New Cases.
found_in:
- 15q11q13_Microduplication_Syndrome-deep-research-falcon.md
References & Deep Research
References
21Deep Research
2Asta Literature Retrieval: Pathophysiology and clinical mechanisms of 15q11q13 Microduplication Syndrome. Core disease mechanisms, molecular and...
This report is retrieval-only and is generated directly from Asta results.
- Papers retrieved: 20
- Snippets retrieved: 20
Relevant Papers
[1] 16p13.11 deletion variants associated with neuropsychiatric disorders cause morphological and synaptic changes in induced pluripotent stem cell-derived neurons
- Authors: E. Buttermore, Nickesha C Anderson, Pin-Fang Chen, N. Makhortova, Kristina H. Kim et al.
- Year: 2022
- Venue: Frontiers in Psychiatry
- URL: https://www.semanticscholar.org/paper/c7feb5856b06514c3e9e70e5f293c829a6117c5d
- DOI: 10.3389/fpsyt.2022.924956
- PMID: 36405918
- PMCID: 9669751
- Summary: Patient-derived, induced pluripotent stem cells provide a platform for investigating the morphological, electrophysiological, and gene-expression changes that result from 16p13.11 CNVs in human-derived neurons and the identification of common phenotypes among neurons derived from patients with overlapping 16p 13.11 deletions will help to improve future treatment options and clinical outcomes.
- Evidence snippets:
- Snippet 1 (score: 0.487) > Mb (10). Since reported clinical phenotypes are heterogeneous, it has been difficult to establish a disease mechanism for how CNVs in this region affect neurodevelopment. > While detailed sequencing analysis of 16p13.11 CNVs have identified genes within affected regions that are commonly associated with NDDs, the relationship between observed genetic mutations and cellular phenotypes for many of the affected genes remains unknown. A recent study using induced pluripotent stem cell (iPSC)-derived neurons found that targeting the NF K B p65 pathway was able to correct proliferation deficits caused by 16p13.11 microduplication, implicating this pathway in the pathogenesis of this syndrome (11). Despite advances in understanding the mechanisms underlying 16p13.11 microduplication, the morphological and synaptic alterations that underlie the clinical phenotypes associated with 16p13.11 deletions have not been characterized. Furthermore, identification of common cellular phenotypes between patients with different 16p13.11 deletion sizes remains largely unknown. Studies directed at elucidating common phenotypes between patients with different mutation sizes provide the opportunity to establish genotype-phenotype relationships for key cell biological features that are responsive to phenotypic screening approaches capable of dissecting the molecular basis of dysregulated pathways in patient neurons. > Using exome sequencing and microarray analyses, we identified a subset of patients with early-and young adult-onset psychosis with heterozygous deletions within chromosome 16p13.11. In this study, we derived human iPSCs from two families with patients harboring 16p13.11 deletions (probands, patients) and familial controls. One of the probands has only interval I deleted, while the other two patients, a father and son pair, have both intervals I and II deleted (4). Patient-derived iPSCs provide a platform to study the cell-autonomous effects of 16p13.11 CNVs on human neurons. > We hypothesized that loss of region I, which includes the genes PDXDC1, NTAN1, and RRN3, would contribute to common cell autonomous phenotypes in iPSC-
[2] Clinical findings and genetic analysis of patients with copy number variants involving 17p13.3 using a single nucleotide polymorphism array: a single-center experience
- Authors: Bin Liang, Donghong Yu, Wantong Zhao, Yan Wang, Xiaoqing Wu et al.
- Year: 2022
- Venue: BMC Medical Genomics
- URL: https://www.semanticscholar.org/paper/c99e28f021fa90801c1054b5872ed4a20e492b66
- DOI: 10.1186/s12920-022-01423-5
- PMID: 36544138
- PMCID: 9773569
- Citations: 3
- Summary: The clinical significance of small duplications including YWHAE and CRK but not PAFAH1B1 remains uncertain, for which parental testing and clinical heterogeneity should be considered in genetic counseling.
- Evidence snippets:
- Snippet 1 (score: 0.477) > Moreover, in group I 17p13.3 microduplication, Curry et al. [23] reported that disruption of ABR and duplication of BHLHA9 were associated with clefts and split hand/foot with long bone deficiency phenotypes, respectively. Capra et al. [26] reported that a boy carrying a maternally inherited 329.5-kb 17p13.3 duplication, including BHLHA9, YWHAE, and CRK, presented with mild dysmorphic phenotype, autism, and mental retardation, while his mother was affected by a bipolar and borderline disorder and was addicted to alcohol. It can be seen that phenotypic heterogeneity existed in the mother and her child. Another report [27] described two patients manifesting distinctive features (patient 1, primary hypothyroidism; patient 2, bilateral cryptorchidism) that were not previously described in the duplication 17p13.3 spectrum. Whether these rare manifestations observed in the two patients were caused by a two-hit event or not is not known. Overall, considering 17p13.3 microduplication showing reduced penetrance, variable expressivity, and lack of a clear pathogenic mechanism, the clinical significance of the microduplication encompassing only YWHAE and CRK, but not PAFAH1B1, requires further investigation. > Interestingly, case 3 also carried a 74.2 Mb mosaic duplication of approximately 3.5 on chromosome 17p13.2q25.3 and a 1.0 Mb deletion in the 17q terminus, in addition to deletion of the MDS region. The SNP data were consistent with that some cells have ring 17 while others have dicentric or interlock ring 17. Given the dosage sensitivity of genes and regions involved in the three CNVs, case 3 may show compound manifestations of these known genomic disorders, such as MDS, Potocki-Lupski syndrome (MIM:610883) [12], Charcot-Marie-Tooth disease, type 1A (CMT1A, MIM:118220) [28,29], 17q11.2
[3] Cytogenomic Abnormalities and Underlying Mechanisms for Intellectual and Developmental Disabilities
- Authors: Peining Li
- Year: 2013
- Venue: Journal of Molecular and Genetic Medicine
- URL: https://www.semanticscholar.org/paper/2578c174427082f0beff180c57c6414783a4601b
- DOI: 10.4172/1747-0862.1000073
- Citations: 1
- Summary: Functional analyses using in vitro cellular phenotyping and in vivo animal modeling have been developed for clinically detected pCNVs and there is urgent demand for rapid transition from diagnostic discovery to study of disease-causing mechanisms and exploration of therapeutic approaches.
- Evidence snippets:
- Snippet 1 (score: 0.471) > For many newly detected pCNVs, little is known about the dosagesensitive genes and their cellular and developmental functions. The limited availability and accessibility of live brain and neuron tissues is the major obstacle in the study of disease-causing mechanisms in human mental development. Recent progress in stem cell technologies has made possible the modeling of human mental diseases using patient derived stem cells. In 2010, Marchetto et al. developed a culture system using induced pluripotent stem cells (iPSCs) from Rett syndrome patients' fibroblasts [8]. These Rett syndrome iPSCs were able to undergo X-inactivation and generate functional neurons. Neurons derived from these iPSCs had fewer synapses, reduced spine density, smaller soma size, altered calcium signaling and electrophysiological defects when compared to controls. This cellular model provided critical evidence of an unexplored developmental window before disease onset and enable direct testing of drug effect in rescuing synaptic defects. > The microdeletion and microduplication at the same genomic locus offer an opportunity to study dosage-sensitive genes, especially for the opposite phenotypes of haploinsufficient and triple-sensitive genes. However, clinical evaluation could be complicated by overlapped phenotypes, variable expressivity, reduced penetrance and lack of longitudinal study of late-onset phenotypes for many genomic disorders. Recent studies observed opposite phenotypes in a few genomic disorders. For example, the microdeletion syndrome at 16p11.2 (OMIM#611913) and the reciprocal microduplication syndrome (OMIM#614671) were initially associated with ASD but a subsequent study revealed mirror body mass index phenotypes. Microdeletion at 16p11.2 is often associated with obesity, macrocephaly and ASD, while the reciprocal microduplication is associated with underweight, microcephaly and schizophrenia [9]. Mouse models of 16p11.2 microdeletion and microduplication detected in vivo brain anomalies and behavior disorders [10]. Overexpression and transcript suppression of the 29 candidate genes from this 16p11.2
[4] Disorders of the genome architecture: a review
- Authors: Dhavendra Kumar
- Year: 2008
- Venue: Genomic Medicine
- URL: https://www.semanticscholar.org/paper/df00164481646356263fd7a235e072cde2e723e1
- DOI: 10.1007/s11568-009-9028-2
- PMID: 19277903
- Citations: 35
- Influential citations: 3
- Summary: Widespread application of high-resolution genome analyses may offer to detect more sporadic phenotypes resulting from genomic rearrangements involving de novo copy number variation.
- Evidence snippets:
- Snippet 1 (score: 0.454) > Genetic diseases are recognized to be one of the major categories of human disease. Traditionally genetic diseases are subdivided into chromosomal (numerical or structural aberrations), monogenic or Mendelian diseases, multifactorial/polygenic complex diseases and mitochondrial genetic disorders. A large proportion of these conditions occur sporadically. With the advent of newer molecular techniques, a number of new disorders and dysmorphic syndromes are delineated in detail. Some of these conditions do not conform to the conventional inheritance patterns and mechanisms are often complex and unique. Examples include submicroscopic microdeletions or microduplications, trinucleotide repeat disorders, epigenetic disorders due to genomic imprinting, defective transcription or translation due to abnormal RNA patterning and pathogenic association with single nucleotide polymorphisms and copy number variations. Among these several apparently monogenic disorders result from non-allelic homologous recombination associated with the presence of low copy number repeats on either side of the critical locus or gene cluster. The term ‘disorders of genome architecture’ is alternatively used to highlight these disorders, for example Charcot-Marie-Tooth type IA, Smith-Magenis syndrome, Neurofibromatosis type 1 and many more with an assigned OMIM number. Many of these so called genomic disorders occur sporadically resulting from largely non-recurrent de novo genomic rearrangements. Locus-specific mutation rates for genomic rearrangements appear to be two to four times greater than nucleotide-specific rates for base substitutions. Recent studies on several disease-associated recombination hotspots in male-germ cells indicate an excess of genomic rearrangements resulting in microduplications that are clinically underdiagnosed compared to microdeletion syndromes. Widespread application of high-resolution genome analyses may offer to detect more sporadic phenotypes resulting from genomic rearrangements involving de novo copy number variation.
[5] Consequences of aneuploidy in human fibroblasts with trisomy 21
- Authors: Sunyoung Hwang, Paola Cavaliere, Rui Li, L. Zhu, Noah E. Dephoure et al.
- Year: 2020
- Venue: Proceedings of the National Academy of Sciences of the United States of America
- URL: https://www.semanticscholar.org/paper/5ae9f7792cd2e4a8e2d6178f5a322da9f96ba3ac
- DOI: 10.1101/2020.08.14.251082
- PMID: 33526671
- PMCID: 8017964
- Citations: 57
- Influential citations: 6
- Summary: It is shown that several aneuploidy-associated phenotypes are present in trisomy 21 cells, including lower viability and increased dependency on serine-driven lipid synthesis, and the lack of evidence for widespread dosage compensation or dysregulation of chromosomal domains in human autosomes is supported.
- Evidence snippets:
- Snippet 1 (score: 0.450) > Significance An abnormal number of chromosomes or aneuploidy accounts for most spontaneous abortions, as missegregation of a single chromosome during development is often lethal. Only individuals with trisomy 21, which causes Down syndrome, can live to adulthood but show cognitive disabilities, increased risk for leukemias, autoimmune disorders, and clinical symptoms associated with premature aging. The mechanisms by which aneuploidy affects cellular function to cause Down syndrome are not understood. Our studies revealed that aneuploidy causes several defects in cells from individuals with Down syndrome. These include increased gene and protein expression, lower viability, and increased dependency on serine to proliferate. Our studies establish a critical role of aneuploidy, independent of triplicated gene identity, in driving cellular defects associated with trisomy 21. An extra copy of chromosome 21 causes Down syndrome, the most common genetic disease in humans. The mechanisms contributing to aneuploidy-related pathologies in this syndrome, independent of the identity of the triplicated genes, are not well defined. To characterize aneuploidy-driven phenotypes in trisomy 21 cells, we performed global transcriptome, proteome, and phenotypic analyses of primary human fibroblasts from individuals with Patau (trisomy 13), Edwards (trisomy 18), or Down syndromes. On average, mRNA and protein levels were increased by 1.5-fold in all trisomies, with a subset of proteins enriched for subunits of macromolecular complexes showing signs of posttranscriptional regulation. These results support the lack of evidence for widespread dosage compensation or dysregulation of chromosomal domains in human autosomes. Furthermore, we show that several aneuploidy-associated phenotypes are present in trisomy 21 cells, including lower viability and increased dependency on serine-driven lipid synthesis. Our studies establish a critical role of aneuploidy, independent of triplicated gene identity, in driving cellular defects associated with trisomy 21.
[6] A Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features
- Authors: O. Palumbo, P. Palumbo, Ester Di Muro, L. Cinque, A. Petracca et al.
- Year: 2020
- Venue: Genes
- URL: https://www.semanticscholar.org/paper/2101a5069af4ecb28806ee1f83f4bf2ab659a02a
- DOI: 10.3390/genes11060707
- PMID: 32604767
- PMCID: 7349372
- Citations: 18
- Influential citations: 1
- Summary: AnKRD11, CDH15, and CTU2 are proposed as candidate genes for explaining the related neurodevelopmental manifestations shared by these patients with overlapping 16.2q24.3 microduplication, providing supporting evidence of an emerging syndrome.
- Evidence snippets:
- Snippet 1 (score: 0.447) > Also, in vitro functional studies showed that mutant proteins result in decreased cell adhesion suggesting that CDH15 alterations, either alone or in combination with other factors, likely play a role in the etiology of ID [21]. Finally, copy number variations (both deletions and duplications) affecting other genes involved in neural cell adhesion molecules have been recently associated with neurodevelopmental disorders [22,23]. Accordingly, 16q24.2q24.3 microduplication can be added to available data corroborating a key role of these cellular pathways in cognitive development. > CTU2 is an additional candidate gene mapping into 16q24.2q24.3 microduplication SRO and encoding a protein involved in the post-transcriptional modification of transfer RNAs (tRNAs). This protein plays a role in thiolation of uridine residue present at the wobble position in a subset of tRNAs, resulting in enhanced codon reading accuracy. Biallelic variants in CTU2 have been associated with a specific syndromic phenotype featuring microcephaly, facial dysmorphism, renal agenesis, and ambiguous genitalia [24,25], and this gene has been recently listed into the Developmental Disorders Genotype-Phenotype Database (DDG2P). > Altogether, the evidence emerging from our study and the current knowledge concerning the proposed candidate genes support our hypothesis that their copy number alteration contribute to the etiology of the clinical phenotype observed in patients with 16q24.2q24.3 microduplication mainly for neurodevelopmental features shared among affected individuals. > For the other genes duplicated in patients discussed in the present study, although none of them seem to be clearly associable with the clinical traits reported, we cannot exclude their involvement in the etiology of the clinical condition. More detailed genetic and/or functional studies, or patients with point mutations/CNVs affecting only one or a few of these genes, are needed to elucidate this possibility.
[7] Exploring pathway interactions to detect molecular mechanisms of disease: 22q11.2 deletion syndrome
- Authors: Woosub Shin, M. Kutmon, Eleni Mina, Therese van Amelsvoort, C. Evelo et al.
- Year: 2023
- Venue: Orphanet Journal of Rare Diseases
- URL: https://www.semanticscholar.org/paper/e7f38266ecbaf1d1da3e525e1969a29f36c1cddc
- DOI: 10.1186/s13023-023-02953-6
- PMID: 37872602
- PMCID: 10594698
- Citations: 3
- Summary: The pathway interaction method was able to detect a molecular network that could possibly explain the development of neuropsychiatric diseases among the 22q11DS patients, and could be used for similar contexts, where complex genetic mechanisms need to be identified to explain the resulting phenotypic plasticity.
- Evidence snippets:
- Snippet 1 (score: 0.440) > Background 22q11.2 Deletion Syndrome (22q11DS) is a genetic disorder characterized by the deletion of adjacent genes at a location specified as q11.2 of chromosome 22, resulting in an array of clinical phenotypes including autistic spectrum disorder, schizophrenia, congenital heart defects, and immune deficiency. Many characteristics of the disorder are known, such as the phenotypic variability of the disease and the biological processes associated with it; however, the exact and systemic molecular mechanisms between the deleted area and its resulting clinical phenotypic expression, for example that of neuropsychiatric diseases, are not yet fully understood. Results Using previously published transcriptomics data (GEO:GSE59216), we constructed two datasets: one set compares 22q11DS patients experiencing neuropsychiatric diseases versus healthy controls, and the other set 22q11DS patients without neuropsychiatric diseases versus healthy controls. We modified and applied the pathway interaction method, originally proposed by Kelder et al. (2011), on a network created using the WikiPathways pathway repository and the STRING protein-protein interaction database. We identified genes and biological processes that were exclusively associated with the development of neuropsychiatric diseases among the 22q11DS patients. Compared with the 22q11DS patients without neuropsychiatric diseases, patients experiencing neuropsychiatric diseases showed significant overrepresentation of regulated genes involving the natural killer cell function and the PI3K/Akt signalling pathway, with affected genes being closely associated with downregulation of CRK like proto-oncogene adaptor protein. Both the pathway interaction and the pathway overrepresentation analysis observed the disruption of the same biological processes, even though the exact lists of genes collected by the two methods were different. Conclusions Using the pathway interaction method, we were able to detect a molecular network that could possibly explain the development of neuropsychiatric diseases among the 22q11DS patients. This way, our method was able to complement the pathway overrepresentation analysis, by filling the knowledge gaps on how the affected pathways are linked to the original deletion on chromosome 22. We expect our pathway interaction method could be used for problems with similar contexts, where complex genetic mechanisms need to be identified to explain the
[8] Investigating the role of NPR1 in dilated cardiomyopathy and its potential as a therapeutic target for glucocorticoid therapy
- Authors: Yaomeng Huang, Tongxin Li, Shichao Gao, Shuyu Li, Xiaoran Zhu et al.
- Year: 2023
- Venue: Frontiers in Pharmacology
- URL: https://www.semanticscholar.org/paper/be229f6f2059faab4c97ec0a04bd055adab9dfe1
- DOI: 10.3389/fphar.2023.1290253
- PMID: 38026943
- PMCID: 10662320
- Citations: 3
- Summary: Natriuretic peptide receptor 1 (NPR1) was identified as a core gene associated with DCM through bioinformatics analysis and led to substantial improvements in cardiac and renal function, accompanied by an upregulation of NPR1 expression.
- Evidence snippets:
- Snippet 1 (score: 0.429) > Multiple pathways and molecules are involved in this process; however, the detailed underlying mechanisms remain unclear. In recent years, with the development of high-throughput sequencing and gene chip technologies, the use of bioinformatics technology to explore the occurrence, development, and prognosis of diseases has become a hot topic for scholars worldwide (Hwang et al., 2018;Nayor et al., 2019;Rinschen et al., 2019;Sturm et al., 2019;Montaner et al., 2020). > The present study aimed to use bioinformatics technology to screen for DCM-related genes and investigate their mechanisms, with the purpose of revealing the pathogenesis of DCM and seeking treatment methods. The GSE3586 dataset, containing expression profiles related to DCM, was selected from the Gene Expression Omnibus (GEO) database. This study aimed to predict the core genes that may play crucial roles in disease progression at the molecular level through the enrichment of relevant molecular pathways associated with DCM. Furthermore, the phenotype of the core genes was validated to further support the results of the bioinformatics analysis through basic and clinical experiments. Additionally, the role of glucocorticoids in DCM treatment is discussed in this article with the purpose of providing a theoretical and experimental basis for exploring the pathogenesis of DCM and elucidating therapeutic methods. This study also provides a theoretical reference for the interpretation, early diagnosis, and treatment of DCM.
[9] Retinoic Acid Induced 1, RAI1: A Dosage Sensitive Gene Related to Neurobehavioral Alterations Including Autistic Behavior
- Authors: P. Carmona-Mora, K. Walz
- Year: 2010
- Venue: Current Genomics
- URL: https://www.semanticscholar.org/paper/fd71900e9fb4ef4a9ae5290a08e485137368bdd1
- DOI: 10.2174/138920210793360952
- PMID: 21629438
- PMCID: 3078685
- Citations: 54
- Influential citations: 5
- Summary: The evidence of RAI1 as a dosage sensitive gene, its relationship with different neuro behavioral traits, gene structure and mutations, and what is known about its molecular and cellular function are discussed, as a first step in the elucidation of the mechanisms that relate dosage sensitive genes with abnormal neurobehavioral outcomes.
- Evidence snippets:
- Snippet 1 (score: 0.421) > Genomic structural changes, such as gene Copy Number Variations (CNVs) are extremely abundant in the human genome. An enormous effort is currently ongoing to recognize and catalogue human CNVs and their associations with abnormal phenotypic outcomes. Recently, several reports related neuropsychiatric diseases (i.e. autism spectrum disorders, schizophrenia, mental retardation, behavioral problems, epilepsy) with specific CNV. Moreover, for some conditions, both the deletion and duplication of the same genomic segment are related to the phenotype. Syndromes associated with CNVs (microdeletion and microduplication) have long been known to display specific neurobehavioral traits. It is important to note that not every gene is susceptible to gene dosage changes and there are only a few dosage sensitive genes. Smith-Magenis (SMS) and Potocki-Lupski (PTLS) syndromes are associated with a reciprocal microdeletion and microduplication within chromosome 17p11.2. in humans. The dosage sensitive gene responsible for most phenotypes in SMS has been identified: the Retinoic Acid Induced 1 (RAI1). Studies on mouse models and humans suggest that RAI1 is likely the dosage sensitive gene responsible for clinical features in PTLS. In addition, the human RAI1 gene has been implicated in several neurobehavioral traits as spinocerebellar ataxia (SCA2), schizophrenia and non syndromic autism. In this review we discuss the evidence of RAI1 as a dosage sensitive gene, its relationship with different neurobehavioral traits, gene structure and mutations, and what is known about its molecular and cellular function, as a first step in the elucidation of the mechanisms that relate dosage sensitive genes with abnormal neurobehavioral outcomes.
[10] New insights into candidate genes for autism spectrum disorder in 8p23.1 duplication syndrome
- Authors: M. M. Côrrea, Thiago Corrêa, C. Santos-Rebouças, Marino Miloca Rodrigues, G. Luca et al.
- Year: 2022
- Venue: Brazilian Journal of Case Reports
- URL: https://www.semanticscholar.org/paper/7549630ec79b57d7221fb427280bd360a35590b2
- DOI: 10.52600/2763-583x.bjcr.2023.3.1.16-23
- Summary: Clinical and cytomolecular findings of an 8p23.1 duplication in a boy with mild facial dysmorphisms, cardiac anomalies and ASD are described, pointing out crucial interactions among BLK, GATA4, PINX1, and TNKS and genes associated with ASD.
- Evidence snippets:
- Snippet 1 (score: 0.418) > The 8p23.1 duplication syndrome is a rare condition, characterized by dysmorphisms, intellectual disability, congenital cardiac anomalies, and autism spectrum disorder (ASD). The current model for explaining the pathogenesis of this condition postulates that few dosage-sensitive genes within the duplication are sufficient for the core clinical features, although the molecular mechanisms leading to the ASD presentation remain to be solved. Herein, we described clinical and cytomolecular findings of an 8p23.1 duplication in a boy with mild facial dysmorphisms, cardiac anomalies and ASD. Therefore, we investigated the influence of duplicated genes on the pathophysiology of ASD in our patient. We identified four duplicated genes (BLK, GATA4, PINX1, TNKS) connected with proteins previously associated with ASD and involved in significant enriched pathways associated with human neurological conditions. Moreover, the candidate genes are highly expressed in brain regions associated to ASD, such as the hippocampus. Taken together, these results point out crucial interactions among BLK, GATA4, PINX1, and TNKS and genes associated with ASD. We indicate cellular networks perturbations encompassing neuronal development pathways related to our patient's condition. Thus, these findings bring new insights into the genetic basis of ASD in patients with 8p23.1 duplication syndrome.
[11] Transcriptional profiling of Hutchinson-Gilford progeria patients identifies primary target pathways of progerin
- Authors: Sandra Vidak, Sohyoung Kim, Tom Misteli
- Year: 2026
- Venue: Nucleus
- URL: https://www.semanticscholar.org/paper/4bd99b0875508364d8672b6da5a50d024d485a53
- DOI: 10.1080/19491034.2025.2611484
- PMID: 41489464
- PMCID: 12773485
- Summary: To probe the clinical relevance of previously implicated cellular pathways and to address the extent of gene expression heterogeneity between patients, transcriptomic analysis of a comprehensive set of HGPS patients finds misexpression of several cellular pathways, including multiple signaling pathways, the UPR and mesodermal cell fate specification.
- Evidence snippets:
- Snippet 1 (score: 0.416) > Oxidative stress represents another key pathogenic mechanism in HGPS, as impaired NRF2 activity or increased reactive oxygen species (ROS) levels are sufficient to recapitulate HGPSassociated phenotypes [17,32,60]. Collectively, these findings underscore the multifactorial nature of HGPS pathogenesis, implicating interconnected signaling cascades involved in inflammation, oxidative stress, proteostasis, and vascular remodeling. Reassuringly, our findings indicate that many of the major pathways that have been described to contribute to HGPS phenotypes in mouse and cellular disease models are also misregulated in progeria patients, and targeting these pathways may provide therapeutic avenues to mitigate disease severity and improve outcomes in HGPS. > Although individuals with HGPS typically exhibit a characteristic set of clinical features, such as craniofacial abnormalities, growth retardation, and cardiovascular complications, there is notable variability in the age of onset, severity, and progression of symptoms between patients [7,9]. At the cellular level, HGPS is associated with several hallmark abnormalities, including nuclear envelope defects, decreased expression of several nuclear proteins and epigenetic marks, mitochondrial dysfunction, and increased cellular senescence [1,11,30,31,61]. These cellular phenotypes also exhibit considerable variation between patients, possibly contributing to differences in clinical outcomes. Our results indicate that even though some degree of transcriptional heterogeneity between the individual patients exists, the majority of patients exhibit misregulation of a set of shared pathways, suggesting that these pathways are universal driver mechanisms in HGPS. Further work is needed to understand the molecular and genetic factors that underlie inter-individual variability in disease expression and progression. > A limitation of pathway analysis of HGPS patient samples is to distinguish the pathways which are directly targeted by the disease-causing progerin protein and the emergence of adaptive secondary response pathways during progression of the disease in patients during their lifetime. The same caveat applies to the use of cell-based models used in the study of HGPS disease mechanisms.
[12] Neurodevelopmental Disorders Associated with Abnormal Gene Dosage: Smith–Magenis and Potocki–Lupski Syndromes
- Authors: Juanita Neira-Fresneda, L. Potocki
- Year: 2015
- Venue: Journal of Pediatric Genetics
- URL: https://www.semanticscholar.org/paper/ae2f935107027507a7fda71a609b1a42c7e12981
- DOI: 10.1055/s-0035-1564443
- PMID: 27617127
- PMCID: 4918721
- Citations: 51
- Influential citations: 1
- Summary: The neurobehavioral phenotypes of SMS and PTLS patients during different life phases are described as well as clinical guidelines for diagnosis and a multidisciplinary approach once diagnosis is confirmed by array comparative genomic hybridization or RAI1 gene sequencing.
- Evidence snippets:
- Snippet 1 (score: 0.413) > The proximal short arm of chromosome 17 is a genomic region that is prone to rearrangements which have been extensively characterized elsewhere. 1,2 Several distinct genomic disorders map to this region including the autosomal dominant peripheral neuropathies such as Charcot-Marie-Tooth disease type 1A (CMT1A, MIM#118220) and hereditary neuropathy with liability to pressure palsies (HNPP, MIM#162500), the chromosomal microduplication/microdeletion syndromes, Potocki-Lupski syndrome (PTLS, MIM#610883), and Smith-Magenis syndrome (SMS, MIM#182290), as well as the newly described PMP22-RAI1 duplication syndrome (Yuan et al, unpublished data, 2015). > Although haploinsufficiency of the single retinoic acidinduced gene (RAI1) is responsible for much of the phenotype in SMS, 3,4 both SMS and PTLS are examples of contiguous gene syndromes (CGS), as the clinical features of each are due to abnormal dosage and variation of physically contiguous yet functionally unrelated genes in the 17p11.2 genomic region. 5 The mechanism leading to genomic rearrangements in common microdeletion syndromes was first elucidated in SMS. 6 Interestingly, the clinical syndrome associated with duplication 17p11.2 (now known as PTLS) was initially defined based on the shared molecular structure among patients, the duplication representing the mechanistically predicted homologous recombination reciprocal of the SMS microdeletion. 7 Keywords ► congenital heart disease ► autism ► intellectual disability ► mirror traits ► gene dosage Abstract Smith-Magenis syndrome (SMS) and Potocki-Lupski syndrome (PTLS) are reciprocal contiguous gene syndromes within the well-characterized 17p11.2 region. Approximately 3.6 Mb microduplication of 17p11.2, known as PTLS, represents the mechanistically predicted homologous recombination reciprocal of the SMS microdeletion, both resulting in multiple congenital anomalies. Mouse model studies have revealed that the retinoic acid-inducible
[13] Identification of molecular signatures and pathways involved in Rett syndrome using a multi-omics approach
- Authors: Ainhoa Pascual-Alonso, Clara Xiol, Dmitrii Smirnov, R. Kopajtich, H. Prokisch et al.
- Year: 2023
- Venue: Human Genomics
- URL: https://www.semanticscholar.org/paper/8a7c3afd9cb1678bc1463754753cf69512f23eaa
- DOI: 10.1186/s40246-023-00532-1
- PMID: 37710353
- PMCID: 10503149
- Citations: 10
- Summary: Background Rett syndrome (RTT) is a neurodevelopmental disorder mainly caused by mutations in the methyl-CpG-binding protein 2 gene ( MECP2 ). MeCP2 is a multi-functional protein involved in many cellular processes, but the mechanisms by which its dysfunction causes disease are not fully understood. The duplication of the MECP2 gene causes a distinct disorder called MECP2 duplication syndrome (MDS), highlighting the importance of tightly regulating its dosage for proper cellular function. Add...
- Evidence snippets:
- Snippet 1 (score: 0.413) > Background Rett syndrome (RTT) is a neurodevelopmental disorder mainly caused by mutations in the methyl-CpG-binding protein 2 gene ( MECP2 ). MeCP2 is a multi-functional protein involved in many cellular processes, but the mechanisms by which its dysfunction causes disease are not fully understood. The duplication of the MECP2 gene causes a distinct disorder called MECP2 duplication syndrome (MDS), highlighting the importance of tightly regulating its dosage for proper cellular function. Additionally, some patients with mutations in genes other than MECP2 exhibit phenotypic similarities with RTT, indicating that these genes may also play a role in similar cellular functions. The purpose of this study was to characterise the molecular alterations in patients with RTT in order to identify potential biomarkers or therapeutic targets for this disorder. Methods We used a combination of transcriptomics (RNAseq) and proteomics (TMT mass spectrometry) to characterise the expression patterns in fibroblast cell lines from 22 patients with RTT and detected mutation in MECP2 , 15 patients with MDS, 12 patients with RTT-like phenotypes and 13 healthy controls. Transcriptomics and proteomics data were used to identify differentially expressed genes at both RNA and protein levels, which were further inspected via enrichment and upstream regulator analyses and compared to find shared features in patients with RTT. Results We identified molecular alterations in cellular functions and pathways that may contribute to the disease phenotype in patients with RTT, such as deregulated cytoskeletal components, vesicular transport elements, ribosomal subunits and mRNA processing machinery. We also compared RTT expression profiles with those of MDS seeking changes in opposite directions that could lead to the identification of MeCP2 direct targets. Some of the deregulated transcripts and proteins were consistently affected in patients with RTT-like phenotypes, revealing potentially relevant molecular processes in patients with overlapping traits and different genetic aetiology. Conclusions The integration of data in a multi-omics analysis has helped to interpret the molecular consequences of MECP2 dysfunction, contributing to the characterisation of the molecular landscape in patients with RTT. The comparison with MDS provides knowledge of MeCP2 direct targets, whilst the correlation with RTT-
[14] The contribution of genetic determinants of blood gene expression and splicing to molecular phenotypes and health outcomes
- Authors: A. Tokolyi, E. Persyn, A. Nath, K. Burnham, J. Marten et al.
- Year: 2025
- Venue: Nature Genetics
- URL: https://www.semanticscholar.org/paper/5435e12fd796fca6db2c0ce1844b6fc252d2d73e
- DOI: 10.1038/s41588-025-02096-3
- PMID: 40038547
- PMCID: 11906350
- Citations: 12
- Summary: This study mapped blood gene expression and splicing quantitative trait loci and uncovered gene-regulatory mechanisms at disease loci with therapeutic implications, such as WARS1 in hypertension, IL7R in dermatitis and IFNAR2 in COVID-19.
- Evidence snippets:
- Snippet 1 (score: 0.413) > The biological mechanisms through which most nonprotein-coding genetic variants affect disease risk are unknown. To investigate gene-regulatory mechanisms, we mapped blood gene expression and splicing quantitative trait loci (QTLs) through bulk RNA sequencing in 4,732 participants and integrated protein, metabolite and lipid data from the same individuals. We identified cis-QTLs for the expression of 17,233 genes and 29,514 splicing events (in 6,853 genes). Colocalization analyses revealed 3,430 proteomic and metabolomic traits with a shared association signal with either gene expression or splicing. We quantified the relative contribution of the genetic effects at loci with shared etiology, observing 222 molecular phenotypes significantly mediated by gene expression or splicing. We uncovered gene-regulatory mechanisms at disease loci with therapeutic implications, such as WARS1 in hypertension, IL7R in dermatitis and IFNAR2 in COVID-19. Our study provides an open-access resource on the shared genetic etiology across transcriptional phenotypes, molecular traits and health outcomes in humans (https://IntervalRNA.org.uk). > The majority of genetic variants associated with common diseases and other complex traits identified through genome-wide association studies (GWAS) lie in nonprotein-coding sequences 1 . Consequently, the molecular mechanisms that underpin many of these genotype-phenotype associations are unclear. Molecular quantitative trait locus (QTL) mapping studies, which identify genetic determinants of transcript, protein or metabolite abundance, can address this knowledge gap by identifying the molecular intermediaries that mediate genetically driven disease risk. These studies can provide specific hypotheses for functional validation experiments 2,3 . > Molecular QTL data can be used for a range of biomedical applications. For example, they have the potential to identify and validate new therapeutic targets and pathways, inform about the biological mechanisms of drug action and safety, highlight new therapeutic indications and reveal clinically relevant biomarkers [4][5][6] . > Many previous studies have carried out QTL mapping within a single molecular domain such as gene or protein expression [7][8][9][10][11][12] .
[15] New therapeutic targets in rare genetic skeletal diseases
- Authors: M. Briggs, Peter A. Bell, M. Wright, K. A. Pirog
- Year: 2015
- Venue: Expert Opinion on Orphan Drugs
- URL: https://www.semanticscholar.org/paper/1363107f71ae6d2d60abca471cddf3da5d13644b
- DOI: 10.1517/21678707.2015.1083853
- PMID: 26635999
- PMCID: 4643203
- Citations: 37
- Influential citations: 1
- Summary: An overview of disease mechanisms that are shared amongst groups of different GSDs and potential therapeutic approaches that are under investigation are described to generate critical mass for the identification and validation of novel therapeutic targets and biomarkers.
- Evidence snippets:
- Snippet 1 (score: 0.410) > proteins of the cartilage ECM such as type II collagen [50]. However, emerging knowledge suggests that the primary genetic defect may be less important than the cells' response to the expression of the mutant gene product [107]. Moreover, the largely overlooked response of a cell (i.e. chondrocyte) to the abnormal extracellular environment is also important for disease progression as illustrated by several GSDs discussed in this review. > It is important that 'omics'-based approaches and technologies are systematically applied to the study of rare GSDs so that definitive reference profiles and disease signatures are generated for each phenotype. These can then be used in a Systems Biology approach to identify both common and dissimilar pathological signatures and disease mechanisms. This approach is entirely dependent upon relevant in vitro and in vivo models (and also novel 'disease-mechanism phenocopies' [107]) for testing new diagnostic and prognostic tools and for determining the molecular mechanisms that underpin the pathophysiology so that effective therapeutic treatments can be developed and validated. This approach will eventually lead to personalized treatments and care strategies centred on shared disease mechanisms with the use of relevant biomarkers to monitor the efficacy of treatment and disease progression. > It is vital that all relevant stakeholders are involved from the outset in defining the appropriate outcomes of any potential therapeutic regime. The perceptions of a successful therapy can differ widely between the clinical academic community and the relevant patient-support groups and it is vital that there is engagement on all these issues. > In summary, the identification of causative genes and mutations for GSDs over the last 20 years, coupled with the generation and in-depth analysis of a plethora of relevant cell and mouse models, has derived new knowledge on disease mechanisms and suggested potential therapeutic targets. The fast-evolving hypothesis that clinically disparate diseases can share common disease mechanisms is a powerful concept that will generate critical mass for the identification and validation of novel therapeutic targets and biomarkers.
[16] Spatiotemporal 7q11.23 Protein Network Implicates the GTF2I-PRKDC-DDR Pathway During Early-Fetal Brain Development in Psychiatric Diseases
- Authors: G. Lin, Liang Chen, Weidi Wang, Wenxiang Cai, Weichen Song et al.
- Year: 2020
- Venue: Unknown venue
- URL: https://www.semanticscholar.org/paper/6a2df6310ac4d8f7f3f76da6f21f8a221ebf1cce
- DOI: 10.21203/rs.3.rs-93461/v1
- Summary: Striatum, hippocampus, and amygdala are crucial regions for establishing connectivity between 7q11.23 proteins and their partners in early and late fetal periods, and the results suggested that GTF2I-PRKDC-DDR and GTF 2I-BRCA1-dDR pathway is crucial for the 7q 11.23 CNV genes to contribute to the pathogenesis of psychiatric diseases.
- Evidence snippets:
- Snippet 1 (score: 0.406) > A different approach of addressing this issue is based on creating animal or cell models to help identify the related molecular and cellular mechanisms. For instance, mice with a heterozygous deletion of GTF2I or GTF2IRD1 show defects in skeletal and craniofacial. [14]. In addition, the embryos of these mice present with a small head; this is consistent with the clinical phenotype of patients carrying a 7q11. 23 deletion. Nevertheless, the signaling pathways affected by this CNV remain unknown. > Replication factor C subunit 2 (RFC2), another 7q11.23 gene, encodes a subunit of the replication factor C (RFC) complex [15] and is known to play a role in ATR signaling [16,17]. Haploinsufficiency for RFC2 leaded to G2/M checkpoint arrest after DNA damage [18]. However, little is known about how genes with the 7q11.23 deletion/duplication may affect the occurrence of neurodevelopmental disorders because these genes are involved not only in multiple developmental stages but also within different tissues. Hence, genes exhibiting 7q11.23 deletion/duplication play different roles in different developmental stages and different anatomic structures. > CNVs have been reported to modulate gene expression, which, ultimately, might affect disease predisposition or clinical phenotypes [19,20]. Several researches have investigated CNV pathogenesis in psychiatric disorders by constructing a static topological network based on a single developmental stage [21]. Within different developmental periods, protein expression can change, as can protein-protein interactions (PPIs) [22]. Nevertheless, protein expression is a dynamic process that can occur in a different manner across different anatomical areas [23,24]. Analyses of molecular networks can reveal biological modularity and complex signaling pathways [25,26]. Previous studies discovered the pathogenesis of CNVs by constructing dynamic protein-protein interaction (PPI) networks according to alterations of protein expression in different anatomical areas and during different developmental periods [27,28]. > In addition, multiple studies mentioned above focused only on one or two genes and were unable to demonstrate how the 7q11.23 CNV is involved in brain development.
[17] Drug repurposing in Rett and Rett-like syndromes: a promising yet underrated opportunity?
- Authors: Claudia Fuchs, P. A. ‛. ’t Hoen, A. Müller, Friederike Ehrhart, C. V. van Karnebeek
- Year: 2024
- Venue: Frontiers in Medicine
- URL: https://www.semanticscholar.org/paper/b00d0859458647edeebf3cf53f9b39c79311d5ed
- DOI: 10.3389/fmed.2024.1425038
- PMID: 39135718
- PMCID: 11317438
- Citations: 1
- Summary: The potential of drug repurposing (DR) as a promising avenue for addressing the unmet medical needs of individuals with RTT and related disorders is explored and Leveraging existing drugs for new therapeutic purposes presents an attractive strategy.
- Evidence snippets:
- Snippet 1 (score: 0.405) > Rigorous preclinical and clinical studies are also crucial for better understanding the complex pathophysiology of these syndromes. To date, the precise molecular mechanisms underlying these complex disorders are still not fully understood; hindering the identification and validation of potential drug targets. This specifically applies to CDD and FOXG1-syndrome: both conditions were identified as distinct clinical entities only recently and it is understandable that research efforts initially focused primarily on "classical" RTT. This discrepancy is reflected also in the very different numbers of repurposing studies highlighted in Figure 1. Continued efforts in pre-clinical (identification of valuable cell and animal models etc.) and clinical research (better understanding of the natural history, clinical manifestations, disease progression, biomarkers etc.) will be essential for advancing our understanding and improving outcomes for individuals affected by these syndromes. In particular, better characterizing the shared symptoms and pathways across these entities, will provide valuable insights into the underlying biology and potentially uncover new common mechanisms and targeted therapies. If the disorders demonstrate convergence in their underlying molecular pathways, this provides an opportunity for designing joint DR 10.3389/fmed.2024.1425038 strategies across RTT and RTT-like disorders. This could reduce the time needed for the development of DR and increase the number of patients benefiting from the treatments, resulting in more attractive business models. > Despite promising DR results in preclinical or early-phase clinical trials for RTT and related disorders in our opinion DR is still underrated and underutilized in this kind of disorders. DR holds immense potential for addressing the unmet medical needs and therapeutic challenges posed by such complex NDDs, and recent advancements screening and computational techniques, offer the unique opportunity to predict drug-disease interactions and prioritize candidate compounds for further investigation. By leveraging existing drugs and repurposing them for new indications, this approach offers a pragmatic and efficient strategy to accelerate the development of treatments for individuals affected by these debilitating conditions.
[18] Chromatin modifiers in neurodevelopment
- Authors: Sarallah Rezazadeh, H. Ji, Cecilia Giulivi
- Year: 2025
- Venue: Frontiers in Molecular Neuroscience
- URL: https://www.semanticscholar.org/paper/7a4d8c063c2b3a908a65bcb637cd818edad8db92
- DOI: 10.3389/fnmol.2025.1551107
- PMID: 40469903
- PMCID: 12133960
- Citations: 2
- Summary: This mini review delves into key chromatin modifiers, including the histone methyl transferases NSD1 and ASH1L, the methyl-CpG-binding repressor MeCP2, and the enzymatic repressor EZH2, and spotlight their pivotal roles in early brain development and neurological disorders.
- Evidence snippets:
- Snippet 1 (score: 0.404) > Therefore, while epigenetic changes are essential for understanding specific aspects of neurodevelopmental disorders, it is crucial to view these mechanisms as part of a larger, more complex system that encompasses genetic, proteomic, and metabolic factors. Few examples underscore that while epigenetic mechanisms-such as DNA methylation and histone modificationsare essential in regulating gene expression and contribute to neurodevelopmental disorders, they do not fully explain the complex pathophysiology of these diseases. In many cases, the genetic mutations, absence of or dysfunction of protein, or toxic protein aggregation (e.g., Fragile X syndrome, HD) that occur in these disorders play a central role in the clinical phenotypes. Therefore, a comprehensive understanding of neurodevelopmental disorders must integrate epigenetic mechanisms and the broader genetic, proteomic, and cellular pathways that contribute to disease. An integrative approach that considers not only the regulation of gene expression but also the functional consequences of these changes at the protein, metabolic and cellular pathway levels will be essential for advancing our understanding of these intricate disorders and developing effective interventions and treatments. . B., Villate, O., Llano, I., Ocio, I., Martí, I., et al. (2020). Targeted next-generation sequencing in patients with suggestive X-linked intellectual disability. Genes 11:51. doi: 10.3390/genes11010051
[19] Precision Therapeutics in Lennox–Gastaut Syndrome: Targeting Molecular Pathophysiology in a Developmental and Epileptic Encephalopathy
- Authors: Debopam Samanta
- Year: 2025
- Venue: Children
- URL: https://www.semanticscholar.org/paper/455479c1bfbea7b90b73c109228f67c813d13888
- DOI: 10.3390/children12040481
- PMID: 40310132
- PMCID: 12025602
- Citations: 19
- Influential citations: 1
- Summary: A narrative review explores precision therapeutic strategies for LGS based on molecular pathophysiology, including channelopathies, receptor and ligand dysfunction, receptor and ligand dysfunction, cell signaling abnormalities, cell signaling abnormalities, synaptopathies, and the repurposing of existing medications with mechanism-specific effects.
- Evidence snippets:
- Snippet 1 (score: 0.401) > A key advantage of disease-modifying therapies is their potential to target pathogenic mechanisms early in the disease course, potentially preventing the progression of some infantile epileptic encephalopathies to LGS. > This narrative review explores precision therapeutic strategies based on specific monogenic causes and disease mechanisms relevant to LGS. A comprehensive literature search (PubMed, MEDLINE, ClinicalTrials.gov, conference abstracts from the American Academy of Neurology and American Epilepsy Society, and gray literature) was conducted through 19 February 2025 to identify established ASMs, repurposed and novel drugs, as well as various gene therapy approaches with potential relevance to LGS. Given that over 900 monogenic causes of DEEs have been identified-implicating diverse cellular components such as ion channels, receptors, synaptic proteins, signaling pathways, metabolic processes, and epigenetic regulators-this review discusses current and emerging precision therapeutics based on shared molecular mechanisms and the pathophysiology of select genes associated with LGS [17] (Table 1).
[20] scGRNom: a computational pipeline of integrative multi-omics analyses for predicting cell-type disease genes and regulatory networks
- Authors: Ting Jin, Peter R Rehani, Mufang Ying, Jiawei Huang, Shuang Liu et al.
- Year: 2020
- Venue: Genome Medicine
- URL: https://www.semanticscholar.org/paper/a81382fef4e3f7cf5b4bec64266e372cf3a52da5
- DOI: 10.1186/s13073-021-00908-9
- PMID: 34044854
- PMCID: 8161957
- Citations: 37
- Summary: A computational pipeline, scGRNom (single-cell Gene Regulatory Network prediction from multi-omics), to predict cell-type disease genes and regulatory networks including transcription factors and regulatory elements, with applications to schizophrenia and Alzheimer’s disease.
- Evidence snippets:
- Snippet 1 (score: 0.399) > Recent genome-wide association studies (GWAS) studies have identified a variety of genetic risk variants associated with multiple brain diseases. For example, a recent study found 109 pleiotropic loci significantly associated with at least two brain disorders [1]. Many cross-disease common genetic risk factors have revealed many shared functional consequences in clinical presentations [2]. Recent studies have also revealed shared symptoms at both psychiatric and physical levels between neurodegenerative and neuropsychiatric diseases [3]. For instance, 97% of Alzheimer's disease patients develop neuropsychiatric symptoms throughout the disease [4]. Besides, additional insights into each disease's progression and causes have further demonstrated the highly interlinked nature of both disease types [5]. However, our understanding of the molecular mechanisms of genetic variants between diseases remains elusive, particularly at the cell-type levels. > Alzheimer's disease (AD) and schizophrenia (SCZ) are neurodegenerative and neuropsychiatric diseases, respectively. Both are significantly associated with genetic variants and have complex underlying cellular and molecular mechanisms from genotype to phenotype [6,7]. Notably, AD is physiologically characterized by accumulations of amyloid beta plaques and neurofibrillary tau protein tangles in the brain [8]. Amyloid beta plaques primarily originate from the apolipoprotein E-encoding gene APOE and its multiple variants. The APOE gene is a single step in the broader amyloidogenic processing pathway (APP), and additional genes involved in the process contribute to the regulation of amyloid beta production [6]. Much work has identified major genes of interest involved in the APP [6]. However, a distinct need still exists to further explore these disease loci to understand better the interplay between their regulatory elements and eventual amyloid beta creation and accumulation. Similarly, neurofibrillary tau tangles are associated with many genetic loci and require a study of the highly complex molecular mechanisms required to achieve disease pathology [8]. Further, the downstream effects from both amyloid beta and neurofibrillary tangles within and between various cell types add additional complexity toward linking specific regulatory events and elements with clinical pathology [9,10].
Notes
- This provider combines
search_papers_by_relevancewithsnippet_search. - No synthesis or second-stage model call is performed.
Question: You are an expert researcher providing comprehensive, well-cited information.
Provide detailed information focusing on: 1. Key concepts and definitions with current understanding 2. Recent developments and latest research (prioritize 2023-2024 sources) 3. Current applications and real-world implementations 4. Expert opinions and analysis from authoritative sources 5. Relevant statistics and data from recent studies
Format as a comprehensive research report with proper citations. Include URLs and publication dates where available. Always prioritize recent, authoritative sources and provide specific citations for all major claims.
Disease Characteristics Research Template
Target Disease
- Disease Name: 15q11q13 Microduplication Syndrome
- MONDO ID: (if available)
- Category: Mendelian
Research Objectives
Please provide a comprehensive research report on 15q11q13 Microduplication Syndrome covering all of the disease characteristics listed below. This report will be used to populate a disease knowledge base entry. Be thorough and cite primary literature (PMID preferred) for all claims.
For each section, suggested databases/resources are listed. These are the first places you should search for information on each topic.
1. Disease Information
Search first: OMIM, Orphanet, ICD-10/ICD-11, MeSH, PubMed
- What is the disease? Provide a concise overview.
- What are the key identifiers? (OMIM, Orphanet, ICD-10/ICD-11, MeSH, Mondo)
- What are the common synonyms and alternative names?
- Is the information derived from individual patients (e.g., EHR) or aggregated disease-level resources?
2. Etiology
- Disease Causal Factors: What are the primary causes? (genetic, environmental, infectious, mechanistic)
- Risk Factors:
Search first: PubMed, Cochrane Library, UpToDate, clinical guidelines, ClinVar, ClinGen, GWAS Catalog, PheGenI, CTD, CDC, WHO, epidemiological databases
- Genetic risk factors (causal variants, susceptibility loci, modifier genes)
- Environmental risk factors (toxins, lifestyle, occupational exposures, age, sex, family history)
- Protective Factors:
Search first: PubMed, Cochrane Library, clinical trial databases, GWAS Catalog, gnomAD, WHO, CDC, nutrition databases
- Genetic protective factors (protective variants, modifier alleles)
- Environmental protective factors (diet, lifestyle, exposures that reduce risk)
- Gene-Environment Interactions: How do genetic and environmental factors interact to influence disease?
Search first: CTD, PubMed, PheGenI, GxE databases
3. Phenotypes
Search first: HPO (Human Phenotype Ontology), OMIM, Orphanet, PubMed, clinicaltrials.gov, MedDRA, SNOMED CT, DECIPHER, LOINC
For each phenotype, provide: - Phenotype type: symptoms, clinical signs, physical manifestations, behavioral changes, or laboratory abnormalities
For symptoms/signs: HPO, OMIM, Orphanet, PubMed For behavioral changes: HPO, DSM, RDoC (Research Domain Criteria), PubMed For laboratory abnormalities: LOINC, SNOMED CT, LabTests Online, PubMed - Phenotype characteristics: Search first: OMIM, Orphanet, HPO, PubMed - Age of symptom onset (neonatal, childhood, adult-onset, late-onset) - Symptom severity (mild, moderate, severe, variable) - Symptom progression (stable, progressive, episodic, fluctuating) - Frequency among affected individuals (percentage or qualitative) - Quality of life impact: Effects on daily functioning and well-being (per-phenotype when possible) Search first: EQ-5D database, SF-36, WHO QOL databases, PubMed - Suggest HPO (Human Phenotype Ontology) terms for each phenotype
4. Genetic/Molecular Information
- Causal Genes: Gene mutations or chromosomal abnormalities responsible for disease (gene symbols, OMIM IDs)
Search first: OMIM, ClinVar, HGMD, Ensembl, NCBI Gene
- Pathogenic Variants:
- Affected genes (gene symbols, HGNC IDs) > Search first: OMIM, NCBI Gene, Ensembl, HGNC, UniProt, GeneCards
- Variant classification (pathogenic, likely pathogenic, VUS per ACMG/AMP guidelines) > Search first: ClinVar, ClinGen, ACMG/AMP guidelines, VarSome
- Variant type/class (missense, frameshift, nonsense, splice-site, structural)
- Allele frequency in population databases > Search first: gnomAD, 1000 Genomes, ExAC, TOPMed, dbSNP
- Somatic vs germline origin > Search first: COSMIC (somatic), ClinVar, ICGC, TCGA
- Functional consequences (loss of function, gain of function, dominant negative)
- Modifier Genes: Genes that modify disease severity or expression
- Epigenetic Information: DNA methylation, histone modifications, chromatin changes affecting disease
Search first: ENCODE, Roadmap Epigenomics, MethBase, DiseaseMeth
- Chromosomal Abnormalities: Large-scale genetic changes (aneuploidy, translocations, inversions)
Search first: DECIPHER, ClinVar, ECARUCA, UCSC Genome Browser
5. Environmental Information
- Environmental Factors: Non-genetic contributing factors (toxins, radiation, pollution, occupational exposure)
Search first: CTD (Comparative Toxicogenomics Database), TOXNET, PubMed, EPA databases
- Lifestyle Factors: Behavioral factors (smoking, diet, exercise, alcohol consumption)
Search first: CDC databases, WHO, PubMed, NHANES
- Infectious Agents: If applicable, pathogens causing or triggering disease (bacteria, viruses, fungi, parasites)
Search first: NCBI Taxonomy, ViPR, BV-BRC, MicrobeDB, GIDEON
6. Mechanism / Pathophysiology
- Molecular Pathways: Specific signaling cascades or biochemical pathways involved (Wnt, MAPK, mTOR, PI3K-AKT, etc.)
Search first: KEGG, Reactome, WikiPathways, PathBank, BioCyc
- Cellular Processes: Cell-level mechanisms (apoptosis, autophagy, cell cycle dysregulation, inflammation, etc.)
Search first: Gene Ontology (GO), Reactome, KEGG, PubMed
- Protein Dysfunction: How protein structure or function is altered (misfolding, aggregation, loss of function, gain of function)
Search first: UniProt, PDB (Protein Data Bank), InterPro, Pfam, AlphaFold
- Metabolic Changes: Alterations in metabolic processes (energy metabolism, lipid metabolism, amino acid metabolism)
Search first: KEGG, BioCyc, HMDB (Human Metabolome Database), BRENDA
- Immune System Involvement: Role of immune response (autoimmunity, immunodeficiency, chronic inflammation)
Search first: ImmPort, Immunome Database, IEDB, Gene Ontology
- Tissue Damage Mechanisms: How tissues/ are injured (oxidative stress, ischemia, fibrosis, necrosis)
Search first: PubMed, Gene Ontology, Reactome
- Biochemical Abnormalities: Specific molecular defects (enzyme deficiencies, receptor dysfunction, ion channel defects)
Search first: BRENDA, UniProt, KEGG, OMIM, PubMed
- Epigenetic Changes: DNA methylation, histone modifications affecting gene expression in disease
Search first: ENCODE, Roadmap Epigenomics, MethBase, DiseaseMeth
- Molecular Profiling (if available):
- Transcriptomics/gene expression changes > Search first: GEO (Gene Expression Omnibus), ArrayExpress, GTEx, Human Cell Atlas, SRA
- Proteomics findings > Search first: PRIDE, ProteomeXchange, Human Protein Atlas, STRING, BioGRID
- Metabolomics signatures > Search first: MetaboLights, Metabolomics Workbench, HMDB, METLIN
- Lipidomics alterations > Search first: LIPID MAPS, SwissLipids, LipidHome, Metabolomics Workbench
- Genomic structural features > Search first: UCSC Genome Browser, Ensembl, NCBI, dbVar, DGV
- Advanced Technologies (if applicable):
- Single-cell analysis findings (cell-type specific mechanisms, cellular heterogeneity) > Search first: Human Cell Atlas, Single Cell Portal, GEO, CELLxGENE
- Spatial transcriptomics findings > Search first: GEO, Spatial Research, Vizgen, 10x Genomics data
- Multi-omics integration results > Search first: TCGA, ICGC, cBioPortal, LinkedOmics, PubMed
- Functional genomics screens (CRISPR, RNAi) > Search first: DepMap, GenomeRNAi, PubMed, BioGRID ORCS
For each mechanism, describe: - The causal chain from initial trigger to clinical manifestation - Which mechanisms are upstream vs downstream - What cell types and biological processes are involved - Suggest GO terms for biological processes and CL terms for cell types
7. Anatomical Structures Affected
- Organ Level:
- Primary organs directly affected
- Secondary organ involvement (complications, secondary effects)
- Body systems involved (cardiovascular, nervous, digestive, respiratory, endocrine, etc.)
Search first: Uberon, FMA (Foundational Model of Anatomy), OMIM, HPO, ICD-11, MeSH, SNOMED CT
- Tissue and Cell Level:
- Specific tissue types affected (epithelial, connective, muscle, nervous)
- Specific cell populations targeted (with Cell Ontology terms)
Search first: Uberon, Human Protein Atlas, Cell Ontology, Human Cell Atlas, CellMarker, PanglaoDB
- Subcellular Level:
- Cellular compartments involved (mitochondria, nucleus, ER, lysosomes) (with GO Cellular Component terms)
Search first: Gene Ontology (Cellular Component), UniProt, Human Protein Atlas
- Localization:
- Specific anatomical sites (with UBERON terms) > Search first: FMA, Uberon, NeuroNames (for brain), SNOMED CT
- Lateralization (unilateral, bilateral, asymmetric) > Search first: HPO, clinical literature, imaging databases
8. Temporal Development
- Onset:
- Typical age of onset (congenital, pediatric, adult, geriatric)
- Onset pattern (acute, subacute, chronic, insidious)
Search first: OMIM, Orphanet, HPO, PubMed
- Progression:
- Disease stages (early, intermediate, advanced, end-stage) > Search first: Cancer Staging Manual (AJCC), WHO classifications, PubMed
- Progression rate (rapid, slow, variable)
- Disease course pattern (episodic, relapsing-remitting, progressive, stable)
- Disease duration (self-limited, chronic lifelong)
Search first: Disease registries, longitudinal cohort databases, natural history studies, PubMed, Orphanet, OMIM
- Patterns:
- Remission patterns (spontaneous, treatment-induced) > Search first: Clinical trial databases, disease registries, PubMed
- Critical periods (time windows of vulnerability or opportunity for intervention) > Search first: PubMed, developmental biology databases, clinical guidelines
9. Inheritance and Population
- Epidemiology:
- Prevalence (cases per 100,000 at given time)
- Incidence (new cases per 100,000 per year)
Search first: Orphanet, CDC, WHO, GBD (Global Burden of Disease), national registries, SEER, disease registries
- For Genetic Etiology:
- Inheritance pattern (AD, AR, X-linked, mitochondrial, multifactorial, polygenic) > Search first: OMIM, Orphanet, ClinVar, GTR (Genetic Testing Registry)
- Penetrance (complete, incomplete, age-dependent) > Search first: ClinVar, OMIM, PubMed, ClinGen
- Expressivity (variable, consistent) > Search first: OMIM, ClinVar, PubMed
- Genetic anticipation (increasing severity in successive generations) > Search first: OMIM, PubMed (especially for repeat expansion disorders)
- Germline mosaicism > Search first: ClinVar, OMIM, genetic counseling literature, PubMed
- Founder effects (population-specific mutations) > Search first: gnomAD, population genetics databases, PubMed
- Consanguinity role > Search first: OMIM, population studies, genetic counseling resources
- Carrier frequency > Search first: gnomAD, carrier screening databases, GeneReviews, GTR
- Population Demographics:
- Affected populations (ethnic or demographic groups with higher prevalence) > Search first: gnomAD, 1000 Genomes, PAGE Study, PubMed, population registries
- Geographic distribution (endemic areas, regional variation) > Search first: WHO, CDC, GBD, Orphanet, geographic epidemiology databases
- Geographic distribution of specific variants
- Sex ratio (male:female) > Search first: Disease registries, OMIM, PubMed, epidemiological databases
- Age distribution of affected individuals > Search first: CDC, disease registries, SEER, Orphanet
10. Diagnostics
- Clinical Tests:
- Laboratory tests (blood, urine, tissue chemistry, specific enzyme assays) > Search first: LOINC, LabTests Online, PubMed
- Biomarkers (proteins, metabolites, genetic markers, circulating biomarkers) > Search first: FDA Biomarker List, BEST (Biomarkers, EndpointS, and other Tools), PubMed
- Imaging studies (X-ray, CT, MRI, PET, ultrasound) > Search first: RadLex, DICOM, Radiopaedia, imaging databases
- Functional tests (pulmonary function, cardiac stress tests) > Search first: LOINC, clinical guidelines, PubMed
- Electrophysiology (EEG, EMG, ECG, nerve conduction studies) > Search first: LOINC, clinical neurophysiology databases, PubMed
- Biopsy findings (histopathology, immunohistochemistry) > Search first: SNOMED CT, College of American Pathologists resources, PubMed
- Pathology findings (microscopic examination) > Search first: SNOMED CT, Digital Pathology databases, PubMed
- Genetic Testing:
Search first: GTR (Genetic Testing Registry), GeneReviews, ClinGen
- Overview of recommended genetic testing approach
- Whole genome sequencing (WGS) utility > Search first: GTR, ClinVar, GEL (Genomics England), gnomAD
- Whole exome sequencing (WES) utility > Search first: GTR, ClinVar, OMIM, GeneMatcher
- Gene panels (which panels, which genes) > Search first: GTR, ClinVar, laboratory-specific databases
- Single gene testing > Search first: GTR, ClinVar, OMIM, GeneReviews
- Chromosomal microarray (CMA) > Search first: DECIPHER, ClinVar, dbVar, ECARUCA
- Karyotyping > Search first: Chromosome Abnormality Database, ClinVar, cytogenetics resources
- FISH > Search first: ClinVar, cytogenetics databases, PubMed
- Mitochondrial DNA testing > Search first: MITOMAP, MSeqDR, ClinVar, GTR
- Repeat expansion testing > Search first: GTR, ClinVar, repeat expansion databases, PubMed
- Omics-Based Diagnostics (if applicable):
- RNA sequencing / transcriptomics > Search first: GEO, ArrayExpress, GTEx, RNA-seq databases
- Proteomics > Search first: PRIDE, ProteomeXchange, FDA Biomarker database
- Metabolomics > Search first: MetaboLights, Metabolomics Workbench, HMDB
- Epigenomics > Search first: GEO, ENCODE, Roadmap Epigenomics, MethBase
- Liquid biopsy > Search first: COSMIC, ClinVar, liquid biopsy databases, PubMed
- Clinical Criteria:
- Standardized diagnostic criteria (DSM, ICD, society guidelines) > Search first: DSM-5, ICD-11, clinical society guidelines, UpToDate
- Differential diagnosis (other conditions to rule out, with distinguishing features) > Search first: DynaMed, UpToDate, clinical decision support systems
- Screening:
- Screening methods for asymptomatic individuals (newborn screening, carrier screening, cascade screening) > Search first: ACMG recommendations, CDC newborn screening, GTR
11. Outcome/Prognosis
- Survival and Mortality:
- Survival rate (5-year, 10-year, overall) > Search first: SEER, cancer registries, disease-specific registries, PubMed
- Life expectancy (with and without treatment if applicable) > Search first: Orphanet, disease registries, actuarial databases, PubMed
- Mortality rate > Search first: CDC, WHO, GBD, national mortality databases
- Disease-specific mortality (deaths directly attributable to disease) > Search first: Disease registries, CDC Wonder, GBD, PubMed
- Morbidity and Function:
- Morbidity (disease-related disability and health impacts) > Search first: GBD, WHO, disability databases, PubMed
- Disability outcomes (long-term functional impairments) > Search first: ICF (International Classification of Functioning), disability registries
- Quality of life measures (EQ-5D, SF-36, PROMIS, disease-specific tools) > Search first: EQ-5D database, SF-36, PROMIS, PubMed
- Disease Course:
- Complications (secondary problems: infections, organ failure, etc.) > Search first: ICD codes, disease registries, clinical databases, PubMed
- Recovery potential (likelihood and extent of recovery, with vs without treatment) > Search first: Natural history studies, rehabilitation databases, PubMed
- Prediction:
- Prognostic factors (age, disease severity, biomarkers, treatment response) > Search first: Prognostic models databases, clinical calculators, PubMed
- Prognostic biomarkers (molecular markers predicting disease course) > Search first: FDA Biomarker database, PubMed, cancer prognostic databases
12. Treatment
- Pharmacotherapy:
- Pharmacological treatments (drug names, drug classes, mechanisms of action) > Search first: DrugBank, RxNorm, ATC classification, DailyMed, FDA databases
- Pharmacogenomics (how genetic variants affect drug metabolism, efficacy, toxicity) > Search first: PharmGKB, CPIC (Clinical Pharmacogenetics), FDA Table of PGx Biomarkers
- Advanced Therapeutics:
- Gene therapy (viral vectors, CRISPR, gene replacement, gene editing) > Search first: ClinicalTrials.gov, FDA gene therapy database, ASGCT resources
- Cell therapy (stem cell transplant, CAR-T, cellular therapeutics) > Search first: ClinicalTrials.gov, FDA cell therapy database, FACT standards
- RNA-based therapies (ASOs, siRNA, mRNA therapies) > Search first: ClinicalTrials.gov, FDA approvals, PubMed
- Targeted therapies (treatments directed at specific molecular targets) > Search first: My Cancer Genome, OncoKB, ClinicalTrials.gov, FDA approvals
- Immunotherapies (checkpoint inhibitors, monoclonal antibodies) > Search first: Cancer Immunotherapy Database, FDA approvals, ClinicalTrials.gov
- Surgical and Interventional:
- Surgical interventions (types of surgery, timing, outcomes) > Search first: CPT codes, surgical registries, clinical guidelines, PubMed
- Supportive and Rehabilitative:
- Supportive care (symptom management, pain control, nutrition) > Search first: Clinical guidelines, Cochrane Library, PubMed
- Rehabilitation (physical therapy, occupational therapy, speech therapy) > Search first: Rehabilitation medicine databases, clinical guidelines, PubMed
- Experimental:
- Experimental treatments in clinical trials (with NCT identifiers if available) > Search first: ClinicalTrials.gov, EU Clinical Trials Register, WHO ICTRP
- Treatment Outcomes:
- Treatment response rates > Search first: Clinical trial databases, FDA reviews, systematic reviews, PubMed
- Side effects and adverse events > Search first: FDA Adverse Event Reporting System (FAERS), MedWatch, PubMed
- Treatment Strategy:
- Treatment algorithms (clinical pathways, decision trees) > Search first: Clinical practice guidelines, NCCN Guidelines, UpToDate
- Combination therapies > Search first: ClinicalTrials.gov, treatment guidelines, PubMed
- Personalized medicine approaches (genotype-guided treatment) > Search first: My Cancer Genome, CIViC, PharmGKB, precision medicine databases
For each treatment, suggest MAXO (Medical Action Ontology) terms where applicable.
13. Prevention
- Prevention Levels:
- Primary prevention (preventing disease occurrence: vaccination, risk factor modification) > Search first: CDC, WHO, USPSTF recommendations, Cochrane Library
- Secondary prevention (early detection and treatment: screening programs, early intervention) > Search first: USPSTF, CDC screening guidelines, WHO
- Tertiary prevention (preventing complications in those with disease) > Search first: Clinical guidelines, disease management protocols, PubMed
- Immunization: Vaccine strategies (if applicable)
Search first: CDC vaccine schedules, WHO immunization, FDA vaccine database
- Screening and Early Detection:
- Screening programs (population-based: newborn screening, cancer screening) > Search first: CDC screening programs, USPSTF, cancer screening databases
- Genetic screening (carrier screening, preimplantation genetic diagnosis, prenatal testing) > Search first: ACMG recommendations, ACOG guidelines, GTR
- Risk stratification (identifying high-risk individuals for targeted prevention) > Search first: Risk prediction models, clinical calculators, PubMed
- Behavioral Interventions: Lifestyle modifications to reduce risk
Search first: CDC, WHO, behavioral intervention databases, Cochrane Library
- Counseling: Genetic counseling (risk assessment, family planning guidance)
Search first: NSGC resources, ACMG guidelines, GeneReviews
- Public Health:
- Public health interventions (sanitation, vector control, health education) > Search first: CDC, WHO, public health databases, PubMed
- Environmental interventions (reducing environmental risk factors) > Search first: EPA databases, WHO environmental health, PubMed
- Prophylaxis: Preventive medications or procedures
Search first: Clinical guidelines, FDA approvals, PubMed
14. Other Species / Natural Disease
- Taxonomy: Species affected (with NCBI Taxon identifiers)
Search first: NCBI Taxonomy
- Breed: Specific breeds affected (with VBO identifiers if applicable)
Search first: VBO (Vertebrate Breed Ontology)
- Gene: Orthologous genes in other species (with NCBI Gene IDs)
Search first: NCBI Gene
- Natural Disease:
- Naturally occurring disease in other species (companion animals, wildlife) > Search first: OMIA (Online Mendelian Inheritance in Animals), VetCompass, PubMed
- Veterinary relevance and importance in animal health > Search first: OMIA, veterinary databases, PubMed
- Comparative Biology:
- Comparative pathology (similarities and differences across species) > Search first: OMIA, comparative pathology databases, PubMed
- Evolutionary conservation of disease mechanisms > Search first: HomoloGene, OrthoMCL, Alliance of Genome Resources
- Transmission (if applicable):
- Zoonotic potential > Search first: CDC zoonotic diseases, WHO zoonoses, GIDEON
- Cross-species susceptibility > Search first: NCBI Taxonomy, veterinary databases, PubMed
15. Model Organisms
- Model Types:
- Model organism type (mammalian, invertebrate, cellular, in vitro) > Search first: Alliance of Genome Resources, model organism databases
- Specific model systems (mouse, rat, zebrafish, Drosophila, C. elegans, yeast, cell lines, organoids, iPSCs) > Search first: MGI, RGD, ZFIN, FlyBase, WormBase, SGD, ATCC, Cellosaurus
- Induced models (drug treatment, surgical intervention, environmental manipulation) > Search first: MGI, model organism databases, PubMed
- Genetic Models:
- Types available (knockout, knock-in, transgenic, conditional, humanized) > Search first: MGI, IMPC, KOMP, EuMMCR, IMSR
- Model Characteristics:
- Phenotype recapitulation (how well model reproduces human disease features) > Search first: Model organism databases, comparative studies, PubMed
- Model limitations (aspects of human disease not captured) > Search first: Model organism databases, PubMed, review articles
- Applications:
- Research applications (what aspects of disease can be studied) > Search first: Model organism databases, PubMed
- Resources:
- Model databases > Search first: MGI, RGD, ZFIN, FlyBase, WormBase, IMSR, EMMA, MMRRC
Citation Requirements
- Cite primary literature (PMID preferred) for all mechanistic and clinical claims
- Prioritize recent reviews and landmark papers
- Include direct quotes from abstracts where possible to support key statements
- Distinguish evidence source types: human clinical, model organism, in vitro, computational
Output Format
Structure your response as a comprehensive narrative organized by the sections above. For each section, provide: - Factual content with specific details (numbers, percentages, gene names, variant nomenclature) - Ontology term suggestions (HPO, GO, CL, UBERON, CHEBI, MAXO, MONDO) where applicable - Evidence citations with PMIDs - Direct quotes from abstracts to support key claims - Clear indication when information is not available or not applicable for this disease
This report will be used to populate a disease knowledge base entry with: - Pathophysiology descriptions with causal chains - Gene/protein annotations (HGNC, GO terms) - Phenotype associations (HP terms) with frequencies - Cell type involvement (CL terms) - Anatomical locations (UBERON terms) - Chemical entities (CHEBI terms) - Treatment annotations (MAXO terms) - Evidence items with PMIDs and exact abstract quotes - Epidemiology, prognosis, diagnostic, and prevention information - Animal model descriptions with phenotype recapitulation details
Comprehensive Research Report: 15q11q13 Microduplication Syndrome (dup15q / 15q11-q13 duplication syndrome)
Executive summary
15q11q13 microduplication syndrome (often discussed under the umbrella term dup15q syndrome) is a neurodevelopmental copy-number variant (CNV) disorder caused by increased copy number of the 15q11.2–q13.1 region. Clinical impact depends strongly on parent of origin because the region contains imprinted genes, especially UBE3A, which is maternally expressed in neurons; maternal duplications tend to be more penetrant and severe than paternal duplications. Recent (2023–2024) work has advanced (i) population-level incidence estimates from genome-wide NIPS, (ii) larger literature-based clinical syntheses of phenotype and inheritance, and (iii) translational biomarkers (beta-band EEG) and mechanistic models (patient-derived neurons, chromosome-engineered mice) that are being leveraged for therapy development and clinical trials. (parijs2024populationscreeningfor pages 1-2, bisba2024chromosome15q11q13duplication pages 1-2, saravanapandian2024sleepeegsignatures pages 2-4, elamin2023theroleof pages 1-2)
Key evidence-at-a-glance
| Category | Finding (concise) | Quantitative detail | Source (first author year) | Publication date (month year) | URL |
|---|---|---|---|---|---|
| Identifiers | Disease identifier | OMIM 608636 for chromosome 15q11-q13 duplication syndrome | Bisba 2024 (bisba2024chromosome15q11q13duplication pages 1-2) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Genetics | Common inheritance pattern among literature cases | Of carriers inheriting from a parent, 62.96% maternal and 37.04% paternal; 80.20% inherited from a parent overall | Bisba 2024 (bisba2024chromosome15q11q13duplication pages 5-7) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Genetics | Postnatal inheritance totals | Table 6 totals: 48 maternal, 29 paternal, 17 de novo, 8 unknown | Bisba 2024 (bisba2024chromosome15q11q13duplication pages 5-7) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Genetics | Prenatal inheritance totals | Table 7 totals: 3 maternal, 1 paternal, 3 de novo, 1 unknown | Bisba 2024 (bisba2024chromosome15q11q13duplication pages 5-7) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Epidemiology | Population incidence from genome-wide NIPS | 23/333,187 = 0.0069% detected 15q11-q13 duplications | Parijs 2024 (parijs2024populationscreeningfor pages 1-2, parijs2024populationscreeningfor pages 3-5) | Apr 2024 | https://doi.org/10.1038/s41431-023-01336-6 |
| Diagnostics | Positive predictive value of NIPS detection | PPV 100% for this CNV in followed cases | Parijs 2024 (parijs2024populationscreeningfor pages 3-5) | Apr 2024 | https://doi.org/10.1038/s41431-023-01336-6 |
| Epidemiology | Estimated general prevalence cited in review | Rare congenital disease; cited prevalence 1 in 30,000 to 1 in 60,000 children worldwide | Bisba 2024 (bisba2024chromosome15q11q13duplication pages 5-7) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Postnatal phenotype distribution: composite phenotype | 62/115 = 53.91% | Bisba 2024 Table 2 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Postnatal phenotype distribution: normal | 15/115 = 13.04% | Bisba 2024 Table 2 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Postnatal phenotype distribution: developmental delay | 15/115 = 13.04% | Bisba 2024 Table 2 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Postnatal phenotype distribution: ASD | 8/115 = 6.95% | Bisba 2024 Table 2 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Postnatal phenotype distribution: epilepsy | 2/115 = 1.74% | Bisba 2024 Table 2 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Postnatal phenotype distribution: behavioral problems | 3/115 = 2.61% | Bisba 2024 Table 2 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Postnatal phenotype distribution: congenital heart defects | 2/115 = 1.74% | Bisba 2024 Table 2 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Prenatal phenotype distribution: normal | 10/14 = 71.43% | Bisba 2024 Table 3 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Prenatal phenotype distribution: congenital heart defects | 3/14 = 21.43% | Bisba 2024 Table 3 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Phenotypes | Prenatal phenotype distribution: IUGR | 1/14 = 7.14% | Bisba 2024 Table 3 (bisba2024chromosome15q11q13duplication pages 3-5) | Oct 2024 | https://doi.org/10.3390/genes15101304 |
| Genetics | Parent-of-origin effect in population screening | Maternal and paternal duplications occurred in approximately equal numbers in screening, but maternal duplications were consistently associated with phenotype; 7 fetuses inherited the duplication among 14 amniocenteses with follow-up | Parijs 2024 (parijs2024populationscreeningfor pages 3-5) | Apr 2024 | https://doi.org/10.1038/s41431-023-01336-6 |
| Phenotypes | Autism burden reported in mechanistic study | Autism reported in 77%–100% of affected individuals | Elamin 2023 (elamin2023theroleof pages 1-2) | Apr 2023 | https://doi.org/10.1016/j.stemcr.2023.02.002 |
| Phenotypes | Seizure burden in idic(15) | Seizures in 63% of individuals with idic(15) | Elamin 2023 (elamin2023theroleof pages 1-2) | Apr 2023 | https://doi.org/10.1016/j.stemcr.2023.02.002 |
| Biomarkers | Human EEG beta biomarker cohort size | N = 41 children, age 9–189 months | Saravanapandian 2020 (saravanapandian2020propertiesofbeta pages 1-2) | Aug 2020 | https://doi.org/10.1186/s11689-020-09326-1 |
| Biomarkers | Beta biomarker stability | Beta power ICC = 0.93; beta peak frequency ICC = 0.92 | Saravanapandian 2020 (saravanapandian2020propertiesofbeta pages 1-2) | Aug 2020 | https://doi.org/10.1186/s11689-020-09326-1 |
| Biomarkers | Clinical reproducibility of EEG biomarker | Research vs clinical EEG beta power ICC = 0.94 | Saravanapandian 2020 (saravanapandian2020propertiesofbeta pages 1-2) | Aug 2020 | https://doi.org/10.1186/s11689-020-09326-1 |
| Biomarkers | Clinical correlates of beta peak frequency | Epilepsy status R² = 0.11, p = 0.038; daily living skills R² = 0.17, p = 0.01 | Saravanapandian 2020 (saravanapandian2020propertiesofbeta pages 1-2) | Aug 2020 | https://doi.org/10.1186/s11689-020-09326-1 |
| Biomarkers | Sleep EEG abnormalities in children | Dup15q n = 15 vs controls n = 12; elevated beta power, reduced spindle density, reduced/absent SWS | Saravanapandian 2021 (saravanapandian2021abnormalsleepphysiology pages 1-2) | Aug 2021 | https://doi.org/10.1186/s13229-021-00460-8 |
| Biomarkers | Mouse sleep EEG translational study size | 35 mice total after exclusions; matDp/+ 9, WT 8; patDp/+ 6, WT 4; Ube3a OE 5, WT 3 | Saravanapandian 2024 (saravanapandian2024sleepeegsignatures pages 2-4) | Jul 2024 | https://doi.org/10.1186/s11689-024-09556-7 |
| Biomarkers | Mouse sleep EEG findings | Maternal duplication mice mirrored elevated beta oscillations; matDp/+ and Ube3a OE had reduced sleep-onset latency; no alterations in NREM sleep in any of the 3 mouse groups | Saravanapandian 2024 (saravanapandian2024sleepeegsignatures pages 2-4) | Jul 2024 | https://doi.org/10.1186/s11689-024-09556-7 |
| Diagnostics | Recommended/used genomic methods in clinical literature | Array-CGH/Affymetrix CytoScan 750K used in large 2024 review cohort; MLPA suggested as cost- and time-effective first-line test in some familial interstitial cases | Bisba 2024; Levandivska 2023 (bisba2024chromosome15q11q13duplication pages 1-2, levandivska2023inherited15qduplication pages 1-2) | Oct 2024; Jun 2023 | https://doi.org/10.3390/genes15101304; https://doi.org/10.32345/2664-4738.2.2023.08 |
| Data infrastructure | LADDER database purpose | Database launched to harmonize data across registries, clinic visits, trials, and studies for AS and dup15q; started collaboration in 2019 | Potter 2024 (potter2024linkingangelmanand pages 3-5) | Jan 2024 | https://doi.org/10.1177/26330040241254122 |
| Trials | Retinoic acid pilot trial | NCT05281965; Early Phase 1; randomized crossover; estimated enrollment 20; ages 6–18 years | ClinicalTrials.gov / Feng et al. listing (NCT05281965 chunk 1) | Mar 2022 posting | https://clinicaltrials.gov/study/NCT05281965 |
| Trials | Basmisanil phase 2 trial | NCT05307679; Phase 2; randomized double-blind placebo-controlled; actual enrollment 7; ages 2–14 years; terminated for sponsor decision not safety | ClinicalTrials.gov / Roche listing (NCT05307679 chunk 1) | Apr 2022 posting; updated Nov 2025 | https://clinicaltrials.gov/study/NCT05307679 |
| Trials | Soticlestat ARCADE study | NCT03694275; Phase 2; open-label/non-randomized; actual enrollment 20; ages 2–55 years; maintenance endpoint weeks 9–20 | ClinicalTrials.gov / Takeda listing (NCT03694275 chunk 1, NCT03694275 chunk 2) | Oct 2018 posting; updated May 2022 | https://clinicaltrials.gov/study/NCT03694275 |
| Trials | All-trans retinoic acid efficacy study | NCT07079696; Phase 2; single-group; estimated enrollment 90; ages 3–7 years; treatment duration 18 months | ClinicalTrials.gov / Zhejiang University listing (NCT07079696 chunk 1) | Jul 2025 posting | https://clinicaltrials.gov/study/NCT07079696 |
Table: This table compiles the main identifiers, epidemiology, inheritance patterns, phenotype frequencies, biomarker statistics, diagnostic approaches, and active/recent clinical trials for 15q11q13 microduplication (dup15q) syndrome from the cited evidence. It is useful as a compact reference for knowledge-base population and evidence tracing.
1. Disease information
1.1 Disease overview (current understanding)
15q11q13 microduplication syndrome refers to pathogenic duplications (or higher copy gains such as triplications/tetrasomies) involving the proximal long arm of chromosome 15 that encompass neurodevelopmentally relevant genes and, in many cases, the Prader–Willi/Angelman critical region. A widely used clinical framing is that dup15q syndrome is “defined as the presence of three or more copies of 15q11.2-q13.1” (cited in a prenatal cohort report) and is associated with developmental delay/intellectual disability, hypotonia, autism spectrum disorder (ASD), epilepsy/seizures, and behavioral problems. (parijs2024populationscreeningfor pages 1-2, bisba2024chromosome15q11q13duplication pages 1-2)
1.2 Key identifiers
- MONDO: MONDO_0012081 (15q11q13 microduplication syndrome). (OpenTargets Search: dup15q syndrome,15q11-q13 duplication syndrome,15q11q13 microduplication syndrome)
- OMIM: 608636 (15q11-q13 duplication syndrome / dup15q). (bisba2024chromosome15q11q13duplication pages 1-2)
Not retrieved in current evidence set: Orphanet/ORDO ID, MeSH ID, ICD-10/ICD-11 code. These typically exist in curated resources but were not available in the retrieved texts.
1.3 Synonyms and alternative names
Commonly used names in the 2023–2024 literature include: - dup15q syndrome / 15q11.2–q13.1 duplication syndrome / chromosome 15q11-q13 duplication syndrome (bisba2024chromosome15q11q13duplication pages 1-2, parijs2024populationscreeningfor pages 1-2) - Cytogenetic-mechanism labels encountered in the literature: interstitial 15q duplication and isodicentric 15 [idic(15)] forms of dup15q (bisba2024chromosome15q11q13duplication pages 1-2, levandivska2023inherited15qduplication pages 1-2)
1.4 Evidence provenance
The evidence used here includes: - Aggregated literature synthesis + new clinical cases (Genes 2024 review/series). (bisba2024chromosome15q11q13duplication pages 1-2, bisba2024chromosome15q11q13duplication pages 3-5) - Population-level screening analysis from genome-wide cfDNA NIPS (European Journal of Human Genetics 2024). (parijs2024populationscreeningfor pages 1-2, parijs2024populationscreeningfor pages 3-5) - Mechanistic human-cell work using patient-derived neurons and CRISPR correction (Stem Cell Reports 2023). (elamin2023theroleof pages 1-2) - Translational mouse biomarker work (Journal of Neurodevelopmental Disorders 2024). (saravanapandian2024sleepeegsignatures pages 2-4) - Rare-disease data infrastructure paper describing a linked database for AS + dup15q natural history and trial readiness (Therapeutic Advances in Rare Disease 2024). (potter2024linkingangelmanand pages 3-5)
2. Etiology
2.1 Disease causal factors
Primary cause: germline copy-number gain (duplication/triplication/tetrasomy) of 15q11.2–q13.1, generated through non-allelic homologous recombination facilitated by low-copy repeats and canonical breakpoints BP1–BP5. (bisba2024chromosome15q11q13duplication pages 1-2, bisba2024chromosome15q11q13duplication media 4f90b336)
Parent-of-origin (imprinting) is a key causal modifier: - In a population-screening cohort, the authors conclude: “maternal duplications are invariably associated with a clinical phenotype … [while] the majority of paternal duplication carriers are phenotypically normal” with some mildly affected phenotypes observed. (parijs2024populationscreeningfor pages 3-5) - Bisba et al. (2024) note many pathogenic presentations are maternally derived and implicate maternally expressed imprinted genes (notably UBE3A) as contributors to ASD/developmental disorders. (bisba2024chromosome15q11q13duplication pages 5-7)
2.2 Risk factors
- Genetic: Presence of a 15q11-q13 duplication (particularly maternally derived) is itself the dominant risk factor for neurodevelopmental phenotypes (ASD/ID/epilepsy). (parijs2024populationscreeningfor pages 1-2, elamin2023theroleof pages 1-2)
- Structural genomic architecture: The segmental duplication/LCR structure and BP1–BP5 breakpoint framework predispose to rearrangements (duplications). (bisba2024chromosome15q11q13duplication pages 1-2, bisba2024chromosome15q11q13duplication media 4f90b336)
Environmental risk/protective factors: not specifically established for this CNV syndrome in the retrieved evidence set.
2.3 Protective factors and gene–environment interactions
No robust protective factors or gene–environment interaction studies specific to dup15q were retrieved in the evidence set.
3. Phenotypes (clinical presentation)
3.1 Phenotype spectrum and frequencies (recent synthesis)
A 2024 combined series/review (Bisba et al.) compiled phenotypic features from postnatal cases (defined across literature + their cases), with a notable fraction recorded as having composite phenotype (multiple neurodevelopmental features). Reported postnatal feature frequencies include: composite phenotype 53.91% (62/115), “normal” 13.04% (15/115), developmental delay 13.04% (15/115), ASD 6.95% (8/115), epilepsy 1.74% (2/115), congenital heart defects 1.74% (2/115). (bisba2024chromosome15q11q13duplication pages 3-5)
Prenatal-case features in the same synthesis (n=14) included “normal” 71.43% (10/14), congenital heart defects 21.43% (3/14), and intrauterine growth restriction (IUGR) 7.14% (1/14) (noting follow-up after birth was often unavailable). (bisba2024chromosome15q11q13duplication pages 3-5)
Important interpretation note: These summary tables aggregate heterogeneous ascertainment (including prenatal referrals and incomplete follow-up), and therefore should not be treated as population penetrance estimates.
3.2 Age of onset, progression, and severity
- Typical onset: congenital/genetic; neurodevelopmental symptoms usually manifest in infancy/early childhood (developmental delay, hypotonia, ASD features), and epilepsy may present early (including infantile spasms, especially in severe subtypes such as idic(15)). (elamin2023theroleof pages 1-2)
- Course: lifelong neurodevelopmental disability is common; severity varies with duplication type and parent-of-origin. (bisba2024chromosome15q11q13duplication pages 1-2, parijs2024populationscreeningfor pages 3-5)
3.3 Key phenotypes and suggested HPO terms
Below are common features with ontology suggestions (frequency varies by subtype and ascertainment): - Developmental delay: HP:0001263 (Global developmental delay) (bisba2024chromosome15q11q13duplication pages 1-2, bisba2024chromosome15q11q13duplication pages 3-5) - Intellectual disability: HP:0001249 (Intellectual disability) (bisba2024chromosome15q11q13duplication pages 1-2, parijs2024populationscreeningfor pages 1-2) - Autism spectrum disorder: HP:0000729 (Autistic behavior) (bisba2024chromosome15q11q13duplication pages 1-2, elamin2023theroleof pages 1-2) - Hypotonia: HP:0001252 (Muscular hypotonia) (parijs2024populationscreeningfor pages 1-2) - Seizures/Epilepsy: HP:0001250 (Seizures), HP:0001270 (Epileptic encephalopathy—when severe) (bisba2024chromosome15q11q13duplication pages 1-2, elamin2023theroleof pages 1-2) - Sleep disturbance: HP:0002360 (Sleep disturbance) supported by sleep-EEG biomarker work (saravanapandian2021abnormalsleepphysiology pages 1-2) - Congenital heart defects: HP:0001627 (Abnormality of the cardiovascular system) (bisba2024chromosome15q11q13duplication pages 3-5)
3.4 Quality of life (QoL) impact
While formal QoL instruments were not retrieved in the evidence set, the need for lifelong care and functional impairment is emphasized in rare-disease infrastructure work linking dup15q and Angelman syndrome datasets to support natural history and trial readiness. (potter2024linkingangelmanand pages 3-5)
4. Genetic / molecular information
4.1 Causal genomic abnormality and duplication classes
- Structural basis: recurrent CNVs mediated by LCRs at BP1–BP5 (BP schematic shown in Bisba et al. Figure 1). (bisba2024chromosome15q11q13duplication media 4f90b336)
- Cytogenetic forms:
- Interstitial duplications (tandem duplications within chromosome 15)
- Isodicentric 15 [idic(15)] supernumerary chromosomes (often increasing dosage further)
These classes are frequently invoked as the two common forms of Dup15q. (mim2024expandingdeepphenotypic pages 1-2, levandivska2023inherited15qduplication pages 1-2)
4.2 Key genes and dosage mechanisms
The region includes: - UBE3A (imprinted; maternally expressed in neurons) – dosage increase implicated in ASD and cellular hyperexcitability phenotypes. (elamin2023theroleof pages 1-2, bisba2024chromosome15q11q13duplication pages 1-2) - A cluster of GABAA receptor subunit genes (e.g., GABRA5, GABRB3, GABRG3) implicated in inhibitory neurotransmission and linked to EEG beta oscillation signatures and seizures. (saravanapandian2020propertiesofbeta pages 1-2, saravanapandian2024sleepeegsignatures pages 2-4) - Non-imprinted genes in BP1–BP2 often highlighted in clinical CNV interpretation: NIPA1, NIPA2, CYFIP1, TUBGCP5. (bisba2024chromosome15q11q13duplication pages 3-5)
4.3 Parent-of-origin and penetrance/expressivity
- Bisba et al. interpret inherited duplications from apparently unaffected parents as evidence of reduced penetrance and variable expressivity. (bisba2024chromosome15q11q13duplication pages 5-7)
- In their literature synthesis of inherited cases, 80.20% of carriers inherited the duplicated region from one parent, with 62.96% maternal and 37.04% paternal among inherited cases. (bisba2024chromosome15q11q13duplication pages 5-7)
4.4 Variant classification and population frequencies
- At the population-screening level, 23 duplications in 333,187 NIPS profiles were found (incidence 0.0069%) and confirmed in maternal DNA where follow-up existed, with positive predictive value 100%: “Hence, the positive predictive value to detect this CNV by NIPS is 100%.” (parijs2024populationscreeningfor pages 3-5)
- Precise allele frequencies by duplication class and size were not derived beyond this incidence estimate in the evidence set.
4.5 Epigenetic information
Genomic imprinting (parent-of-origin gene expression) is an epigenetic mechanism central to the 15q11–q13 locus; this is explicitly emphasized in 2024 work addressing structural variants overlapping the imprinting region and in population-screening context. (mim2024expandingdeepphenotypic pages 1-2, parijs2024populationscreeningfor pages 1-2)
5. Environmental information
No specific environmental toxins, lifestyle factors, or infectious triggers were identified in the retrieved evidence set as contributors to dup15q clinical expression.
6. Mechanism / pathophysiology
6.1 Causal chain (high-level)
- Copy-number gain of 15q11.2–q13.1 occurs via NAHR at BP1–BP5. (bisba2024chromosome15q11q13duplication media 4f90b336)
- Gene-dosage imbalance results, including increased dosage of maternally expressed UBE3A (in maternal duplications) and increased dosage of GABAA receptor subunit genes. (elamin2023theroleof pages 1-2, saravanapandian2020propertiesofbeta pages 1-2)
- Circuit-level consequences include altered excitation/inhibition dynamics, reflected in a robust EEG phenotype with excess beta oscillations (12–30 Hz). (saravanapandian2020propertiesofbeta pages 1-2, saravanapandian2024sleepeegsignatures pages 2-4)
- Downstream manifestations include ASD/ID, epilepsy, hypotonia, sleep disruption, and other neurodevelopmental impairments. (bisba2024chromosome15q11q13duplication pages 1-2, saravanapandian2021abnormalsleepphysiology pages 1-2)
6.2 Human cellular evidence (patient-derived neurons; 2023)
In patient-derived neurons with CRISPR-corrected isogenic controls, Dup15q was associated with neuronal hyperexcitability (increased excitatory synaptic event frequency/amplitude and increased action potential firing). Normalizing UBE3A levels (via antisense oligonucleotide approaches) generally prevented hyperexcitability; UBE3A overexpression recapitulated many phenotypes, supporting a causal role for UBE3A dosage while also leaving room for contributions from other duplicated genes. (elamin2023theroleof pages 1-2)
Statistics extracted from this mechanistic study’s clinical context: autism is reported in 77–100% and seizures in 63% of individuals with idic(15). (elamin2023theroleof pages 1-2)
6.3 Systems biomarkers: EEG beta phenotype and sleep physiology
A key translational biomarker is the beta-band EEG phenotype. - In a cohort study, beta power and beta peak frequency were highly stable across visits (ICC ~0.92–0.93) and comparable between research and clinical EEG (ICC 0.94), supporting use as a clinical trial biomarker. (saravanapandian2020propertiesofbeta pages 1-2) - Sleep physiology abnormalities in children with Dup15q include “elevated beta power, reduced spindle density, and reduced or absent SWS” in overnight EEG comparisons. (saravanapandian2021abnormalsleepphysiology pages 1-2)
6.4 Translational mouse models (2024)
A 2024 sleep-EEG study in chromosome-engineered mice found that maternal duplication mice mirrored the elevated beta oscillation phenotype observed clinically and concluded that this supports translational validity of the beta EEG biomarker for preclinical drug-target studies. (saravanapandian2024sleepeegsignatures pages 2-4)
6.5 Pathways and ontology suggestions
- Suggested GO Biological Process terms (conceptual mapping from evidence):
- GABAergic synaptic transmission (GO:0007268-related; mechanism implied by GABAA modulation–like EEG phenotype) (saravanapandian2020propertiesofbeta pages 1-2)
- Regulation of membrane potential / neuronal excitability (GO:0042391 family) (elamin2023theroleof pages 1-2)
- Synapse organization / synaptogenesis (GO:0050808-related; discussed with CYFIP1/NIPA genes) (bisba2024chromosome15q11q13duplication pages 5-7)
- Suggested Cell Ontology terms (CL; conceptual mapping):
- Cortical excitatory neuron (glutamatergic neuron) and GABAergic interneuron (inferred from E/I and GABAA involvement). (elamin2023theroleof pages 1-2, saravanapandian2020propertiesofbeta pages 1-2)
7. Anatomical structures affected
7.1 Primary systems
- Nervous system / brain (dominant clinical manifestations and EEG biomarkers). (saravanapandian2021abnormalsleepphysiology pages 1-2, saravanapandian2020propertiesofbeta pages 1-2)
- Occasional cardiac involvement (congenital heart defects noted in prenatal/postnatal summaries). (bisba2024chromosome15q11q13duplication pages 3-5)
7.2 Suggested UBERON terms
- Brain: UBERON:0000955
- Cerebral cortex: UBERON:0000956
- Heart: UBERON:0000948 (for CHD cases)
7.3 Subcellular / GO Cellular Component (conceptual)
UBE3A is an E3 ubiquitin ligase; mechanistic implications include altered protein turnover pathways, but specific subcellular compartments were not directly specified in the retrieved evidence. (elamin2023theroleof pages 1-2)
8. Temporal development
- Onset: congenital (genetic CNV); clinical recognition typically in early childhood when developmental delay/ASD/hypotonia become evident. (bisba2024chromosome15q11q13duplication pages 1-2)
- Progression/course: chronic/lifelong neurodevelopmental disorder with variable severity; epilepsy can be persistent and disabling in severe cases. (elamin2023theroleof pages 1-2)
9. Inheritance and population
9.1 Incidence and prevalence (recent data)
- Population incidence estimate from screening: In genome-wide NIPS profiles, 15q11-q13 duplications were detected in 23 of 333,187 pregnancies (0.0069%). (parijs2024populationscreeningfor pages 3-5, parijs2024populationscreeningfor pages 1-2)
- Prevalence estimate cited in a 2024 review: “affecting 1 in 30,000 to 1 in 60,000 children worldwide” (noting this is a cited estimate within the review, not newly measured there). (bisba2024chromosome15q11q13duplication pages 5-7)
9.2 Inheritance pattern and counseling implications
- The disorder is often described as autosomal dominant in CNV inheritance terms, but with reduced penetrance/variable expressivity (especially paternal or smaller duplications) and a strong parent-of-origin effect. (bisba2024chromosome15q11q13duplication pages 1-2, parijs2024populationscreeningfor pages 3-5)
- In Bisba’s literature synthesis, among postnatal cases with known inheritance patterns, the totals were 48 maternal, 29 paternal, 17 de novo, 8 unknown. (bisba2024chromosome15q11q13duplication pages 5-7)
10. Diagnostics
10.1 Recommended / real-world testing approaches reflected in 2023–2024 evidence
- Chromosomal microarray (CMA / array-CGH) is used as a primary diagnostic modality in clinical series; Bisba et al. diagnosed cases using Affymetrix CytoScan 750K array-CGH. (bisba2024chromosome15q11q13duplication pages 1-2)
- Genome-wide NIPS (cfDNA) can detect maternal 15q11-q13 duplications at population scale; Parijs et al. report that in their dataset, duplications detected in cfDNA were confirmed in maternal DNA (when follow-up existed) and report PPV 100%. (parijs2024populationscreeningfor pages 3-5)
- MLPA and karyotyping are used to confirm interstitial duplications and characterize structure; a family series recommends MLPA as cost/time effective “in cases of Dup15q suspicion.” (levandivska2023inherited15qduplication pages 1-2)
10.2 Differential diagnosis (conceptual, based on locus overlap)
Given locus complexity and overlap with imprinting disorders: - Prader–Willi syndrome and Angelman syndrome (loss of paternal vs maternal expression within 15q11–q13) are key differentials in the same region. (parijs2024populationscreeningfor pages 1-2, mim2024expandingdeepphenotypic pages 1-2)
10.3 Ontology suggestions
- Diagnostic procedure (MAXO-like mapping for actions): chromosomal microarray analysis, genetic counseling, prenatal cfDNA screening.
Not retrieved in current evidence set: explicit ACMG/ClinGen CNV interpretation criteria text, and GTR test listings.
11. Outcome / prognosis
Robust survival and mortality statistics were not retrieved in the evidence set. Clinical burden is driven by neurodevelopmental disability and epilepsy severity; the 2024 population-screening paper emphasizes counseling complexity due to variable phenotype even within families. (parijs2024populationscreeningfor pages 3-5)
12. Treatment
12.1 Standard-of-care (supportive; evidence limitations)
The retrieved 2023–2024 sources emphasize symptom domains (ASD, epilepsy, sleep disturbance) but do not provide comprehensive, guideline-grade management algorithms. Supportive neurodevelopmental interventions and seizure management are implied as key care components, and the field is increasingly focused on objective biomarkers (EEG beta) to support trials. (saravanapandian2021abnormalsleepphysiology pages 1-2, potter2024linkingangelmanand pages 3-5)
12.2 Clinical trials and emerging therapeutics (real-world implementation)
Several interventional studies on ClinicalTrials.gov illustrate active drug-repurposing/targeted strategies:
- Soticlestat (TAK-935/OV935) – Phase 2, open-label signal-finding trial in Dup15q or CDKL5 deficiency disorder (ARCADE).
- NCT 03694275, enrollment 20 (actual); evaluates percent change in motor seizure frequency during maintenance weeks 9–20. (NCT03694275 chunk 1)
- Trial record includes a linked publication (PMID 37011526) in Epilepsy & Behavior (2023) referenced in the registry entry. (NCT03694275 chunk 2)
-
MAXO suggestion: antiseizure therapy; cholesterol 24S-hydroxylase inhibitor therapy (mechanism per keyword list). (NCT03694275 chunk 1)
-
Basmisanil (GABAA receptor subtype negative allosteric modulation hypothesis)
- NCT 05307679, Phase 2 randomized double-blind placebo-controlled; enrollment 7 (actual); terminated “due to sponsor decision and not related to safety or tolerability.” (NCT05307679 chunk 1)
-
MAXO suggestion: GABA receptor modulator therapy; clinical trial participation. (NCT05307679 chunk 1)
-
Retinoic acid / all-trans retinoic acid (ATRA) strategies (UBE3A-related mechanism suggested in trial descriptions)
- NCT 05281965: randomized crossover early phase 1 study of retinoic acid in dup15q; estimated enrollment 20; ages 6–18 years; outcomes include ADOS-based social reciprocity scores. (NCT05281965 chunk 1)
- NCT 07079696: Phase 2 single-group ATRA study; estimated enrollment 90; ages 3–7 years; includes EEG/fMRI and proteomics sampling in protocol summary. (NCT07079696 chunk 1)
- MAXO suggestion: retinoid therapy; behavioral assessment; EEG biomarker monitoring. (NCT05281965 chunk 1, NCT07079696 chunk 1)
Adverse events / response rates: not extractable from the evidence set here (except termination rationale for basmisanil). (NCT05307679 chunk 1)
13. Prevention
No primary prevention is available for a germline CNV disorder aside from reproductive options and counseling. - Secondary prevention / early detection: genome-wide NIPS can detect maternal duplications and prompts confirmatory testing and counseling. Parijs et al. discuss reporting and counseling guidance, noting that “Following these guidelines, 15q11-q13 duplications should be reported as maternal secondary findings” with invasive testing and genetic counseling recommended. (parijs2024populationscreeningfor pages 3-5)
MAXO suggestion: genetic counseling; prenatal genetic screening. (parijs2024populationscreeningfor pages 3-5)
14. Other species / natural disease
No naturally occurring non-human disease equivalent was retrieved in the evidence set.
15. Model organisms and experimental models
15.1 Mouse models (2024)
Sleep-EEG phenotyping was performed in chromosome-engineered mice modeling maternal vs paternal inheritance and in Ube3a overexpression mice. The study supports translational validity of the beta oscillation biomarker and notes nuanced divergence from human NREM abnormalities. (saravanapandian2024sleepeegsignatures pages 2-4)
15.2 Patient-derived cellular models (2023)
CRISPR-corrected patient-derived neurons provide an isogenic system to attribute electrophysiological phenotypes to dosage (especially UBE3A), supporting preclinical target validation approaches (ASO-based normalization, gene-editing controls). (elamin2023theroleof pages 1-2)
15.3 Model limitations
Mouse studies reported preserved NREM sleep and recovery post-deprivation, contrasting with human sleep abnormalities; this highlights species differences and the need for multi-model triangulation. (saravanapandian2024sleepeegsignatures pages 2-4, saravanapandian2021abnormalsleepphysiology pages 1-2)
Visual evidence (breakpoints and phenotype frequency)
Bisba et al. include (i) a breakpoint schematic (BP1–BP5) and (ii) tables summarizing phenotype frequencies and duplication spans; these were retrieved as cropped images and support the breakpoint architecture and the frequency summaries cited above. (bisba2024chromosome15q11q13duplication media 4f90b336, bisba2024chromosome15q11q13duplication media 5c40aa4c, bisba2024chromosome15q11q13duplication media a29eeba0, bisba2024chromosome15q11q13duplication media d7b953db)
Evidence gaps and limitations (for knowledge-base curation)
- Curated database IDs (Orphanet/MeSH/ICD) were not available in the retrieved evidence set and would require direct queries to those resources.
- Penetrance and true population prevalence remain challenging because clinical cohorts are biased and even population screening is limited by inclusion (pregnant women only) and incomplete fetal follow-up. (parijs2024populationscreeningfor pages 3-5)
- Standard-of-care guidelines and systematic reviews of therapies specific to dup15q were not retrieved; the most actionable treatment evidence in this set comes from clinical trial registries and biomarker work.
URLs and publication dates (key 2023–2024 sources)
- Bisba et al. “Chromosome 15q11-q13 Duplication Syndrome: A Review of the Literature and 14 New Cases.” Oct 2024. https://doi.org/10.3390/genes15101304 (bisba2024chromosome15q11q13duplication pages 1-2)
- Parijs et al. “Population screening for 15q11-q13 duplications…” Apr 2024. https://doi.org/10.1038/s41431-023-01336-6 (parijs2024populationscreeningfor pages 1-2)
- Saravanapandian et al. “Sleep EEG signatures in mouse models of 15q11.2-13.1 duplication (Dup15q) syndrome.” Jul 2024. https://doi.org/10.1186/s11689-024-09556-7 (saravanapandian2024sleepeegsignatures pages 2-4)
- Elamin et al. “The role of UBE3A … using patient-derived, CRISPR-corrected neurons.” Apr 2023. https://doi.org/10.1016/j.stemcr.2023.02.002 (elamin2023theroleof pages 1-2)
- Potter et al. “Linking Angelman and dup15q data for expanded research (LADDER) database…” Jan 2024. https://doi.org/10.1177/26330040241254122 (potter2024linkingangelmanand pages 3-5)
Appendix: key 2023–2024 expert interpretation statements (direct excerpts)
- Population screening interpretation: “the incidence of the 15q11-q13 duplications is 0.0069%.” and “Hence, the positive predictive value to detect this CNV by NIPS is 100%.” (parijs2024populationscreeningfor pages 3-5)
- Parent-of-origin impact: “maternal duplications are invariably associated with a clinical phenotype … [while] the majority of paternal duplication carriers are phenotypically normal” (parijs2024populationscreeningfor pages 3-5)
- Sleep biomarkers in children: “Children with Dup15q syndrome showed abnormal sleep physiology with elevated beta power, reduced spindle density, and reduced or absent SWS” (saravanapandian2021abnormalsleepphysiology pages 1-2)
References
-
(parijs2024populationscreeningfor pages 1-2): Ilse Parijs, Nathalie Brison, Leen Vancoillie, Machteld Baetens, Bettina Blaumeiser, Sébastien Boulanger, Julie Désir, Boyan Dimitrov, Nathalie Fieremans, Katrien Janssens, Sandra Janssens, Axel Marichal, Björn Menten, Colombine Meunier, Kim Van Berkel, Ann Van Den Bogaert, Koenraad Devriendt, Kris Van Den Bogaert, and Joris Robert Vermeesch. Population screening for 15q11-q13 duplications: corroboration of the difference in impact between maternally and paternally inherited alleles. European Journal of Human Genetics, 32:31-36, Apr 2024. URL: https://doi.org/10.1038/s41431-023-01336-6, doi:10.1038/s41431-023-01336-6. This article has 9 citations and is from a domain leading peer-reviewed journal.
-
(bisba2024chromosome15q11q13duplication pages 1-2): Maria Bisba, Christina Malamaki, Pantelis Constantoulakis, and Spiros Vittas. Chromosome 15q11-q13 duplication syndrome: a review of the literature and 14 new cases. Genes, 15:1304, Oct 2024. URL: https://doi.org/10.3390/genes15101304, doi:10.3390/genes15101304. This article has 21 citations.
-
(saravanapandian2024sleepeegsignatures pages 2-4): Vidya Saravanapandian, Melika Madani, India Nichols, Scott Vincent, Mary Dover, Dante Dikeman, Benjamin D. Philpot, Toru Takumi, Christopher S. Colwell, Shafali Jeste, Ketema N. Paul, and Peyman Golshani. Sleep eeg signatures in mouse models of 15q11.2-13.1 duplication (dup15q) syndrome. Journal of Neurodevelopmental Disorders, Jul 2024. URL: https://doi.org/10.1186/s11689-024-09556-7, doi:10.1186/s11689-024-09556-7. This article has 1 citations and is from a peer-reviewed journal.
-
(elamin2023theroleof pages 1-2): Marwa Elamin, Aurelie Dumarchey, Christopher Stoddard, Tiwanna M. Robinson, Christopher Cowie, Dea Gorka, Stormy J. Chamberlain, and Eric S. Levine. The role of ube3a in the autism and epilepsy-related dup15q syndrome using patient-derived, crispr-corrected neurons. Stem Cell Reports, 18:884-898, Apr 2023. URL: https://doi.org/10.1016/j.stemcr.2023.02.002, doi:10.1016/j.stemcr.2023.02.002. This article has 25 citations and is from a domain leading peer-reviewed journal.
-
(bisba2024chromosome15q11q13duplication pages 5-7): Maria Bisba, Christina Malamaki, Pantelis Constantoulakis, and Spiros Vittas. Chromosome 15q11-q13 duplication syndrome: a review of the literature and 14 new cases. Genes, 15:1304, Oct 2024. URL: https://doi.org/10.3390/genes15101304, doi:10.3390/genes15101304. This article has 21 citations.
-
(parijs2024populationscreeningfor pages 3-5): Ilse Parijs, Nathalie Brison, Leen Vancoillie, Machteld Baetens, Bettina Blaumeiser, Sébastien Boulanger, Julie Désir, Boyan Dimitrov, Nathalie Fieremans, Katrien Janssens, Sandra Janssens, Axel Marichal, Björn Menten, Colombine Meunier, Kim Van Berkel, Ann Van Den Bogaert, Koenraad Devriendt, Kris Van Den Bogaert, and Joris Robert Vermeesch. Population screening for 15q11-q13 duplications: corroboration of the difference in impact between maternally and paternally inherited alleles. European Journal of Human Genetics, 32:31-36, Apr 2024. URL: https://doi.org/10.1038/s41431-023-01336-6, doi:10.1038/s41431-023-01336-6. This article has 9 citations and is from a domain leading peer-reviewed journal.
-
(bisba2024chromosome15q11q13duplication pages 3-5): Maria Bisba, Christina Malamaki, Pantelis Constantoulakis, and Spiros Vittas. Chromosome 15q11-q13 duplication syndrome: a review of the literature and 14 new cases. Genes, 15:1304, Oct 2024. URL: https://doi.org/10.3390/genes15101304, doi:10.3390/genes15101304. This article has 21 citations.
-
(saravanapandian2020propertiesofbeta pages 1-2): Vidya Saravanapandian, Joel Frohlich, Joerg F. Hipp, Carly Hyde, Aaron W. Scheffler, Peyman Golshani, Edwin H. Cook, Lawrence T. Reiter, Damla Senturk, and Shafali S. Jeste. Properties of beta oscillations in dup15q syndrome. Journal of Neurodevelopmental Disorders, Aug 2020. URL: https://doi.org/10.1186/s11689-020-09326-1, doi:10.1186/s11689-020-09326-1. This article has 25 citations and is from a peer-reviewed journal.
-
(saravanapandian2021abnormalsleepphysiology pages 1-2): Vidya Saravanapandian, Divya Nadkarni, Sheng-Hsiou Hsu, Shaun A. Hussain, Kiran Maski, Peyman Golshani, Christopher S. Colwell, Saravanavel Balasubramanian, Amos Dixon, Daniel H. Geschwind, and Shafali S. Jeste. Abnormal sleep physiology in children with 15q11.2-13.1 duplication (dup15q) syndrome. Molecular Autism, Aug 2021. URL: https://doi.org/10.1186/s13229-021-00460-8, doi:10.1186/s13229-021-00460-8. This article has 26 citations and is from a peer-reviewed journal.
-
(levandivska2023inherited15qduplication pages 1-2): S. H. Levandivska, M. I. Dushar, O. V. Tyshchenko, N. L. Huleyuk, E. Y. Patskun, and H. V. Makukh. Inherited 15q duplication in three not related ukrainian families. Medical Science of Ukraine (MSU), 19:58-65, Jun 2023. URL: https://doi.org/10.32345/2664-4738.2.2023.08, doi:10.32345/2664-4738.2.2023.08. This article has 0 citations.
-
(potter2024linkingangelmanand pages 3-5): Sarah Nelson Potter, Elizabeth Reynolds, Katherine C. Okoniewski, Anne Edwards, Julia Gable, Christine Hill, Vesselina Bakalov, Stephanie Zentz, Carolyne Whiting, Emily Cheves, Katie Garbarini, Elizabeth Jalazo, Carrie Howell, Amanda Moore, and Anne Wheeler. Linking angelman and dup15q data for expanded research (ladder) database: a model for advancing research, clinical guidance, and therapeutic development for rare conditions. Therapeutic Advances in Rare Disease, Jan 2024. URL: https://doi.org/10.1177/26330040241254122, doi:10.1177/26330040241254122. This article has 4 citations.
-
(NCT05281965 chunk 1): A Clinical Study Evaluating the Efficacy and Safety of Retinoic Acid in Patients With 15q11-q13 Duplication Syndrome. Second Affiliated Hospital, School of Medicine, Zhejiang University. 2022. ClinicalTrials.gov Identifier: NCT05281965
-
(NCT05307679 chunk 1): A Study to Evaluate the Safety and Efficacy of Basmisanil Treatment in Children Aged 2-14 Years With Dup15q Syndrome. Hoffmann-La Roche. 2022. ClinicalTrials.gov Identifier: NCT05307679
-
(NCT03694275 chunk 1): A Multicenter, Open-label, Pilot Study of Soticlestat (TAK-935/OV935) in Participants With 15Q Duplication Syndrome (Dup 15q) or Cyclin-Dependent Kinase-Like 5 (CDKL5) Deficiency Disorder (ARCADE STUDY). Takeda. 2018. ClinicalTrials.gov Identifier: NCT03694275
-
(NCT03694275 chunk 2): A Multicenter, Open-label, Pilot Study of Soticlestat (TAK-935/OV935) in Participants With 15Q Duplication Syndrome (Dup 15q) or Cyclin-Dependent Kinase-Like 5 (CDKL5) Deficiency Disorder (ARCADE STUDY). Takeda. 2018. ClinicalTrials.gov Identifier: NCT03694275
-
(NCT07079696 chunk 1): Investigating the Therapeutic Efficacy of All-trans Retinoic Acid in Autism Spectrum Disorder Patients With 15q11-13 Duplication Syndrome. Second Affiliated Hospital, School of Medicine, Zhejiang University. 2025. ClinicalTrials.gov Identifier: NCT07079696
-
(OpenTargets Search: dup15q syndrome,15q11-q13 duplication syndrome,15q11q13 microduplication syndrome): Open Targets Query (dup15q syndrome,15q11-q13 duplication syndrome,15q11q13 microduplication syndrome, 15 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(bisba2024chromosome15q11q13duplication media 4f90b336): Maria Bisba, Christina Malamaki, Pantelis Constantoulakis, and Spiros Vittas. Chromosome 15q11-q13 duplication syndrome: a review of the literature and 14 new cases. Genes, 15:1304, Oct 2024. URL: https://doi.org/10.3390/genes15101304, doi:10.3390/genes15101304. This article has 21 citations.
-
(mim2024expandingdeepphenotypic pages 1-2): Rabeya Akter Mim, Anjana Soorajkumar, Noor Kosaji, Muhammad Mizanur Rahman, Shaoli Sarker, Noushad Karuvantevida, Tamannyat Binte Eshaque, Md Atikur Rahaman, Amirul Islam, Mohammod Shah Jahan Chowdhury, Nusrat Shams, K. M. Furkan Uddin, Hosneara Akter, and Mohammed Uddin. Expanding deep phenotypic spectrum associated with atypical pathogenic structural variations overlapping 15q11–q13 imprinting region. Brain and Behavior, Apr 2024. URL: https://doi.org/10.1002/brb3.3437, doi:10.1002/brb3.3437. This article has 9 citations and is from a peer-reviewed journal.
-
(bisba2024chromosome15q11q13duplication media 5c40aa4c): Maria Bisba, Christina Malamaki, Pantelis Constantoulakis, and Spiros Vittas. Chromosome 15q11-q13 duplication syndrome: a review of the literature and 14 new cases. Genes, 15:1304, Oct 2024. URL: https://doi.org/10.3390/genes15101304, doi:10.3390/genes15101304. This article has 21 citations.
-
(bisba2024chromosome15q11q13duplication media a29eeba0): Maria Bisba, Christina Malamaki, Pantelis Constantoulakis, and Spiros Vittas. Chromosome 15q11-q13 duplication syndrome: a review of the literature and 14 new cases. Genes, 15:1304, Oct 2024. URL: https://doi.org/10.3390/genes15101304, doi:10.3390/genes15101304. This article has 21 citations.
-
(bisba2024chromosome15q11q13duplication media d7b953db): Maria Bisba, Christina Malamaki, Pantelis Constantoulakis, and Spiros Vittas. Chromosome 15q11-q13 duplication syndrome: a review of the literature and 14 new cases. Genes, 15:1304, Oct 2024. URL: https://doi.org/10.3390/genes15101304, doi:10.3390/genes15101304. This article has 21 citations.